

Abstract
Immunotherapy is a promising advancement in the treatment of non-small-cell lung carcinoma (NSCLC), although much of how lung tumors interact with the immune system in the natural course of disease remains unknown. We investigated the impact of the expression of immune-centric genes and pathways in tumors on patient survival to reveal novel candidates for immunotherapeutic research. Tumor transcriptomes and detailed clinical characteristics were obtained from patients with NSCLC who were participants of either the Inflammation, Health and Lung Epidemiology (INHALE) (discovery, N = 280) or The Cancer Genome Atlas (TCGA) Lung (replication, N = 1026) studies. Expressions of 2253 genes derived from 48 major immune pathways were assessed for association with patient prognosis using a multivariable Cox model and pathway effects were assessed with an in-house implementation of the Gene Set Enrichment Analysis (GSEA) algorithm. Prognosis-guided gene and pathway analysis of immune-centric expression in tumors revealed significant survival enrichments across both cohorts. The ‘Interleukin Signaling’ pathway, containing 430 genes, was found to be statistically and significantly enriched with prognostic signal in both the INHALE (P = 0.008) and TCGA (P = 0.039) datasets. Subsequent leading-edge analysis identified a subset of genes (N = 23) shared between both cohorts, driving the pathway enrichment. Cumulative expression of this leading-edge gene signature was a strong predictor of patient survival [discovery: hazard ratio (HR) = 1.59, P = 3.0 × 10–8; replication: HR = 1.29, P = 7.4 × 10–7]. These data demonstrate the impact of immune-centric expression on patient outcomes in NSCLC. Furthermore, prognostic gene effects were localized to discrete immune pathways, of which Interleukin Signaling had the greatest impact on overall survival and the subset of genes driving these effects have promise for future therapeutic intervention.
Analysis of immune-centric RNA profiling in association with natural variation in patient outcomes reveals new immunotherapeutic gene and pathway candidates for research in lung cancer.
Introduction
Lung cancer is the leading cause of cancer mortality in the USA (1). Comparatively, lung tumors are highly mutated and contain more somatic mutations than most other cancer types, and as such are estimated to be rich in neoantigen signatures (2). Lung cancer is an excellent candidate for immunotherapeutic treatment because of its high mutation and neoantigen signature, yet immunotherapy has had limited success in the treatment of non-small-cell lung carcinoma (NSCLC) and only PD-1/PD-L1 agents are approved for use in this disease (3). There have been numerous NSCLC immunotherapy trials with agents previously proven successful in the treatment of other high mutation/neoantigen signature cancers such as melanoma and renal cell carcinoma; however, only PD-1 axis agents have shown success warranting the US Food and Drug Administration approval (4). To identify novel immune candidates specific to lung cancer, we conducted an investigation into the ways in which transcriptome profiles within the tumor-immune microenvironment modulate immune interactions and impact patient survival.
Gene and network expression signatures have been successfully used to predict patient survival in lung cancer from primary tumor-specimen RNA expression profiles (5,6). These signatures can be useful to predict response to interventions or can be used as early risk stratification tools (7). Very little research has been conducted to specifically study the link between NSCLC immune expression and patient prognosis outside of global, cancer site agnostic studies such as the prediction of clinical outcomes from genomic profiles (PRECOG) study (8). The PRECOG study evaluated the impact of gene expression signatures and infiltrating immune cells across multiple cancer sites but did not focus on any single cancer site and did not perform gene/pathway network analysis. A lung-cancer-specific focus paired with an immune-centric pathway analysis should reveal the relevant prognostic immune networks in lung tumors, which may prove useful in immunotherapy discovery research. We hypothesized that NSCLC-specific mechanisms of immune modulation can be detected by leveraging tumor transcriptomic profiling paired with patient outcomes data to identify the immune-centric gene networks that impact patient prognosis from primary tumor specimens.
Methods
Cohort descriptions
Discovery cohort (N = 280)
All procedures used to collect and process participant information were approved by the Wayne State University, Henry Ford Health System and McLaren Health Care institutional review boards, and written informed consent was obtained from all subjects prior to participation. Patients with pathologically confirmed NSCLC were enrolled from the greater Detroit region under the inflammation, health and lung epidemiology (INHALE) case–control study. The INHALE study has been described previously (9). Participants meeting enrollment criteria, aged 21–89 years, had never taken amiodarone and had never been diagnosed with bronchiectasis or cystic fibrosis, were asked to complete an interview; provide saliva, blood and tumor tissue; and complete a low-dose chest computerized tomography scan. Primary tumor specimens were sectioned from clinical formalin-fixed paraffin-embedded (FFPE) tissues after review by a board-certified pathologist and corresponding medical records were abstracted from patient medical records to obtain the clinical covariates. Participants were actively followed by the Metropolitan Detroit Cancer Surveillance System (MDCSS), a founding participant in the National Cancer Institute’s Surveillance Epidemiology and End Results program to determine outcome status.
