

Abstract
In response to genotoxic stress, the tumor suppressor protein p73 induces apoptosis and cell cycle arrest. Despite extensive studies on p73-mediated apoptosis, little is known about the cytoplasmic apoptotic function of p73. Here, using H1299 lung cancer cells and diverse biochemical approaches, including colony formation, DNA fragmentation, GST pulldown, and apoptosis assays along with NMR spectroscopy, we show that p73 induces transcription-independent apoptosis via its transactivation domain (TAD) through a mitochondrial pathway and that this apoptosis is mediated by the interaction between p73-TAD and the anti-apoptotic protein B-cell lymphoma-extra large (Bcl-XL or BCL2L1). This binding disrupted an interaction between Bcl-XL and the pro-apoptotic protein BH3-interacting domain death agonist (Bid). In particular, we found that a 16-mer p73-TAD peptide motif (p73-TAD16) mediates transcription-independent apoptosis, accompanied by cytochrome c release from the mitochondria, by interacting with Bcl-XL. Interestingly, the structure of the Bcl-XL–p73-TAD16 peptide complex revealed a novel mechanism of Bcl-XL recognition by p73-TAD. We observed that the α-helical p73-TAD16 peptide binds to a noncanonical site in Bcl-XL, comprising the BH1, BH2, and BH3 domains in an orientation opposite to those of pro-apoptotic BH3 peptides. Taken together, our results indicate that the cytoplasmic apoptotic function of p73 is mediated through a noncanonical mode of Bcl-XL recognition. This finding sheds light on a critical transcription-independent, p73-mediated mechanism for apoptosis induction, which has potential implications for anticancer therapy.
Keywords: apoptosis, protein-protein interaction, protein structure, p73, tumor suppressor gene, cancer, Bcl-XL, BCL2L1, BH3-interacting domain death agonist, transactivation domain
Introduction
p73 is a member of the p53 family of transcription factors that plays key roles in tumor suppression (1–4). Apart from its unique function in neuronal development and differentiation (1, 5), p73 induces apoptosis and cell cycle arrest through the transactivation of p53-responsive genes under conditions of genotoxic stress (1–4). These p53-like activities of p73 are based on the presence of highly homologous domains including transactivation domain (TAD),5 proline-rich domain, DNA-binding domain, and oligomerization domain (Fig. S1A). p73 exists in multiple isoforms, including a pro-apoptotic, transcriptionally active p73 isoform with TAD and an anti-apoptotic, delta-N p73 isoform lacking TAD (3).
p73-mediated apoptosis was attributed to its nuclear transactivation of pro-apoptotic target genes, such as PUMA, Bax, and death receptor CD95 (6, 7). However, p73 is also found to localize to the cytoplasm (8) or the mitochondria (9–11) during apoptosis, suggesting a transcription-independent pro-apoptotic function of p73. The tumor suppressor Wwox caused the redistribution of p73 from the nucleus to cytoplasm, resulting in its pro-apoptotic activity through interaction between them (8). During TRAIL-induced apoptosis, p73 and its fragments, generated by caspase-3 and caspase-8 cleavage, were localized to the mitochondria, and the transcription-inactive TAp73 mutant enhanced TRAIL-induced apoptosis (11). In addition, the scaffolding protein RanBP9 was reported to physically interact with p73 and to increase mitochondrial p73 levels at both the transcriptional and post-translational modification levels, resulting in the cooperative induction of apoptosis at the mitochondria (9). Although evidence for the involvement of p73 in transcription-independent apoptosis has been accumulated (4), a direct apoptogenic role of p73 has not been elucidated.
In recent years, p53 has been shown to directly induce mitochondrial apoptosis in the absence of its transcriptional activity (12–15). In response to apoptotic stimuli, p53 translocates to the mitochondria and activates the apoptotic effectors Bak or Bax, triggering the apoptotic signaling process via mitochondrial outer membrane permeabilization and cytochrome c release from the mitochondria. This transcription-independent apoptosis is inhibited by the sequestration of cytoplasmic p53 by anti-apoptotic Bcl-2 family member Bcl-XL (15, 16). Recently, the p53 DNA-binding domain was shown to bind to Bcl-XL via a binding surface other than the B-cell lymphoma-2 homology 3 (BH3) peptide-binding groove (17). Consistent with that only the p53 N-terminal domain (18) and the caspase-cleaved N-terminal fragment of p53 (residues 1–186) induced transcription-independent apoptosis (19), our previous studies also demonstrated that the p53-TAD interacts with diverse anti-apoptotic Bcl-2 family proteins via the p53-TAD1 or p53-TAD2 motifs (20–22). Although structural modeling studies of p53-TAD in complex with anti-apoptotic Bcl-2 family proteins were previously conducted (20–22), the exact structural information of the complexes remains unknown.
The Bcl-2 protein family is a critical regulator of the mitochondrial apoptosis pathway by controlling the permeability of the outer mitochondrial membrane and cytochrome c release (23, 24). Anti-apoptotic Bcl-2 family proteins play their anti-apoptotic role by sequestering pro-apoptotic BH3-only proteins such as PUMA, Bid, and Bim, which can activate the effectors Bak and Bax. This apoptotic regulation is mediated by the formation of inhibitory complexes between anti-apoptotic Bcl-2 family proteins and the BH3 domains of pro-apoptotic BH3-only proteins. Because blocking the interaction between anti-apoptotic and pro-apoptotic Bcl-2 family proteins is an attractive strategy for anticancer therapy (25), the structures of the pro-apoptotic BH3 peptides bound to anti-apoptotic Bcl-2 family proteins have served as important templates for designing novel anticancer therapeutic agents such as ABT-737 (26).
In this study, we showed that p73 induces transcription-independent apoptosis via its TAD by a mitochondrial pathway and that this apoptotic activity is modulated by anti-apoptotic Bcl-XL. We also demonstrated that a 16-mer p73-TAD peptide (referred to as p73-TAD16) mediates transcription-independent apoptosis through direct interaction with Bcl-XL. Using NMR spectroscopy, we determined the solution structure of the Bcl-XL–p73-TAD16 peptide complex. Interestingly, the complex structure exhibited a novel binding mode of Bcl-XL, which differs from a canonical binding mode between Bcl-XL and the pro-apoptotic BH3 peptides. Thus, our results provide a molecular basis for the transcription-independent p73-mediated apoptosis through Bcl-XL.
Results
p73-TAD induces transcription-independent apoptosis via a mitochondrial pathway
Previously, p53-TAD was found to be involved in the transcription-independent apoptosis of cytoplasmic p53 (18, 19), which prompted us to test whether p73 can directly induce transcription-independent apoptosis via its TAD. We carried out a cell viability assay using H1299 lung cancer cells (27, 28). When H1299 cells were transfected with full-length p73 (TAp73) and p73-TAD (residues 1–67), only ∼50% of the cells expressing TAp73 or p73-TAD were viable compared with cells transfected with a control vector (Fig. 1a). The effects of TAp73 or p73-TAD on cell viability were further confirmed by a colony-formation assay. The overexpression of p73-TAD in H1299 cells suppressed colony formation, albeit to a lesser extent than TAp73 (Fig. S2A). As shown in the DNA fragmentation assay data (Fig. S2B), analysis on the sub-G1 population of H1299 cells transfected with either TAp73 or p73-TAD, which is indicative of apoptotic cells, showed that both constructs induced ∼3-fold more apoptotic cell death than the control vector. In addition, results from the terminal deoxynucleotidyltransferase-mediated nick-end labeling (TUNEL) assay (Fig. 1b), annexin V-APC staining by FACS analysis (Fig. 1c), and caspase-3/7 activity assay (Fig. 1d) confirmed that the cell death induced by TAp73 and p73-TAD is caspase-3/7–dependent apoptosis.
Figure 1.

p73-TAD induces mitochondrial apoptosis in a transcription-independent manner. a, cell viability assay with TAp73 and p73-TAD. H1299 cells were transiently transfected with peGFP-C3-TAp73, peGFP-C3-p73-TAD, TAp73(A156V), or a control vector, and viable cells were measured using a WST-1 assay (n = 6; p < 0.0001, compared with control). b, TUNEL assay with TAp73 and p73-TAD. H1299 cells transfected with the indicated plasmids were stained by TUNEL, and images of TUNEL-positive cells (red fluorescence) were captured by a confocal microscope. c, annexin V–APC apoptosis assay with TAp73 and p73-TAD. Apoptosis of H1299 cells transfected with the indicated plasmids was assessed by annexin V–APC and 7-AAD staining, followed by flow cytometry analysis. Percentages indicate the portion of cells in each quadrant that defines annexin V–APC– or 7-AAD–positive or –negative cells. d, caspase-3/7 activity measurements with TAp73 and p73-TAD. Caspase-3/7 activities were measured in H1299 cells transfected with peGFP–C3–TAp73, peGFP–C3–p73-TAD, or a control vector (n = 3; p < 0.0005, compared with control). e, caspase-3/7 activity measurements with a transcriptionally inactive mutant, TAp73(A156V) (n = 6; p < 0.0001, compared with control). f, cytochrome c release assay of TAp73 and p73-TAD. H1299 cells were transfected with the indicated plasmids, and the cytosolic and mitochondrial fractions from cell lysates were immunoblotted with either anti-cytochrome c or anti-GFP antibodies. GAPDH and VDAC1 were used as cytosol and mitochondrial fraction markers, respectively. The graphs in the lower panel represent relative band intensities. Each band intensity was normalized with respect to the GAPDH or VDAC1 band intensity in the same blot (n = 4; p < 0.05, compared with control). All data are represented as means ± S.D., and statistics were performed using one-way ANOVA test.
