
Figure 1.
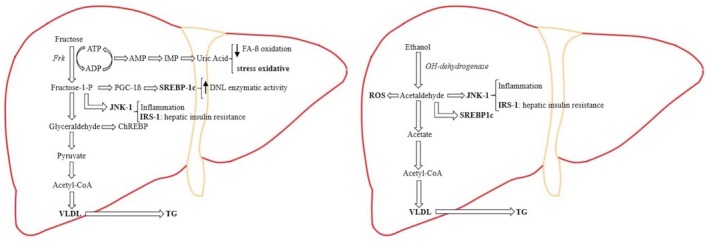
In hepatic fructose metabolism, the major part of fructose is metabolized directly to pyruvate. Due to the liver mitochondria cannot metabolizing the substrate excess, there is an overproduction of DNL and VLDL (very‐low‐density lipoprotein), which is exported out of the liver to contribute to fructose‐induced hypertriglyceridemia. Fructose induces substrate‐dependent phosphate depletion, which increases uric acid contributing to the decrease of FA ß‐oxidation, leading to intrahepatic lipid accumulation, and it is also primarily responsible for the oxidative stress. Fructose also stimulates the overexpression of key transcription factors, which lead to up‐regulation of DNL as ChREBP (carbohydrate response element binding protein) and PGC‐1ß (peroxisomal proliferator‐activated receptor‐γ coactivator‐1ß). PGC‐1ß is a transcriptional coactivator for SREBP‐1c (sterol regulatory element binding protein‐1c), which accentuates DNL enzymatic activity, and JNK‐1 (c‐jun N‐terminal kinase), which, once is induced, begins the inflammation cascade. As part of its inflammatory action, JNK‐1 activation induces IRS‐1 (insulin receptor substrate‐1), which promotes hepatic insulin resistance. In hepatic alcohol metabolism, ethanol is metabolized to acetaldehyde, which, because of its free aldehyde, can generate ROS (reactive oxygen species) formation and toxic damage. Furthermore, acetaldehyde stimulates SREBP‐1c, activating the enzymes of DNL and JNK‐1, promoting the inflammation process and hepatic insulin resistance by inducing IRS‐1. In the ethanol metabolization process there is an excess of the product intermediaries, which stimulates an overproduction of DNL, resulting in further intrahepatic lipid build‐up. By suppressing the activity of the protein that mediates the VLDL production, ethanol intake contributes to hypertriglyceridemia.