Replication cohort (N = 1026)
The provisional TCGA Lung Squamous Cell Carcinoma and TCGA Lung Adenocarcinoma clinical datasets were obtained from the cBioPortal datasets page (10). Consent and acquisition of The Cancer Genome Atlas (TCGA) specimens and clinical data were described previously (11,12). Compiled, de-identified clinical and demographic data were openly available to download for research purposes. TCGA participant identifiers were used to merge across datasets.
Specimen selection and processing
Primary, FFPE specimens were collected per the INHALE protocol and subsequently reviewed by a pathologist. Tumor content and purity were assessed via histopathological confirmation. Tumor-containing regions were identified by the pathologist and successively dissected from the archival paraffin blocks for subblock generation. Five 5 µm curls were then cut from each tumor subblock and RNA was isolated from curls using the Qiagen FFPE RNeasy kit. Isolated RNA was quantified and assessed for purity using a NanoDrop spectrophotometer. Sample concentrations ranged from 15 to 1250 ng/µl with a median concentration of 257 ng/µl and a median absorbance at 260/280 and 260/230 was 1.99 (normal ~2.0) and 2.15 (normal ~2.0 to 2.2), respectively. RNA integrity was assessed using a bio-analyzer; low-concentration or low-integrity samples were not carried forward for transcriptome quantification. Total RNA was then processed using the GeneChip Whole Transcriptome Pico Reagent kit for use with the Affymetrix Whole Transcriptome 2.1 Human Gene array. The University of Michigan core facilities conducted the sample assessment and array processing.
As described previously, TCGA clinical specimens were obtained fresh-frozen and processed for next-generation RNA sequencing analysis according to the Biospecimen Core Resource Center (11,12).
Gene expression quantification and analysis
INHALE RNA expression was quantified using the Affymetrix Whole Transcriptome 2.1 Human Gene array. The array consists of 1.3 million probes, 33 000 coding transcripts, 6500 non-coding transcripts and 11 000 long non-coding RNAs. The density of probe intensities was compared across all arrays before gene-level data were extracted to ensure samples were comparable. Probe expression values were converted into gene-level expression values using a robust multi-array average as described previously (13). An empirical Bayes methodology known as ComBat was used to adjust for batch-level corrections at the gene level across tumor samples (14). Once corrections were complete, gene expression values were log transformed prior to analysis. For genes measured with multiple probes, the probe with the highest median expression was chosen to represent that gene’s expression effect.
TCGA Lung RNA-seq data were downloaded from the cBioPortal datasets page. RNA-seq samples (522 lung adenocarcinoma and 504 lung squamous cell carcinoma tumors) with associated clinical data were available for use. RNA-seq data were preprocessed under the TCGA version 2 best-practices pipeline, which used MapSplice and RSEM for mapping, quantification and normalization of gene-level expression (15,16). Expression data were further log transformed prior to analysis to reduce skew.
All statistical analyses were conducted in R (version 3.4.3). Gene expression, after batch-correction, normalization and transformation, was measured for association with patient survival using a Cox proportional-hazards regression model adjusting for tumor stage and histology. The survival library was used to conduct all survival analyses (version 2.41.3). Tumor stage at diagnosis was determined from MDCSS or through medical record abstracting and was collapsed and coded additively (1–4). Histological subtype was also determined from MDCSS and medical record abstracting.
Base survival model diagnostics
To test the immune-centric gene and pathway expression for association with patient survival, a parsimonious base survival model was constructed to adjust gene effects for clinical covariates. A base survival model was constructed consisting of stage at diagnosis and histological subtype. Clinical variables such as age, race, tumor grade and primary treatment regimen were assessed using the Likelihood-ratio test for potential inclusion into the gene-level model. Each covariate was independently added to the base model containing stage and histology to examine overall model fit, significance, additional degrees of freedom and total missing entries for both cohorts using the Likelihood-ratio test (Supplementary Table 1, available at Carcinogenesis Online).