To exclude the effect of transcription-dependent apoptosis induction via the transactivation of TAp73, we performed cell viability assay and caspase-3/7 activity assays with a transcriptionally inactive TAp73 mutant, TAp73(A156V) (29). The TAp73(A156V) mutant was competent to induce cell death but to a slightly lesser extent than that by TAp73 (Fig. 1e and Fig. S2E), confirming that the cell death effects of p73 are largely independent of transcription. Analysis of cytochrome c release from the mitochondria demonstrated that the transcription-independent apoptosis mediated by TAp73 and p73-TAD occurs via a mitochondrial pathway (Fig. 1f). Taken together, our results showed that p73-TAD mediates transcription-independent apoptosis through a mitochondrial pathway.
p73-TAD physically associates with anti-apoptotic Bcl-XL
To test whether p73-TAD is involved in interactions with anti-apoptotic Bcl-2 family proteins, we performed a GST pulldown assay to probe the complex formation between p73-TAD and anti-apoptotic Bcl-2 family proteins (Fig. 2a). When HEK293T cells were co-transfected with GST-tagged p73-TAD along with anti-apoptotic Bcl-2 family proteins Bcl-XL, Bcl-2, or Bcl-w, the protein complexes were pulled down with GSH-agarose beads, indicating the complex formation between p73-TAD and these anti-apoptotic Bcl-2 family proteins. In addition, immunoprecipitation data showed that specific Bcl-XL–TAp73 and Bcl-XL–p73-TAD complexes were formed when H1299 cells were co-transfected with Bcl-XL and either TAp73 or p73-TAD (Fig. 2b). TAp73 overexpressed in the same cells was also found to bind to endogenous Bcl-XL (Fig. 2c). To further confirm whether such protein complexes are formed between endogenous proteins, we conducted immunoprecipitation experiments using the same cells treated with the DNA-damaging agent etoposide (Fig. 2d). Indeed, TAp73 formed specific endogenous interactions with Bcl-XL and Bcl-2. Conversely, endogenous TAp73 was not associated with the pro-apoptotic BH3-only proteins Bad, PUMA, Bik, Bid, and Bim (Fig. S3).
Figure 2.

p73-TAD binds to anti-apoptotic Bcl-XL. a, the interactions of p73-TAD and Bcl-XL, Bcl-2, and Bcl-w as determined by a GST pulldown assay. Lysates from HEK293T cells transfected with GST-tagged p73-TAD, and the indicated FLAG-tagged plasmids were subjected to GST–p73-TAD pulldown followed by immunoblotting with an anti-FLAG antibody. b, the interaction between TAp73 or p73-TAD and Bcl-XL. H1299 cells transfected with the indicated plasmids were immunoprecipitated (IP) with an anti-FLAG antibody and probed with an anti-p73 antibody. c, the interaction between overexpressed TAp73 or p73-TAD and endogenous Bcl-XL. Lysates from GFP-TAp73 overexpressed H1299 cells were immunoprecipitated with an anti–Bcl-XL antibody and probed with an anti-GFP antibody. The same input sample was divided into two fractions, each of which was then used for immunoprecipitation in lanes 1 and 2 (IgG) or lanes 3 and 4 (Bcl-XL). d, the interaction between endogenous TAp73 and Bcl-XL or Bcl-2. H1299 cells were treated with 50 μm etoposide, and then the cell lysates were immunoprecipitated with an anti–Bcl-XL or anti-Bcl-2 antibody and probed with an anti-p73 antibody. e, p73-TAD liberates a pro-apoptotic BH3-only protein Bid from Bcl-XL. Immunoprecipitation experiments were performed to detect the competition of TAp73 with Bid for binding to endogenous Bcl-XL. HEK293T cells were transfected with pCMV–Myc–Bid and peGFP–C3–TAp73. The cell lysates were immunoprecipitated with an anti–Bcl-XL antibody and probed with the indicated antibodies. f, immunoprecipitation experiments were performed to detect the competition of p73-TAD with Bid for binding to endogenous Bcl-XL. HEK293T cells were transfected with pCMV–Myc–Bid and peGFP–C3–p73-TAD.
To investigate the effect of Bcl-XL overexpression on the transcription-independent p73-mediated apoptosis, we performed cell viability and DNA fragmentation assays using H1299 cells transfected with Bcl-XL and either TAp73 or p73-TAD. The cell viability assay and DNA fragmentation assay data showed that the transcription-independent apoptosis induced by TAp73 or p73-TAD was blocked by overexpression of Bcl-XL (Figs. S4A and S2C). In addition, the release of mitochondrial cytochrome c induced by TAp73 or p73-TAD was significantly prevented by excess Bcl-XL (Fig. S4C). Thus, our results indicated that Bcl-XL overexpression could rescue cells from apoptosis induced by TAp73 and p73-TAD.
p73-TAD liberates pro-apoptotic Bid from Bcl-XL
Based on the previous finding that Bcl-XL interacts directly with the pro-apoptotic BH3-only protein Bid (23, 30), we hypothesized that p73 may compete with Bid for Bcl-XL binding. To test this hypothesis, we performed competition experiments using immunoprecipitation with endogenous Bcl-XL, Myc-Bid, and GFP–TAp73 or GFP–p73-TAD (Fig. 2, e and f). Bid was associated with endogenous Bcl-XL, but not with TAp73 (Fig. S3). Notably, we found that both TAp73 and p73-TAD disrupted the Bcl-XL–Bid complex by associating with Bcl-XL, releasing Bid in a dose-dependent manner. Thus, our results suggest that p73-TAD triggers transcription-independent mitochondrial apoptosis by liberating the direct apoptotic activator Bid from Bcl-XL sequestration.
p73-TAD16 mediates transcription-independent apoptosis via Bcl-XL binding
Despite the low sequence identity among p53 family TADs, the FXXφWXXL motif (where φ indicates a bulky hydrophobic residue, and X indicates any residue, respectively) within these domains is highly conserved (Fig. S1B). Our recent studies showed that p53-TAD binds to anti-apoptotic Bcl-2 family proteins via the FXXφWXXL motif (20–22). To test whether the homologous motif in p73-TAD is responsible for Bcl-XL binding, we performed immunoprecipitation experiments with a GFP-tagged 16-mer p73-TAD peptide (residues 10–25, referred to as p73-TAD16) containing the FXXφWXXL motif. The results showed that GFP–p73-TAD16 bound specifically to Bcl-XL (Fig. 3a).
Figure 3.

p73-TAD16 mediates transcription-independent apoptosis via interaction with Bcl-XL. a, the physical association between p73-TAD16 and endogenous Bcl-XL determined by immunoprecipitation (IP) experiments. Cell lysates from H1299 cells transfected with peGFP–C3–p73-TAD16 were immunoprecipitated and immunoblotted with the indicated antibodies. The same input sample was divided into two fractions, each of which was then used for immunoprecipitation in lanes 1 and 2 (IgG) or lanes 3 and 4 (Bcl-XL). b, cell viability assay with p73-TAD16. c, caspase-3/7 activity measurement with p73-TAD16 (n = 3; p < 0.001, compared with control). d, cytochrome (Cyt) c release assay of H1299 cells transfected with peGFP–C3–p73-TAD16 (n = 3; p < 0.005, compared with control). e, caspase-3/7 activity was measured in H1299 cells transfected with peGFP–C3–TAp73(Δ1–25), peGFP–C3–p73-TAD(Δ1–25), peGFP–C3–TAp73, or peGFP–C3–p73–TAD (n = 3; p < 0.0005, compared with control in b and e). The data are represented as means ± S.D., and statistics were performed using one-way ANOVA test (b, c, and e) or Student's t test (d).
To examine whether the Bcl-XL–binding motif alone is competent to induce transcription-independent apoptosis, we performed cell viability and apoptosis assays with p73-TAD16. Cell viability, caspase-3/7 activity, and DNA fragmentation assays demonstrated that p73-TAD16 can trigger transcription-independent apoptosis at a comparable level to p73-TAD (Fig. 3, b and c) and that this apoptosis can be blocked by overexpression of Bcl-XL (Figs. S4B and S2D). In addition, p73-TAD16 induced the release of cytochrome c from the mitochondria (Fig. 3d), suggesting that it induces transcription-independent apoptosis via a mitochondrial pathway.
To further analyze whether binding of p73-TAD16 to Bcl-XL is required to mediate transcription-independent p73-induced apoptosis, we generated TAp73 and p73-TAD mutants lacking p73-TAD16 to disrupt the Bcl-XL binding of p73-TAD16 and tested the ability of the mutants to trigger apoptosis using a caspase-3/7 activity assay. The TAp73(Δ1–25) and p73-TAD(Δ1–25) mutants caused a substantial loss of apoptotic activity relative to WT TAp73 and p73-TAD (Fig. 3e). This indicated that the interaction between p73-TAD16 and Bcl-XL is critical for the induction of transcription-independent p73-mediated apoptosis. Collectively, these results suggested that p73-TAD16 mediates transcription-independent apoptosis via direct interaction with Bcl-XL.
Solution structure of the Bcl-XL/p73-TAD16 peptide complex
To characterize the molecular interaction between Bcl-XL and the p73-TAD16 peptide, we monitored the chemical shift perturbations of Bcl-XL titrated with the p73-TAD16 peptide using NMR spectroscopy (Fig. 4a and Fig. S5). The 2D 1H-15N HSQC spectra showed that a majority of the Bcl-XL cross-peaks moved significantly upon binding to the p73-TAD16 peptide (Fig. S6). The chemical shift perturbations of Bcl-XL induced by p73-TAD16 peptide binding were mapped primarily to the α2–α5 and α8 helices, corresponding to the BH1, BH2, and BH3 domains (Fig. 4a), indicating that this region is involved in binding to the p73-TAD16 peptide.
Figure 4.

Solution structure of the Bcl-XL–p73-TAD16 peptide complex. a, the overlaid 2D 1H-15N HSQC spectra for 15N-labeled Bcl-XL in the absence (black) or presence (red) of the p73-TAD16 peptide (left panel). The Bcl-XL residues showing chemical shift changes of ΔCS > 0.15 ppm and 0.1 ppm < ΔCS < 0.15 ppm are colored red and yellow, respectively (right panel). The α-helices and BH domains are indicated in the bottom and top of the graph, respectively. b, the ensemble of the final 20 lowest-energy NMR structures of the Bcl-XL–p73-TAD16 peptide complex (left) and a representative structure (right). The Bcl-XL and p73-TAD16 peptide are colored purple and light green, respectively. c, the helical wheel diagram of the p73-TAD16 peptide bound to Bcl-XL (left) and the corresponding region in the structure (right). The view is from the N terminus. The helical region of the p73-TAD16 peptide, encompassing residues Phe15–Glu23, is shown. The bulky hydrophobic residues in the p73-TAD16 peptide (Phe15, Leu18, Trp19, and Leu22) (orange) face the hydrophobic groove of Bcl-XL (purple), whereas the remaining hydrophilic residues (cyan) are exposed to the solvent. d, the binding interface between the Bcl-XL and p73-TAD16 peptide. The p73-TAD16 residues involved in the interactions with Bcl-XL are shown in green, and the Bcl-XL residues that are involved in the hydrophobic and electrostatic interactions with the p73-TAD16 peptide are shown in purple and blue, respectively (left panel). The two intermolecular π-stacking interactions between Phe97 and Tyr195 in Bcl-XL and Trp19 and Phe15 in the p73-TAD16 peptide, respectively, are shown (right panel). Ave., average.
To gain structural insights into the molecular interaction between Bcl-XL and the p73-TAD16 peptide, we determined the solution structure of the Bcl-XL–p73-TAD16 peptide complex using NMR spectroscopy (Fig. 4b). The structural statistics of an ensemble of the final 20 lowest-energy NMR structures are shown in Table 1. Intermolecular NOEs in the complex are listed in Table 2, and a strip of the 13C, 15N-filtered, edited NOESY spectra is shown in Fig. S7.
Table 1.
NMR and refinement statistics for the structure of Bcl-XL–p73-TAD16 peptide complex
| NMR distance and dihedral constraints | |
| Distance constraints | |
| Total NOE | 2165 |
| Intraresidue | 503 (121)a |
| Inter-residue | 1448 (93) |
| Sequential (|i − j| = 1) | 721 (64) |
| Medium-range (|i − j| < 4) | 484 (29) |
| Long-range (|i − j| > 5) | 218 (0) |
| Intermolecular | 25 |
| Hydrogen bonds | 57 (4) |
| Total dihedral angle restraints | |
| φ | 126 (9) |
| ψ | 128 (9) |
| Structure statistics | |
| Violations (means ± S.D.) | |
| Distance constraints (Å) | 0.054 ± 0.002 |
| Dihedral angle constraints (°) | 0.462 ± 0.192 |
| Maximum dihedral angle violation (°) | 4.37 |
| Maximum distance constraint violation (Å) | 0.48 |
| Deviations from idealized geometry | |
| Bond lengths (Å) | 0.014 ± 0.000 |
| Bond angles (°) | 0.966 ± 0.016 |
| Impropers (°) | 2.192 ± 0.072 |
| Average pairwise root-mean-square deviation (Å)b | |
| Heavy | 1.32 ± 0.20 |
| Backbone | 0.88 ± 0.12 |
a The numbers in parentheses are for p73-TAD16 peptide.
b Pairwise root-mean-square deviations were calculated for residues 3–19 and 44–194 of Bcl-XL and residues 15–23 of p73-TAD16 among 20 refined structures.
Table 2.
The intermolecular NOEs between Bcl-XL and p73-TAD16 peptide
| Bcl-XL | p73-TAD16 peptide | ||
|---|---|---|---|
| Phe97 | HB2 | Trp19 | HA |
| Phe97 | HB2 | Leu22 | HG |
| Val141 | HB | Trp19 | HH2 |
| Val141 | HB | Trp19 | HD1 |
| Val141 | HG1 | Trp19 | HD1 |
| Val141 | HG2 | Trp19 | HH2 |
| Val141 | HG2 | Trp19 | HD1 |
| Leu194 | HB2 | Trp19 | HD1 |
| Leu194 | HB2 | Leu18 | HD2 |
| Leu194 | HB2 | Leu18 | HD1 |
| Leu194 | HB2 | Thr14 | HA |
| Leu194 | HB2 | Thr14 | HG2 |
| Leu194 | HB3 | Thr14 | HA |
| Leu194 | HB3 | Thr14 | HG2 |
| Leu194 | HB3 | Trp19 | HD1 |
| Leu194 | HBa | Leu18 | HB |
| Tyr195 | HDa | Phe15 | HA |
| Tyr195 | HDa | Thr14 | HB |
| Tyr195 | HDa | Thr14 | HG2 |
| Tyr195 | HDa | Leu18 | HD1 |
| Tyr195 | HDa | Leu18 | HD2 |
| Tyr195 | HEa | Phe15 | HA |
| Tyr195 | HEa | Thr14 | HG2 |
| Tyr195 | HEa | Leu18 | HD1 |
| Tyr195 | HEa | Leu18 | HD2 |
a Ambiguous stereospecific assignment.
Consistent with the chemical shift perturbations observed (Fig. 4a), the p73-TAD16 peptide binds to a hydrophobic groove of Bcl-XL, in which the BH1, BH2, and BH3 domains and α3 helix are clustered together. Although the p73-TAD16 peptide is intrinsically disordered in the free state (31), the residues Phe15–Leu22 of the peptide adopt an amphipathic α-helix upon binding to Bcl-XL (Fig. S8), with the hydrophobic residues facing the hydrophobic groove of Bcl-XL and the remaining hydrophilic residues being exposed to the solvent (Fig. 4c). Structural comparison between the Bcl-XL–p73-TAD16 peptide and Bcl-XL–BH3 peptides (in Bak (32), Bad (33), Bim (34), and PUMA (35)) complexes showed that the overall fold of Bcl-XL bound to the p73-TAD16 peptide is similar to those of the Bcl-XL–BH3 peptide complexes (Fig. S9).
Binding interface between Bcl-XL and p73-TAD16 peptide
As observed in the Bcl-XL–Bad BH3 peptide (33) and Bcl-XL–Bim BH3 peptide (34) complexes, the Bcl-XL–p73-TAD16 peptide complex is formed through extensive hydrophobic interactions (between Phe15, Leu18, Trp19, and Leu22 in the p73-TAD16 peptide and Ala93, Phe97, Val141, Ala142, Phe191, Leu194, and Tyr195 in Bcl-XL) and electrostatic interactions (between Glu23 and Asp25 in p73-TAD16 peptide and Arg100 and Arg139 in Bcl-XL) (Fig. 4d). In particular, Phe97 and Tyr195 in Bcl-XL are involved in π-stacking interactions with Trp19 and Phe15 in the p73-TAD16 peptide, respectively (Fig. 4d and Fig. S6C). Consistent with the intermolecular π-stacking interaction observed in the structure of the Bcl-XL–PUMA BH3 peptide complex (35), these interactions may significantly contribute to the stability of the protein complex.