Gene set enrichment analysis
Major immune pathways (N = 48) were assembled from the Reactome pathway database (R-HAS-168256.6), a pathway database that features curated and reviewed biological pathways assembled from multiple bioinformatics resources such as KEGG, Ensembl, UniProt, NCBI as well as from a prior inflammation and lung cancer pathway study (17,18). A total of 2253 genes were contained within the 48 immune pathways that served as the focus of our immune-centric gene and pathway prognostic analysis. A custom implementation of the Gene Set Enrichment Analysis (GSEA) was used to conduct a prognostic pathway analysis on the discovery and replication cohorts to measure aggregate gene effects within each immune pathway as described previously (19). Briefly, genes in the dataset were ranked and weighted by the independent gene-level Wald statistics and a maximum running sum was calculated for each pathway/gene set with a Kolmogorov-Smirnov-like test. Prognostic gene effects were incorporated into GSEA using the Wald statistic from the Cox proportional-hazards regression adjusting for stage and histology to rank and weight the enrichment analysis. To determine any given pathway’s enrichment significance, 1000 phenotype-based permutations were computed and the GSEA statistics were calculated for each permutation across all pathways. Phenotype-permutation testing maintains the complex, gene-correlation architecture of transcriptome data and serves to generate a null distribution of pathway test statistics. Normalization of the observed and permutated pathway enrichment statistics was performed by subtracting the mean enrichment score of each pathway across all permutations followed by dividing by the standard deviation of the permuted test statistics to account for deviations in GSEA test statistics in association with pathway size. A one-sided, empirical P value was calculated for each pathway using the normalized observed and permuted enrichment values.
Post Gene Set Enrichment Analysis
To identify the subset of genes driving a given pathway’s prognostic significance, a leading-edge analysis was conducted as described previously (19). Leading-edge analysis takes advantage of the assumption that not all genes within any given pathway are expected to contribute greatly to a pathway’s significance for any given biological phenomenon. Gene-level prognostic effects of the leading-edge gene sets, within significant pathways, were cross-referenced and a consensus list was constructed requiring genes to maintain moderate model significance (P < 0.15) as well as retain a concordant prognostic effect direction in both the discovery and the replication cohorts. To further visualize these leading-edge consensus members, the protein–protein interactions of the consensus gene set members were assessed, networked and visualized using STRING, a database of direct and indirect protein associations. Additionally, we assessed the cumulative effects of these gene set members on prognosis by generating a gene–pathway signature score for each cohort. The Interleukin Signaling leading-edge signature score was generated through a cumulative summation of each leading-edge gene’s expression in each patient, weighted by the individual gene’s Z-score from the multivariable COX model. Gene–pathway signature scores were then subsequently tested for cumulative prognostic significance in the study cohorts, and hazard ratios (HRs) were reported as per a standard deviation increase/decrease in the signature score.
Results
Baseline prognostic predictors
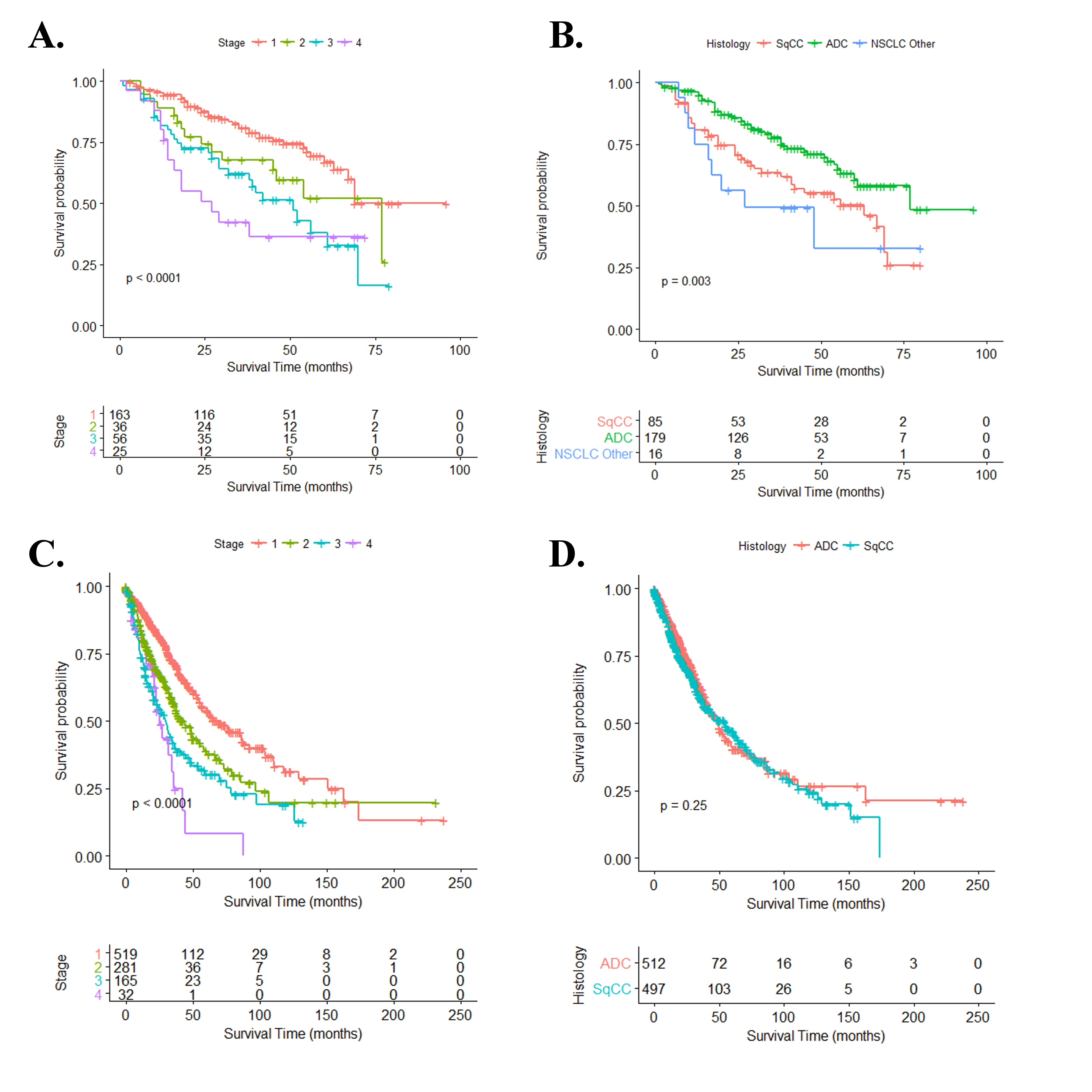
To enable the gene and pathway-based survival analysis in the two independent cohorts, we first developed baseline survival models containing relevant clinical prognostic factors to limit expression-phenotype confounding. The discovery analysis leveraged RNA expression and clinical follow-up from 280 NSCLC cases enrolled in the INHALE study, and the replication analysis used RNA expression and clinical follow-up from 1026 NSCLC cases enrolled in the TCGA Lung study as detailed in Table 1. As expected, stage was a strong, independent predictor of overall survival in both discovery and replication cohorts (discovery: HR = 1.50, P = 3.75 × 10–6; replication: HR = 1.50, P = 1.08 × 10–13) (Supplementary Figure 1A, available at Carcinogenesis Online). Histological subtype was also associated with overall survival, with adenocarcinoma faring better than both squamous cell carcinoma and other NSCLC subtypes (HR = 0.55, P = 0.005) in the discovery cohort only (Supplementary Figure 1B, available at Carcinogenesis Online). Accordingly, these variables were included in the multivariable gene-level prognostic model to adjust for differences in outcome as well as gene expression across all stages and histological types of NSCLC. Other clinical variables such as age, race, tumor grade and primary treatment regime were tested for potential inclusion into the gene-level model but were found to generate less informative models, require greater degrees of freedom and contain a greater number of missing entries when compared to the base model containing only stage and histology (Supplementary Table 1, available at Carcinogenesis Online).
Table 1.
Clinical characteristics of the NSCLC INHALE (discovery) and TCGA (replication) cohorts
| Characteristic | INHALE (N = 280) | TCGA Lung (N = 1026) |
|---|---|---|
| Age (mean, SD), years | 63 (9.4) | 66 (9.4) |
| Specimen source | FFPE | Fresh-frozen |
| Transcriptome source | Affymetrix Whole Transcriptome 2.1 Array | TCGA RNA-seq version 2 RSEM |
| Sex (n, %) | ||
| Male | 128 (46) | 614 (60) |
| Female | 152 (54) | 410 (40) |
| Race (n, %) | ||
| White | 155 (5) | 743 (72) |
| Black | 125 (4) | 85 (8) |
| Unknown | 0 (0) | 198 (19) |
| Stage (n, %) | ||
| 1 | 163 (58) | 525 (51) |
| 2 | 36 (13) | 286 (28) |
| 3 | 56 (20) | 169 (16) |
| 4 | 25 (9) | 32 (3) |
| Unknown | 0 (0) | 14 (1) |
| Histology (n, %) | ||
| Adenocarcinoma | 179 (64) | 522 (51) |
| Squamous cell carcinoma | 85 (30) | 504 (49) |
| NSCLC other | 16 (6) | 0 (0) |
SD, standard deviation.