To identify the major determinants of the binding of the p73-TAD16 peptide to Bcl-XL, we performed NMR-binding experiments using mutant p73-TAD16 peptides with alanine substitutions (F15A, L18A, W19A, and L22A). As shown in the 2D 1H-15N HSQC spectra of 15N-labeled Bcl-XL with the mutant peptides (Fig. S10A), the alanine mutations at Phe15, Leu18, Trp19, and Leu22 of the p73-TAD16 peptide completely abolished the NMR chemical shift perturbations observed in the binding with the WT peptide. These data indicate that the bulky hydrophobic residues in the FXXφWXXL motif of p73-TAD are critical for the interaction between Bcl-XL and the p73-TAD16 peptide. Previous p73-TAD mutagenesis results showed that mutations at F15A, W19A, and L22A are the most disruptive to MDM2 binding (36). Thus, the major binding determinants for Bcl-XL and p73-TAD are identical to those for MDM2 and p73-TAD.
To confirm whether binding of p73-TAD16 to Bcl-XL is required for transcription-independent p73-induced apoptosis, we generated a TAp73(4A) mutant containing (F15A/L18A/W19A/L22A) mutations and carried out a caspase-3/7 activity assay. The TAp73(4A) mutant resulted in a substantial loss of apoptotic activity relative to WT TAp73 (Fig. S10B).
A modified mode of Bcl-XL recognition by p73-TAD
Despite the overall similarity between them (Fig. S9), the structure of the Bcl-XL–p73-TAD16 peptide complex exhibits noticeable differences with those of Bcl-XL–BH3 peptide complexes (32–35). The p73-TAD16 peptide binds to a unique site in Bcl-XL (Fig. 5, a and b), which is different from the canonical binding groove of pro-apoptotic BH3 peptides. Previously, pro-apoptotic BH3 peptides in Bak (32), Bad (33), Bid (30), and PUMA (35) were shown to bind to a canonical groove formed by the BH1 and BH3 domains in Bcl-XL (Fig. 5, c–f). In contrast, the p73-TAD16 peptide binds to a noncanonical site comprising the BH1, BH2, and BH3 domains in Bcl-XL (Fig. 5a). At the binding interface, the nine Bcl-XL residues involved in the interaction with the six residues of the p73-TAD16 peptide (Phe15, Leu18, Trp19, Leu22, Glu23, and Asp25) are evenly distributed in the BH1 (Arg139, Val141, and Ala142), BH2 (Phe191, Leu194, and Tyr195), and BH3 domains (Ala93, Phe97, and Arg100) (Fig. 5b). Additionally, the p73-TAD16 peptide binds to Bcl-XL in an opposite orientation to those of pro-apoptotic BH3 peptides in Bak (32), Bad (33), Bid (30), and PUMA (35) (Fig. 5).
Figure 5.
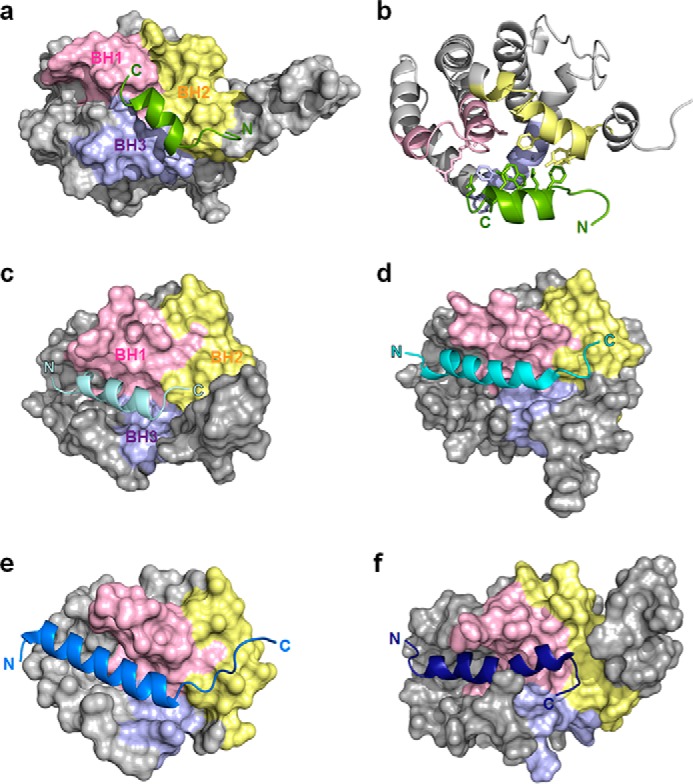
A modified mode of Bcl-XL recognition by p73-TAD. a, structure of the p73-TAD16 peptide bound to Bcl-XL. The p73-TAD16 peptide, shown in light green, is located at the center of the BH1, BH2, and BH3 domains, allowing it to interact with the residues from all three BH domains. b, the p73-TAD16 peptide is equally involved in the interaction with the Bcl-XL residues from the BH1, BH2, and BH3 domains. c, structures of Bcl-XL in complex with the pro-apoptotic BH3 peptides of Bak (PDB code 1BXL). The α1 helix, which is not involved in any peptide binding, is not shown for clarity. d, structure of Bcl-XL in complex with the pro-apoptotic BH3 peptides of Bad (PDB code 1G5J). e, structure of Bcl-XL in complex with the pro-apoptotic BH3 peptides of Bid (PDB code 4QVE). f, structure of Bcl-XL in complex with the pro-apoptotic BH3 peptides of PUMA (PDB code 2M04).
Although our studies mainly focused on the p73–Bcl-XL interaction to compare with the previously reported mechanism for the p53–Bcl-XL interaction (17, 20, 22), the GST pulldown assay data (Fig. 2a) suggested that p73-TAD might also interact with Bcl-2 or Bcl-w to promote apoptosis. We further performed NMR-binding experiments of Bcl-2 or Bcl-w with p73-TAD16 peptide. Significant NMR chemical shift perturbations demonstrated that the p73-TAD16 peptide directly binds to both Bcl-2 and Bcl-w (Fig. S11, A and C). The binding site mapping showed that the p73-TAD16 peptide-binding site on Bcl-2 overlaps well with the pro-apoptotic BH3 peptide-binding site (Fig. S11B). These results suggest that p73-TAD inhibits Bcl-2 or Bcl-w through a direct interaction, in a similar manner as observed with Bcl-XL.
Discussion
Because past studies have extensively focused on transcription-dependent apoptosis of p73, the cytoplasmic apoptotic function of p73 is poorly understood. In this study, we demonstrated that in response to apoptotic stimuli, p73 directly induces transcription-independent apoptosis, accompanied by cytochrome c release from the mitochondria, via a mitochondrial pathway. Our results also showed that induction of transcription-independent apoptosis by p73 is mediated by the interaction between p73-TAD and Bcl-XL. Competition experiments using immunoprecipitation suggest a possible mechanism of how the interaction between p73 and Bcl-XL triggers transcription-independent apoptosis. The interaction between p73-TAD and Bcl-XL displaces an inhibitory interaction between the anti-apoptotic Bcl-XL and the pro-apoptotic BH3-only protein Bid. As shown previously (37, 38), the released Bid is then free to directly activate pro-apoptotic effector proteins, Bax and Bak, leading to cytochrome c release from mitochondria. Collectively, we propose that p73 acts as a sensitizer (or derepressor) BH3-only protein by releasing a direct activator Bid from anti-apoptotic Bcl-XL and thereby triggering mitochondrial apoptosis.
Furthermore, the substantial loss of apoptosis with p73 mutants lacking p73-TAD16 showed that binding of p73-TAD16 to Bcl-XL is critical for the induction of transcription-independent apoptosis, and structure determination of the Bcl-XL–p73-TAD16 peptide complex revealed a molecular basis for the binding of Bcl-XL by p73. Despite the lack of BH3 domain in p73, the p73-TAD16 peptide folds into an amphipathic α-helix, which binds to a hydrophobic groove within Bcl-XL. Structural comparison with the Bcl-XL–Bid BH3 peptide complex (30) showed that the Bcl-XL–binding sites for the p73-TAD16 and Bid BH3 peptides overlap significantly (Fig. 5), supporting that p73-TAD could compete with Bid for Bcl-XL binding.
The suggested mechanism for transcription-independent p73-mediated apoptosis is similar to that of p53 (12–15). Under pro-apoptotic conditions, p53 facilitates the release of pro-apoptotic Bid and Bax by associating with Bcl-XL and Bcl-2 (12). p53 is able to directly permeabilize the outer mitochondrial membrane, through the pore formation of Bax and Bak, and induces cytochrome c release from the mitochondria, which activates the caspase cascade and eventually results in transcription-independent apoptosis. This suggests a high degree of evolutionary conservation between p53 and p73 in cytoplasmic apoptotic function.