Immune gene and pathway associations with overall survival
We next assembled an extensive and curated immune-centric gene (N = 2253) and pathway (N = 48) dataset to assess the impact of immune expression in NSCLC tumors on patient prognosis (Supplementary Table 2, available at Carcinogenesis Online). Immune-centric gene and pathway expression was extracted from whole-transcriptome profiles of 280 NSCLC discovery cohort tissues to identify the components of immune pathway-related expression affecting patient outcomes. In total, 48 immune-centric pathways containing 2253 genes were tested for survival association using the GSEA-weighted Kolmogorov-Smirnov-like test coupled with a phenotype-based permutation strategy. Pathway testing of the discovery cohort elucidated three immune pathways as significantly enriched for survival association: ‘Major Histocompatibility Complex Class II (MHC II) Antigen Presentation’ (enrichment score (ES) = 0.51, P = 0.004), Interleukin Signaling (ES = 0.44, P = 0.008), and ‘Advanced Glycosylation End-product Receptor Signaling’ (ES = 0.66, P = 0.025) (Table 2). Next, we performed a leading-edge analysis for each significant pathway to determine the subset of pathway genes that substantially contributed to the observed pathway significance. This analysis identified 41, 109 and 5 genes within the MHC II Antigen Presentation, Interleukin Signaling and Advanced Glycosylation End-product Receptor Signaling pathways, respectively. These gene subsets were the strongest prognostic predictors in each pathway and cumulatively contributed to each pathway’s maximum enrichment score and significance.
Table 2.
Significant immune pathway prognostic effects
| Pathways (N = 48) | Genes | INHALE GSEA | TCGA Lung GSEA | ||
|---|---|---|---|---|---|
| Enrichment score | P valuea | Enrichment score | P valuea | ||
| MHC Class II Family | 121 | 0.51 | 0.004 | 0.39 | 0.504 |
| Interleukin Signaling | 430 | 0.44 | 0.008 | 0.44 | 0.039 |
| Advanced Glycosylation Receptor Signaling | 13 | 0.66 | 0.025 | 0.39 | 0.733 |
aGSEA was ranked and weighted by each gene-level Wald statistic; significance was assessed with 1000 phenotype permutations.
To confirm the prognostic immune pathway findings, we used the clinical and transcriptomic data from 1026 patients with NSCLC enrolled in the TCGA Lung study. Individual gene effects, characterized with a Cox proportional-hazards regression model adjusting for stage and histology, were incorporated into the immune-centric pathway analysis mirroring the discovery analysis. Pathway analysis within the TCGA cohort validated the prognostic significance of the Interleukin Signaling pathway (ES = 0.44, P = 0.039), as identified in the prior discovery analysis (Figure 1 and Supplementary Table 2, available at Carcinogenesis Online). The Interleukin Signaling pathway (R-HSA-449147) consists of 430 genes that mediate biological responses initiated by cytokines, extracellular molecules secreted by or acting upon leukocytes. To further determine the subset of genes responsible for driving the observed pathway survival effects in the replication cohort, we conducted a leading-edge analysis on the Interleukin Signaling pathway that identified 170 leading-edge genes. The leading-edge genes within the Interleukin Signaling pathway comprise 22.2% (INHALE) and 21.6% (TCGA Lung) of the top 500 prognosis ranked immune-centric genes within the 48 major immune pathways tested. Forty-seven leading-edge Interleukin Signaling genes were shared between the leading-edge analyses in the discovery and replication cohorts. To remove the shared leading-edge candidates with spurious gene-level associations between the two cohorts, gene candidates were filtered based upon their cross-cohort, gene-level effect direction and significance. After filtering for gene-level model statistics, 23 Interleukin Signaling leading-edge genes were confirmed as concordant drivers of the Interleukin Signaling pathway enrichment as seen in Figure 2A and B. To interrogate network connectivity between the 23 validated Interleukin Signaling leading-edge genes, protein–protein interactions were visualized using the STRING database (Figure 2C). Within the 23 leading-edge genes, 83% participated in at least one interaction with another leading-edge gene, and interactions were found to be loosely grouped between three main families of genes, complement/coagulation cascade, cytokine–cytokine interactions and general cell proliferation.
Figure 1.

GSEA plots of the prognostic gene effects within the Interleukin Signaling pathway. The prognosis-weighted running-sum enrichment score is designated by a line with dots representing genes within the pathway of interest out of all possible genes in the dataset. (A) INHALE (discovery) cohort Interleukin Signaling pathway findings. (B) TCGA Lung (replication) cohort Interleukin Signaling pathway findings.
Figure 2.

Prognostic effects across both lung cohorts among the validated Interleukin Signaling leading-edge genes. The validated leading-edge subset ranked by their Cox proportional-hazards Z-scores and color shaded by significance values for the INHALE (A) and TCGA Lung (B) cohorts. (C) Network connectivity between leading-edge genes illustrating shared protein–protein interactions according to STRING. Gene families are colored according to KEGG membership: blue, cytokine–cytokine receptor interactions; red, Ras signaling and green, platelet activation/coagulation cascade. Kaplan–Meier plot of the estimated cumulative prognostic effects of the expression of the Interleukin Signaling leading edge divided into high and low according to the median value of the 23 gene signature in the INHALE (D) and TCGA Lung (E) cohorts.
Finally, we estimated the cumulative prognostic effects of the Interleukin Signaling leading-edge subset in each cohort, retrospectively, constructing a cumulative leading-edge pathway signature score for each NSCLC specimen based upon their cumulative leading-edge gene expression weighted by the Z-score of the gene-level multivariable regression coefficients. The Interleukin Signaling leading-edge signature score was strongly associated with patient survival in both discovery (HR = 1.59, P = 3.0 × 10–8) and replication (HR = 1.29, P = 7.4 × 10–7) cohorts and the median-split high/low univariate Kaplan–Meier plots are displayed in Figure 2D and E. The expressions of the 23 genes comprising the Interleukin Signaling signature for the discovery and replication cohorts are illustrated in Figure 3 alongside each individual’s signature score and relevant clinical covariates such as stage at diagnosis. These gene and pathway data suggest that tumor-immune expression at diagnosis is associated with patient prognosis and the majority of effects are localized to specific pathways of which Interleukin Signaling expression had the most substantial impact on overall survival.
Figure 3.

Heatmap of the expression of the 23 genes comprising the Interleukin Signaling leading-edge signature plotted alongside clinical covariates in the INHALE (A) and TCGA Lung (B) cohorts. Individual clinical covariates for each of the samples were plotted from left to right, African American status (black), stage at diagnosis (blue) and their scaled Interleukin Signaling gene signature score (green, low; gray, interquartile; red, high). Expression of each of the 23 genes was scaled and subsequent clustering was performed on the gene expression in the INHALE cohort to order the genes in both cohorts according to similarity. Individuals (y-axis) were ordered according to the similarity of their gene expression profile within three clusters separately by the quartiles of the Interleukin Signaling gene signature score: low (green; survival benefit), interquartile (gray) and high (red; survival detriment).
Discussion
Immunotherapy, directed at immune checkpoint inhibition, has resulted in significant survival improvements in the treatment of lung cancer; however, overall outcomes among patients with NSCLC remain poor. Studying and harnessing the naturally occurring variation in the immune signatures of lung tumors that predispose patients for better or worse outcomes to guide future molecular and therapeutic research holds promise to advance precision immunotherapy. Yet, the relevant immune-centric genes and pathways affecting prognosis, in NSCLC specifically, remain unknown. To address this, we investigated the expression of immune genes and pathways in primary tumor specimens in connection with patient outcomes. Our findings, within a large discovery and replication cohort, highlight an immune-centric component within NSCLC tumors that strongly influence patient survival and should become the focus of mechanistic studies and therapeutic interventions in lung cancer.