Despite the similarity in the Bcl-XL–binding mechanism between p73-TAD and pro-apoptotic BH3 peptides, the structure of the Bcl-XL–p73-TAD16 peptide complex revealed a novel recognition mode of anti-apoptotic Bcl-2 family proteins. First, the p73-TAD16 peptide binds to a unique site in Bcl-XL, which is different from the canonical BH3 peptide-binding groove. Most of the residues in the pro-apoptotic BH3 peptides of Bak (32), Bad (33), Bim (34), and PUMA (35) hardly make any contact with residues from the BH2 domain, interacting only with residues from the BH1 and BH3 domains in Bcl-XL. However, the p73-TAD16 peptide equally contacts the nine residues from the BH1, BH2, and BH3 domains (three residues from each BH domain) in Bcl-XL (Fig. 5b). This difference may arise from the fact that the p73-TAD16 peptide is shifted toward the C terminus of Bcl-XL, compared with the BH3 peptides, allowing it to localize at the center of the BH1, BH2, and BH3 domains. Second, the binding orientation of the p73-TAD16 peptide toward Bcl-XL is opposite to that of the pro-apoptotic BH3 peptides. This is consistent with the orientation of the p53-TAD1 peptide (residues 15–29) bound to Bcl-XL suggested previously by modeling studies (22). This noncanonical mode of Bcl-XL recognition by p73-TAD could be an alternative way of target protein recognition by anti-apoptotic Bcl-2 family proteins, which provides a novel structural rationale for the design of anticancer therapeutics against anti-apoptotic Bcl-2 family proteins.
Within the nucleus, p73 is known to exert transcription-dependent apoptosis by transactivating pro-apoptotic genes such as Bax and PUMA (6). This transcriptional activity of nuclear p73 is negatively regulated by MDM2 binding (39). Recently, our previous study showed that the p73-TAD16 peptide also acts as a binding motif for MDM2 (36). In combination with mutagenesis data (Fig. S10A), the structural comparison between MDM2–p73-TAD16 peptide (36) and Bcl-XL–p73-TAD16 peptide complexes revealed that MDM2 and Bcl-XL share a remarkably similar mode of interaction with the p73-TAD16 peptide, despite the large difference in their global folds (Fig. S12). The same key hydrophobic residues in the p73-TAD16 peptide (Phe15, Trp19, and Leu22 in FXXφWXXL motif), which are aligned on one face of the amphipathic α-helix, are critical binding determinants for both MDM2 and Bcl-XL. In particular, the aromatic ring of Trp19 binds tightly into the hydrophobic grooves in MDM2 and Bcl-XL, making the largest contribution to the forming both of the complexes. Taken together, the interaction of p73 with Bcl-XL closely mimics that of p73 with MDM2. Hence, α-helical p73-TAD peptide motif-based interactions represent a highly conserved mechanism for apoptosis regulation of p73 in the transcription-dependent and transcription-independent pathways.
Both MDM2 and Bcl-XL are well-known to be attractive targets for anticancer therapy (25, 40), because they are involved in the apoptotic regulation of cancer cells. Because concomitant inhibition of MDM2 and an anti-apoptotic Bcl-2 family protein induces a synergistic effect on apoptosis in acute myeloid leukemia (41), multitargeting of MDM2 and anti-apoptotic Bcl-2 family proteins can be an effective strategy for anticancer therapy. A high similarity between the binding modes of the p73-TAD16 peptide to MDM2 and Bcl-XL suggests the possibility to design a single peptidomimetic drug targeting both MDM2 and anti-apoptotic Bcl-2 family proteins. Thus, the complex structure revealed here will serve as a useful template for the rational design of multitargeting anticancer therapeutics.
In summary, we demonstrated a novel transcription-independent apoptotic function of p73 in the mitochondrial pathway. We showed that this apoptosis is mediated by the interaction between p73-TAD and anti-apoptotic Bcl-XL. The complex structure revealed a modified mode of Bcl-XL recognition by the p73-TAD16 peptide, which is distinguished from the canonical binding mode of anti-apoptotic Bcl-2 family proteins and pro-apoptotic BH3 domains. Our studies will contribute to molecular understanding of transcription-independent p73-mediated apoptosis, which may have important implications in anticancer therapy.
Experimental procedures
Cell culture and plasmid constructs
The p53-null human lung cancer cell line H1299 was grown in Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum. HEK293T cells were cultured in Dulbecco's modified Eagle's medium with 10% fetal bovine serum, penicillin (100 units/ml), and streptomycin (100 μg/ml). The cells were maintained at 37 °C in a humidified 5% CO2 atmosphere. All p73 constructs, including TAp73, p73-TAD (residues 1–67), and p73-TAD16 (residues 10–25), were subcloned into the peGFP-C3 vector. FLAG-tagged Bcl-XL, Bcl-2, and Bcl-w were generated by inserting each construct into the pCMV2-FLAG vector. GST-tagged p73-TAD and Myc-tagged Bid were inserted into the pEBG and pCMV-Myc vectors, respectively.
Immunoblotting
Cell lysates were boiled in SDS sample buffer, resolved by SDS-PAGE (10 or 15% polyacrylamide), and transferred to polyvinylidene fluoride membranes. After transfer, the membranes were blocked in 5% milk in TBS and Tween 20 (TBST; 10 mm Tris-HCl, pH 8.0, 150 mm NaCl, and 0.05% Tween 20) for 60 min and incubated with primary antibody in 5% milk in TBST for 2 h at room temperature. The membranes were washed three times with TBST and incubated for 1 h at room temperature in TBST containing horseradish peroxidase-linked anti-IgG. After three washes in TBST, immunoreactive products were detected by chemiluminescence with an ECL system (Thermo Scientific). Antibodies specific for Bcl-XL (1:1000 dilution, Cell Signaling Technology, clone 54H6), TAp73 (1:1000 dilution, Bethyl Laboratories Inc., catalog no. A300-126A; 1:500 dilution, Abcam, catalog no. ab137797), Bid (1:1000 dilution, Cell Signaling Technology, catalog no. 2002S), Bad (1:1000 dilution, Cell Signaling Technology, clone: D24A9), PUMA (1:1000 dilution, Cell Signaling Technology, catalog no. 4976), Bik (1:1000 dilution, Cell Signaling Technology, catalog no. 4592), Bim (1:1000 dilution, Cell Signaling Technology, clone: C34C5), cytochrome c (1:2000 dilution, BD Pharmingen, catalog no. 556433), GFP (1:1000 dilution, Santa Cruz Biotechnology, clone: FL), normal rabbit IgG (1:10000 dilution, Thermo Scientific, catalog no. 31460), and normal mouse IgG (1:10000 dilution, Thermo Scientific, catalog no. 31430) were purchased.
Immunoprecipitation
For endogenous protein complexes, cell lysates from HEK293T cells were immunoprecipitated with 1 μg of antibody and immunoblotted. Briefly, whole-cell lysates obtained using a radioimmunoprecipitation assay were immunoprecipitated with rabbit IgG and anti–Bcl-XL antibodies and incubated with 20 μl of antibody plus protein-Sepharose beads (GE Healthcare) for 2 h. The beads were washed three times with buffer (150 mm NaCl, 20 mm Tris·HCl, pH 7.5, 0.5% Nonidet P-40). To verify that equivalent amounts of cell lysate were used in the immunoprecipitates, a sample representing 0.01% of the total lysate (input) was included.
Cell viability assay
The cells were seeded in 96-well plates, allowed to attach for 24 h, and then transfected with a p73-expressing plasmid. 24 h after transfection, the number of viable cells was determined using a tetrazolium salt reduction assay (EZ-Cytox cell proliferation assay, lipopolysaccharide (LPS) solution) according to the manufacturer's protocol. Three replicates were performed for each treatment.
Colony formation assay
Clonogenic cell survival was evaluated in a colony-formation assay. Ten thousand cells were seeded in 6-well dishes 1 day before transient transfection. The cells were then transfected using Lipofectamine 2000 (Invitrogen) transfection reagent and allowed to grow and form colonies under selection with G418 sulfate (200 μg/μl). After 2 weeks of incubation, the cells were fixed with 3.7% formaldehyde and stained with crystal violet. Colonies larger than 0.6 mm in diameter were counted. Three replicates were performed for each treatment.
DNA fragmentation assay
The H1299 cells of 2 × 105 were seeded in 12-well plates; transfected with the TAp73, p73-TAD, and p73-TAD16 plasmid constructs; and incubated for 24 h. Then the cells were collected for fixation with 70% ethanol and kept in the fixative for at least 12 h. The apoptotic cells were determined using a DNA fragmentation assay (solution 3 (1 μg/ml 4′6-diamidino-2-phenyldole, 0.1% Triton X-100 in PBS); Chemometech) according to the manufacturer's protocol.
Apoptosis assay
For annexin V–APC staining by FACS analysis, APC-conjugated annexin V (BD Biosciences) and 7-aminoactinomycin (7-AAD) were used to determine the percentage of cells undergoing apoptosis. Flow cytometry data were collected using a FACSCalibur 4 color flow cytometer (BD Biosciences, San Jose, CA) and CellQuestTM software version 6. The H1299 cells transfected with TAp73 and p73-TAD were stained by TUNEL using an in situ cell death detection kit, TMR red (Roche) according to the manufacturer's instructions and visualized by a confocal microscope (Zeiss LSM800). Caspase-3/7 activity was measured using Caspase-Glo® 3/7 assay kit (Promega) according to the manufacturer's instructions.