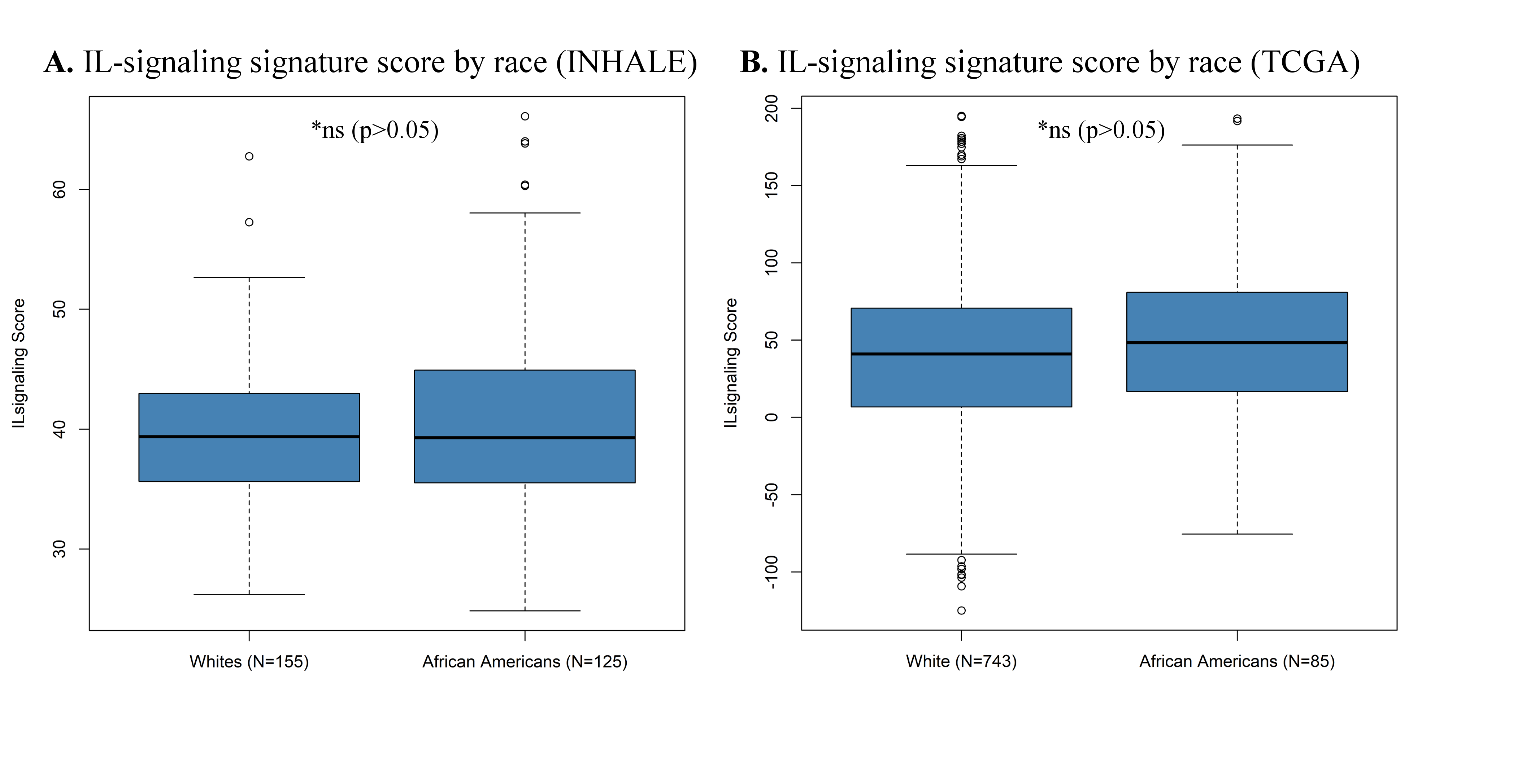
We observed a statistically significant enrichment of prognostic gene signatures within the Interleukin Signaling pathway in two independent NSCLC cohorts, when prognostic gene modeling was paired with pathway analysis. The strongest cross-cohort, gene–pathway associations were localized to 23 genes in the Interleukin Signaling leading edge. As our study cohort contained a considerable number of lung cancer cases in African Americans, we also interrogated whether there were distinct differences in the expression of these key Interleukin Signaling pathway genes in association with survival by race (Supplementary Figure 2, available at Carcinogenesis Online). Among the reduced Interleukin Signaling leading-edge genes, no discernable differences could be detected between NSCLC tumors obtained from African Americans as compared to those obtained from Caucasians.
The inclusion of FFPE tissues paired with array-based transcriptome profiling in the discovery cohort and fresh-frozen specimens paired with RNA-seq profiling in the replication cohort presents a potential issue when comparing genetic signatures between cohorts. Prior studies have interrogated, in depth, the intersection at which sequencing-based and array-based transcriptome profiling on fresh-frozen and FFPE tumor specimens reside. When comparing paired, non-TCGA FFPE and fresh-frozen tumor specimens, gene-pair correlations ranged from 0.83 to 0.94 when comparing these tumor tissue sources on the RNA-seq platform (20). Likewise, a paired-analysis of TCGA tumors using a multitude of tissue sources and sequencing techniques demonstrated that the expression of gene-pairs derived from FFPE array and fresh-frozen RNA-seq were highly correlated (correlation coefficient = 0.87) (21). These findings suggest that we should expect the introduction of only minor variance in gene signatures based upon technological differences alone when comparing the discovery and replication cohorts.
The reduced Interleukin Signaling gene set contains the interleukins/cytokines IL11, IL16, IL22RA1, IL28A and LIF/LIFR; coagulation cascade members FGA, FGB and FGG; and a remaining group of general signaling transduction/cascade members involved in growth, proliferation and differentiation such as KRAS, FGF4/19, GAB2, DUSP4 and PIK3R1. The differential survival detected for tumors with higher or lower expression of the cytokine and coagulation members is the most immediately relevant finding in the context of immunomodulatory tumor interactions, although it should be noted that several of the cell signaling transduction genes (IRAK1, PELI1 and KITLG) are known to directly play a role in immune-related cellular cascades.