Cytochrome c release assay
The H1299 cells were co-transfected with peGFP-C3-TAp73, peGFP-C3-p73-TAD, or peGFP-C3 control vector with or without pCMV2-FLAG-Bcl-XL using Lipofectamine 2000. Cytochrome c release was detected 16 h after transfection, using a cytochrome c apoptosis detection kit (Thermo Scientific) according to the manufacturer's instructions.
GST pulldown assay
48 h after co-transfection, HEK293T cells were harvested, washed in PBS, and lysed in a lysis buffer containing 20 mm Tris-HCl (pH 7.5), 150 mm NaCl, 1% Nonidet P-40, and 1% glycerol, supplemented with Complete-MINI (Roche). The resultant lysates were centrifuged at 15,000 × g for 15 min, and the supernatants were subjected to a GST pulldown assay. For the GST pulldown assay, 500 μg of lysates was mixed with 50 μl of GSH agarose beads (GE Healthcare) at 4 °C for 4 h with gentle shaking. The protein complexes were collected by centrifugation, washed three times with lysis buffer, and then boiled in SDS sample buffer for 5 min to elute protein complexes from the GSH-agarose beads. The reducing agent was added back to the eluted samples before they were processed for SDS-PAGE and subsequent immunoblot analysis. For immunoblotting, proteins separated by SDS-PAGE were transferred to polyvinylidene fluoride membranes (Millipore) and immunoblotted with antibodies according to the manufacturer's instructions. Primary antibodies were detected with either anti-rabbit or anti-mouse antisera conjugated to horseradish peroxidase (Santa Cruz Biotechnology) and then visualized using electrochemiluminescence. The antibodies against α-FLAG and α-GST were purchased from Sigma.
Protein expression and purification
For structure determination, a Bcl-XL construct lacking the α1-α2 loop (residues 45–84) and the C-terminal transmembrane domain (residues 212–233) was expressed and purified as previously described (22). For NMR experiments, isotope-labeled Bcl-XL proteins were grown in M9 medium containing 1 g of 15N-NH4Cl for 15N-labeled Bcl-XL or 1 g of 15N-NH4Cl and 2 g of 13C-glucose for 13C, 15N-labeled Bcl-XL.
Peptide synthesis
Various versions of p73-TAD–derived peptides were purchased from Peptron, Inc. (Daejeon, Republic of Korea). The peptides were synthesized and purified by HPLC at a purity of >95%. The peptide sequences were as follows: WT p73-TAD16, DGGTTFEHLWSSLEPD; p73-TAD13, TTFEHLWSSLEPD; p73-TAD16(F15A), DGGTTAEHLWSSLEPD; p73-TAD16(L18A), DGGTTFEHAWSSLEPD; p73-TAD16(W19A), DGGTTFEHLASSLEPD; and p73-TAD16(L22A), DGGTTFEHLWSSAEPD.
NMR spectroscopy
All NMR data were acquired at 25 °C on Bruker Avance II 800 spectrometers equipped with cryogenic probes at Korea Basic Science Institute (Ochang, Republic of Korea). All NMR samples were prepared in a buffer containing 20 mm sodium phosphate (pH 6.5), 150 mm NaCl, 2 mm DTT, and 10% (v/v) D2O. The interaction of Bcl-XL with p73-TAD16 peptide was monitored by measuring a series of 2D 1H-15N HSQC spectra of 1 mm 15N-labeled Bcl-XL in the absence or presence of unlabeled p73-TAD16 peptide up to a molar ratio of 1:4. Although full saturation could not be reached because of limitation in the protein solubility, the NMR titration experiments showed that a molar ratio of 1:2 (Bcl-XL:p73-TAD16 peptide) was close to a saturation point (Fig. S6B). The weighted average chemical shift perturbations of the complex at a molar ratio of 1:2 were calculated using the equation ΔCS = {[Δδ (1H)]2 + 0.2*[Δδ (15N)]2}1/2. The sequence-specific backbone assignments were accomplished using 3D HNCACB and CBCA(CO)NH, and side-chain assignments were made using 3D HCCH-TOCSY and (H)CCH-TOCSY experiments (42) with 1 mm 15N,13C-labeled Bcl-XL in complex with 2 mm unlabeled p73-TAD16 peptide. Distance constraints were collected from simultaneous 3D 13C/15N-edited NOESY-HSQC spectra (43) (τm = 250 ms). The distance restraints of the p73-TAD16 peptide bound to Bcl-XL were obtained using 2D transferred NOESY (44) experiment at a molar ratio of 10:1 (τm = 250 ms). The intermolecular NOEs between Bcl-XL and the p73-TAD16 peptide were collected from 15N/13C-edited, 15N/13C-filtered 3D NOESY (45) (τm = 250 ms). NMR data were processed with NMRPipe (46) and analyzed using CARA (47).
Structure calculation
The complex structures of Bcl-XL and the p73-TAD16 peptide were calculated separately via a simulated annealing protocol using the Xplor-NIH program (48). For the bound Bcl-XL, the TALOS program (49) was used to predict ϕ and ψ dihedral angles. The secondary structure of the bound Bcl-XL was predicted based on the artificial neural network–predicted secondary structure from the chemical shifts within the TALOS+ program and the medium-range NOE patterns. Generic hydrogen bond restraints were employed for the residues at well-defined α-helical regions. The interproton distances were obtained from the NOE peaks from NOESY experiments based on their intensities. For the bound p73-TAD16 peptide, the secondary structures were predicted from the secondary Hα chemical shift using the three different sets of random coil values (50–52) and the NOE connectivities (Fig. S8). Generic hydrogen bond and dihedral angle restraints were used for the residues at the well-defined α-helical region. Peak intensities from transferred NOESY spectra were translated into the interproton distance restraints. The structure of p73-TAD16 peptide bound to Bcl-XL was calculated using distance restraints derived from the transferred NOESY spectra (Fig. S8) (44). The intermolecular NOEs were obtained from 13C,15N-filtered, edited NOESY spectra of 13C,15N-labeled Bcl-XL bound to unlabeled p73-TAD16 peptide. The solution structure of the Bcl-XL–p73-TAD16 peptide complex was calculated using these intermolecular NOEs. The initial complex structure was obtained by docking the p73-TAD16 peptide to Bcl-XL using conjoined rigid body/torsion angle dynamics (53) and performing further simulated annealing refinement against all of the experimental restraints listed above. Finally, the structures were refined using CNS (54) energy minimization with explicit water. An ensemble of the 20 lowest-energy structures was used for validation with PROCHECK-NMR (55).
Statistical analysis
The data are represented as means ± S.D., and statistics were performed using one-way analysis of variance (ANOVA) or Student's t test. Means ± S.D. for n ≥ 3 are shown in figures. A p value of less than 0.05 was considered statistically significant.
Author contributions
M.-K. Y., B.-Y. K., J.-Y. L., J.-H. H., S. A. K., D.-H. L., M.-S. L., M.-K. L., and J. S. C. data curation; M.-K. Y., B.-Y. K., J.-Y. L., J.-H. H., S. A. K., D.-H. L., M.-S. L., M.-K. L., J. S. C., J. H. C., J.-H. K., S. K., J. S., S. G. P., B. C. P., K.-H. B., S. U. C., and S.-W. C. formal analysis; M.-K. Y. and B.-Y. K. writing-original draft; J. S., K.-H. B., S. U. C., and S.-W. C. writing-review and editing; S. U. C. and S.-W. C. conceptualization; S. U. C., M.-K. L., and S.-W. C. funding acquisition.
Supplementary Material
Acknowledgment
We are grateful to Dr. Eun-Woo Lee for useful discussion.
This work was supported by Grants NRF-2017R1E1A1A01074403, NRF-2017M3A9C4092979, and NRF-2017R1A2B4006378 from the National Research Foundation of Korea funded by the Korean Government (MSIT) and by the KRIBB Research Initiative Program. This work was also supported by Grant 10038744 from the Technology Innovation Program of Korea Evaluation Institute of Industrial Technology funded by Ministry of Trade, Industry & Energy, Republic of Korea. The authors declare that they have no conflicts of interest with the contents of this article.
This article contains Figs. S1–S12.
The atomic coordinates and structure factors (code 6IJQ) have been deposited in the Protein Data Bank (http://wwpdb.org/).
The NMR chemical shift assignments and restraints used for structure calculation of the Bcl-XL–p73-TAD16 peptide complex have been deposited in the Biological Magnetic Resonance Bank database under accession number 36133.
- TAD
- transactivation domain
- HSQC
- heteronuclear single quantum coherence
- Bcl-XL
- B-cell lymphoma-extra large
- MDM2
- mouse double minute 2
- ANOVA
- analysis of variance
- TUNEL
- terminal deoxynucleotidyltransferase-mediated nick-end labeling
- GST
- glutathione S-transferase
- 7-AAD
- 7-aminoactinomycin
- PDB
- Protein Data Bank
- APC
- allophycocyanin
- TOCSY
- total correlation spectroscopy.
References
- 1. Dötsch V., Bernassola F., Coutandin D., Candi E., and Melino G. (2010) p63 and p73, the ancestors of p53. Cold Spring Harb. Perspect. Biol. 2, a004887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Jost C. A., Marin M. C., and Kaelin W. G. Jr. (1997) p73 is a simian [correction of human] p53-related protein that can induce apoptosis. Nature 389, 191–194 10.1038/38298 [DOI] [PubMed] [Google Scholar]
- 3. Melino G., De Laurenzi V., and Vousden K. H. (2002) p73: friend or foe in tumorigenesis. Nat. Rev. Cancer 2, 605–615 10.1038/nrc861 [DOI] [PubMed] [Google Scholar]
- 4. Yoon M. K., Ha J. H., Lee M. S., and Chi S. W. (2015) Structure and apoptotic function of p73. BMB Rep. 48, 81–90 10.5483/BMBRep.2015.48.2.255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Yang A., Walker N., Bronson R., Kaghad M., Oosterwegel M., Bonnin J., Vagner C., Bonnet H., Dikkes P., Sharpe A., McKeon F., and Caput D. (2000) p73-deficient mice have neurological, pheromonal and inflammatory defects but lack spontaneous tumours. Nature 404, 99–103 10.1038/35003607 [DOI] [PubMed] [Google Scholar]
- 6. Melino G., Bernassola F., Ranalli M., Yee K., Zong W. X., Corazzari M., Knight R. A., Green D. R., Thompson C., and Vousden K. H. (2004) p73 induces apoptosis via PUMA transactivation and Bax mitochondrial translocation. J. Biol. Chem. 279, 8076–8083 10.1074/jbc.M307469200 [DOI] [PubMed] [Google Scholar]
- 7. Schilling T., Schleithoff E. S., Kairat A., Melino G., Stremmel W., Oren M., Krammer P. H., and Müller M. (2009) Active transcription of the human FAS/CD95/TNFRSF6 gene involves the p53 family. Biochem. Biophys. Res. Commun. 387, 399–404 10.1016/j.bbrc.2009.07.063 [DOI] [PubMed] [Google Scholar]
- 8. Aqeilan R. I., Pekarsky Y., Herrero J. J., Palamarchuk A., Letofsky J., Druck T., Trapasso F., Han S. Y., Melino G., Huebner K., and Croce C. M. (2004) Functional association between Wwox tumor suppressor protein and p73, a p53 homolog. Proc. Natl. Acad. Sci. U.S.A. 101, 4401–4406 10.1073/pnas.0400805101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Liu T., Roh S. E., Woo J. A., Ryu H., and Kang D. E. (2013) Cooperative role of RanBP9 and P73 in mitochondria-mediated apoptosis. Cell Death Dis. 4, e476 10.1038/cddis.2012.203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Min B., Ryu J., Chi S. W., and Yi G. S. (2015) Ubiquitination-dependent degradation of p73 by the mitochondrial E3 ubiquitin ligase Hades. Biochem. Biophys. Res. Commun. 467, 316–321 10.1016/j.bbrc.2015.09.163 [DOI] [PubMed] [Google Scholar]
- 11. Sayan A. E., Sayan B. S., Gogvadze V., Dinsdale D., Nyman U., Hansen T. M., Zhivotovsky B., Cohen G. M., Knight R. A., and Melino G. (2008) p73 and caspase-cleaved p73 fragments localize to mitochondria and augment TRAIL-induced apoptosis. Oncogene 27, 4363–4372 10.1038/onc.2008.64 [DOI] [PubMed] [Google Scholar]
- 12. Chipuk J. E., Kuwana T., Bouchier-Hayes L., Droin N. M., Newmeyer D. D., Schuler M., and Green D. R. (2004) Direct activation of Bax by p53 mediates mitochondrial membrane permeabilization and apoptosis. Science 303, 1010–1014 10.1126/science.1092734 [DOI] [PubMed] [Google Scholar]
- 13. Leu J. I., Dumont P., Hafey M., Murphy M. E., and George D. L. (2004) Mitochondrial p53 activates Bak and causes disruption of a Bak-Mcl1 complex. Nat. Cell Biol. 6, 443–450 10.1038/ncb1123 [DOI] [PubMed] [Google Scholar]
- 14. Mihara M., Erster S., Zaika A., Petrenko O., Chittenden T., Pancoska P., and Moll U. M. (2003) p53 has a direct apoptogenic role at the mitochondria. Mol. Cell 11, 577–590 10.1016/S1097-2765(03)00050-9 [DOI] [PubMed] [Google Scholar]
- 15. Green D. R., and Kroemer G. (2009) Cytoplasmic functions of the tumour suppressor p53. Nature 458, 1127–1130 10.1038/nature07986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Chipuk J. E., Bouchier-Hayes L., Kuwana T., Newmeyer D. D., and Green D. R. (2005) PUMA couples the nuclear and cytoplasmic proapoptotic function of p53. Science 309, 1732–1735 10.1126/science.1114297 [DOI] [PubMed] [Google Scholar]
- 17. Follis A. V., Llambi F., Ou L., Baran K., Green D. R., and Kriwacki R. W. (2014) The DNA-binding domain mediates both nuclear and cytosolic functions of p53. Nat. Struct. Mol. Biol. 21, 535–543 10.1038/nsmb.2829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Chipuk J. E., Maurer U., Green D. R., and Schuler M. (2003) Pharmacologic activation of p53 elicits Bax-dependent apoptosis in the absence of transcription. Cancer Cell 4, 371–381 10.1016/S1535-6108(03)00272-1 [DOI] [PubMed] [Google Scholar]
- 19. Sayan B. S., Sayan A. E., Knight R. A., Melino G., and Cohen G. M. (2006) p53 is cleaved by caspases generating fragments localizing to mitochondria. J. Biol. Chem. 281, 13566–13573 10.1074/jbc.M512467200 [DOI] [PubMed] [Google Scholar]
- 20. Ha J. H., Shin J. S., Yoon M. K., Lee M. S., He F., Bae K. H., Yoon H. S., Lee C. K., Park S. G., Muto Y., and Chi S. W. (2013) Dual-site interactions of p53 protein transactivation domain with anti-apoptotic Bcl-2 family proteins reveal a highly convergent mechanism of divergent p53 pathways. J. Biol. Chem. 288, 7387–7398 10.1074/jbc.M112.400754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Ha J. H., Won E. Y., Shin J. S., Jang M., Ryu K. S., Bae K. H., Park S. G., Park B. C., Yoon H. S., and Chi S. W. (2011) Molecular mimicry-based repositioning of nutlin-3 to anti-apoptotic Bcl-2 family proteins. J. Am. Chem. Soc. 133, 1244–1247 10.1021/ja109521f [DOI] [PubMed] [Google Scholar]
- 22. Xu H., Ye H., Osman N. E., Sadler K., Won E. Y., Chi S. W., and Yoon H. S. (2009) The MDM2-binding region in the transactivation domain of p53 also acts as a Bcl-X(L)-binding motif. Biochemistry 48, 12159–12168 10.1021/bi901188s [DOI] [PubMed] [Google Scholar]
- 23. Youle R. J., and Strasser A. (2008) The BCL-2 protein family: opposing activities that mediate cell death. Nat. Rev. Mol. Cell Biol. 9, 47–59 10.1038/nrm2308 [DOI] [PubMed] [Google Scholar]
- 24. Moldoveanu T., Follis A. V., Kriwacki R. W., and Green D. R. (2014) Many players in BCL-2 family affairs. Trends Biochem. Sci. 39, 101–111 10.1016/j.tibs.2013.12.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Lessene G., Czabotar P. E., and Colman P. M. (2008) BCL-2 family antagonists for cancer therapy. Nat. Rev. Drug Discov. 7, 989–1000 10.1038/nrd2658 [DOI] [PubMed] [Google Scholar]
- 26. Oltersdorf T., Elmore S. W., Shoemaker A. R., Armstrong R. C., Augeri D. J., Belli B. A., Bruncko M., Deckwerth T. L., Dinges J., Hajduk P. J., Joseph M. K., Kitada S., Korsmeyer S. J., Kunzer A. R., Letai A., et al. (2005) An inhibitor of Bcl-2 family proteins induces regression of solid tumours. Nature 435, 677–681 10.1038/nature03579 [DOI] [PubMed] [Google Scholar]
- 27. Nyman U., Sobczak-Pluta A., Vlachos P., Perlmann T., Zhivotovsky B., and Joseph B. (2005) Full-length p73alpha represses drug-induced apoptosis in small cell lung carcinoma cells. J. Biol. Chem. 280, 34159–34169 10.1074/jbc.M500394200 [DOI] [PubMed] [Google Scholar]
- 28. John K., Alla V., Meier C., and Pützer B. M. (2011) GRAMD4 mimics p53 and mediates the apoptotic function of p73 at mitochondria. Cell Death Differ. 18, 874–886 10.1038/cdd.2010.153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. De Laurenzi V., Raschellá G., Barcaroli D., Annicchiarico-Petruzzelli M., Ranalli M., Catani M. V., Tanno B., Costanzo A., Levrero M., and Melino G. (2000) Induction of neuronal differentiation by p73 in a neuroblastoma cell line. J. Biol. Chem. 275, 15226–15231 10.1074/jbc.275.20.15226 [DOI] [PubMed] [Google Scholar]
- 30. Rajan S., Choi M., Baek K., and Yoon H. S. (2015) Bh3 induced conformational changes in Bcl-Xl revealed by crystal structure and comparative analysis. Proteins 83, 1262–1272 10.1002/prot.24816 [DOI] [PubMed] [Google Scholar]
- 31. Burge S., Teufel D. P., Townsley F. M., Freund S. M., Bycroft M., and Fersht A. R. (2009) Molecular basis of the interactions between the p73 N terminus and p300: effects on transactivation and modulation by phosphorylation. Proc. Natl. Acad. Sci. U.S.A. 106, 3142–3147 10.1073/pnas.0900383106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Sattler M., Liang H., Nettesheim D., Meadows R. P., Harlan J. E., Eberstadt M., Yoon H. S., Shuker S. B., Chang B. S., Minn A. J., Thompson C. B., and Fesik S. W. (1997) Structure of Bcl-xL-Bak peptide complex: recognition between regulators of apoptosis. Science 275, 983–986 10.1126/science.275.5302.983 [DOI] [PubMed] [Google Scholar]
- 33. Petros A. M., Nettesheim D. G., Wang Y., Olejniczak E. T., Meadows R. P., Mack J., Swift K., Matayoshi E. D., Zhang H., Thompson C. B., and Fesik S. W. (2000) Rationale for Bcl-XL/Bad peptide complex formation from structure, mutagenesis, and biophysical studies. Protein Sci. 9, 2528–2534 10.1110/ps.9.12.2528,10.1017/S096183680000331X [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Lee E. F., Sadowsky J. D., Smith B. J., Czabotar P. E., Peterson-Kaufman K. J., Colman P. M., Gellman S. H., and Fairlie W. D. (2009) High-resolution structural characterization of a helical alpha/beta-peptide foldamer bound to the anti-apoptotic protein Bcl-xL. Angew. Chem. Int. Ed. Engl. 48, 4318–4322 10.1002/anie.200805761 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Follis A. V., Chipuk J. E., Fisher J. C., Yun M. K., Grace C. R., Nourse A., Baran K., Ou L., Min L., White S. W., Green D. R., and Kriwacki R. W. (2013) PUMA binding induces partial unfolding within BCL-xL to disrupt p53 binding and promote apoptosis. Nat. Chem. Biol. 9, 163–168 10.1038/nchembio.1166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Shin J. S., Ha J. H., Lee D. H., Ryu K. S., Bae K. H., Park B. C., Park S. G., Yi G. S., and Chi S. W. (2015) Structural convergence of unstructured p53 family transactivation domains in MDM2 recognition. Cell Cycle 14, 533–543 10.1080/15384101.2014.998056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Desagher S., Osen-Sand A., Nichols A., Eskes R., Montessuit S., Lauper S., Maundrell K., Antonsson B., and Martinou J. C. (1999) Bid-induced conformational change of Bax is responsible for mitochondrial cytochrome c release during apoptosis. J. Cell Biol. 144, 891–901 10.1083/jcb.144.5.891 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Moldoveanu T., Grace C. R., Llambi F., Nourse A., Fitzgerald P., Gehring K., Kriwacki R. W., and Green D. R. (2013) BID-induced structural changes in BAK promote apoptosis. Nat. Struct. Mol. Biol. 20, 589–597 10.1038/nsmb.2563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Zeng X., Chen L., Jost C. A., Maya R., Keller D., Wang X., Kaelin W. G. Jr, Oren M., Chen J., and Lu H. (1999) MDM2 suppresses p73 function without promoting p73 degradation. Mol. Cell Biol. 19, 3257–3266 10.1128/MCB.19.5.3257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Vassilev L. T. (2007) MDM2 inhibitors for cancer therapy. Trends Mol. Med. 13, 23–31 10.1016/j.molmed.2006.11.002 [DOI] [PubMed] [Google Scholar]
- 41. Kojima K., Konopleva M., Samudio I. J., Schober W. D., Bornmann W. G., and Andreeff M. (2006) Concomitant inhibition of MDM2 and Bcl-2 protein function synergistically induce mitochondrial apoptosis in AML. Cell Cycle 5, 2778–2786 10.4161/cc.5.23.3520 [DOI] [PubMed] [Google Scholar]
- 42. Sattler M., Schleucher J., and Griesinger C. (1999) Heteronuclear multidimensional NMR experiments for the structure determination of proteins in solution employing pulsed field gradients. Prog. Nucl. Magc. Reson. Sp. 34, 93–158 10.1016/S0079-6565(98)00025-9 [DOI] [Google Scholar]
- 43. Pascal S. M., Muhandiram D. R., Yamazaki T., Formankay J. D., and Kay L. E. (1994) Simultaneous acquisition of 15N- and 13C-edited NOE spectra of proteins dissolved in H2O. J. Magn. Reson. Ser. B 103, 197–201 10.1006/jmrb.1994.1031 [DOI] [Google Scholar]
- 44. Clore G. M., and Gronenborn A. M. (1983) Theory of the time dependent transferred nuclear Overhauser effect: applications to structural analysis of ligand–protein complexes in solution. J. Magn. Reson. 53, 423–442 [Google Scholar]
- 45. Zwahlen C., Legault P., Vincent S. J. F., Greenblatt J., Konrat R., and Kay L. E. (1997) Methods for measurement of intermolecular NOEs by multinuclear NMR spectroscopy: application to a bacteriophage N-peptide/boxB RNA complex. J. Am. Chem. Soc. 117, 711–721 [Google Scholar]
- 46. Delaglio F., Grzesiek S., Vuister G. W., Zhu G., Pfeifer J., and Bax A. (1995) NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR 6, 277–293 [DOI] [PubMed] [Google Scholar]
- 47. Keller R. (2004) The Computer Aided Resonance Assignment Tutorial, CANTINA Verlag, Goldau, Switzerland [Google Scholar]
- 48. Schwieters C. D., Kuszewski J. J., Tjandra N., and Clore G. M. (2003) The Xplor-NIH NMR molecular structure determination package. J. Magn. Reson. 160, 65–73 10.1016/S1090-7807(02)00014-9 [DOI] [PubMed] [Google Scholar]
- 49. Cornilescu G., Delaglio F., and Bax A. (1999) Protein backbone angle restraints from searching a database for chemical shift and sequence homology. J. Biomol. NMR 13, 289–302 10.1023/A:1008392405740 [DOI] [PubMed] [Google Scholar]
- 50. Schwarzinger S., Kroon G. J., Foss T. R., Chung J., Wright P. E., and Dyson H. J. (2001) Sequence-dependent correction of random coil NMR chemical shifts. J. Am. Chem. Soc. 123, 2970–2978 10.1021/ja003760i [DOI] [PubMed] [Google Scholar]
- 51. Tamiola K., Acar B., and Mulder F. A. (2010) Sequence-specific random coil chemical shifts of intrinsically disordered proteins. J. Am. Chem. Soc. 132, 18000–18003 10.1021/ja105656t [DOI] [PubMed] [Google Scholar]
- 52. Zhang H., Neal S., and Wishart D. S. (2003) RefDB: a database of uniformly referenced protein chemical shifts. J. Biomol. NMR 25, 173–195 10.1023/A:1022836027055 [DOI] [PubMed] [Google Scholar]
- 53. Clore G. M., and Schwieters C. D. (2003) Docking of protein-protein complexes on the basis of highly ambiguous intermolecular distance restraints derived from 1H/15N chemical shift mapping and backbone 15N–1H residual dipolar couplings using conjoined rigid body/torsion angle dynamics. J. Am. Chem. Soc. 125, 2902–2912 10.1021/ja028893d [DOI] [PubMed] [Google Scholar]
- 54. Brünger A. T., Adams P. D., Clore G. M., DeLano W. L., Gros P., Grosse-Kunstleve R. W., Jiang J. S., Kuszewski J., Nilges M., Pannu N. S., Read R. J., Rice L. M., Simonson T., and Warren G. L. (1998) Crystallography & NMR system: a new software suite for macromolecular structure determination. Acta Crystallogr. D Biol. Crystallogr. 54, 905–921 10.1107/S0907444998003254,10.1107/S0108767398011465 [DOI] [PubMed] [Google Scholar]
- 55. Laskowski R. A., Rullmannn J. A., MacArthur M. W., Kaptein R., and Thornton J. M. (1996) AQUA and PROCHECK-NMR: programs for checking the quality of protein structures solved by NMR. J. Biomol. NMR 8, 477–486 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.