Interleukins are under investigation as therapeutic tools to induce adaptive immune responses against tumor antigens, or as biomarkers for early detection/diagnosis (22,23). The use of interleukin therapy was approved in 1998 for use in advanced stage melanoma. Today, numerous interleukins are being investigated in either the preclinical or the clinical setting. Likewise, more recent research in lung cancer has highlighted the usefulness of monitoring serum interleukin levels, primarily IL6 and IL8, as risk markers and their detection may aid in early diagnosis of lung cancer development in at-risk populations (23–25). We observed that primary NSCLC specimens with higher expression of IL11, IL22RA1, IL28A and LIF and lower expression of IL16 and LIFR demonstrated worse overall survival in both cohorts. We predict that interventions, likely in combination regimens, that target the activation and inhibition of these interleukins within the tumor microenvironment will have immense value for tumors that display unfavorable activation of the Interleukin Signaling pathway, as evidenced by the poor survival phenotype observed in individuals with high pathway signature scores (Figure 2). Promising translational therapeutic research is already underway for several of these molecules. IL11 is a member of the IL6 family of cytokines and its aberrant expression in multiple epithelial cancer types such as gastric, colorectal and pancreatic malignancies has been linked to increased tumor grade and metastatic propensity (26). Moreover, translational research aimed at inhibiting IL11 in gastrointestinal carcinoma demonstrated a reduction in tumor burden and an increase in survival in mouse models when treated with an IL11 antagonist (27). LIF/LIFR expression has also been implicated in prognostic and molecular studies in breast cancer and is believed to play a role in IL6 family of cytokines mediating invasiveness and metastatic formation and reducing disease-free survival (28,29). Fewer studies have explored the roles of the IL10 family (IL22R, IL28) in cancer. This family of cytokines was initially thought to be purely immunosuppressive in nature, but is now believed to be a major regulator of CD-8+ T-cell antigen surveillance and may play a role in eliciting antigen presentation for subsequent recognition and elimination within tumors (30–32). Likewise, IL16 likely modulates T-helper cells acting as a chemoattractant, and some evidence exists to support its role in tumor prognosis in prostate, breast and ovarian carcinoma (33–35). We also observed poor survival among NSCLC tumors expressing high levels of the fibrinogen chains FGA, FGB and FGG. Exactly how fibrinogen and platelets interact with tumor cells to promote poor prognosis is less clear, although proposed mechanisms highlight a role in distant metastasis formation through vascular regulation, which theoretically could be useful as a potential biomarker for anti-VEGF therapy (36,37).
Our study has explored the prognostic potential of immune-centric gene and pathway expression within two distinct NSCLC cohorts. Investigating the expression of immune factors in lung tumors provides valuable insight into the ways in which tumors and the immune system interact to promote better or worse survival in patients with this disease. The most valuable prognostic signals were localized to a single pathway, Interleukin Signaling, and within this pathway, 23 genes were identified which drive the pathway’s significance. We propose these gene–pathway candidates as future targets for therapeutic and mechanistic studies to advance immunotherapy in lung cancer.
Funding
National Institutes of Health (R01CA141769, P30CA022453, T32CA009531).
Conflict of Interest Statement: None declared.
Supplementary Material
{kind=link}
{kind=link}
Abbreviations
- ES
Enrichment score
- FEPE
formalin fixed paraffin embedded
- GSEA
Gene Set Enrichment Analysis
- HR
hazard ratio
- INHALE
Inflammation, Health and Lung Epidemiology
- NSCLC
non-small-cell lung carcinoma
- TCGA
The Cancer Genome Atlas
References
- 1. Siegel R.L., et al. (2018)Cancer statistics, 2018. CA. Cancer J. Clin., 68, 7–30. [DOI] [PubMed] [Google Scholar]
- 2. Chalmers Z.R., et al. (2017)Analysis of 100,000 human cancer genomes reveals the landscape of tumor mutational burden. Genome Med., 9, 34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Rizvi N.A., et al. (2015)Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science, 348, 124–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Khalil D.N., et al. (2016)The future of cancer treatment: immunomodulation, CARs and combination immunotherapy. Nat. Rev. Clin. Oncol., 13, 273–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Director’s Challenge Consortium for the Molecular Classification of Lung Adenocarcinoma. et al. (2008)Gene expression–based survival prediction in lung adenocarcinoma: a multi-site, blinded validation study. Nat. Med., 14, 822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Beer D.G., et al. (2002)Gene-expression profiles predict survival of patients with lung adenocarcinoma. Nat. Med., 8, 816–824. [DOI] [PubMed] [Google Scholar]
- 7. Zhu C.Q., et al. (2010)Prognostic and predictive gene signature for adjuvant chemotherapy in resected non-small-cell lung cancer. J. Clin. Oncol., 28, 4417–4424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Gentles A.J., et al. (2015)The prognostic landscape of genes and infiltrating immune cells across human cancers. Nat. Med., 21, 938–945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Schwartz A.G., et al. (2016)Risk of lung cancer associated with COPD phenotype based on quantitative image analysis. Cancer Epidemiol. Biomarkers Prev., 25, 1341–1347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Cerami E., et al. (2012)The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov., 2, 401–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Cancer Genome Atlas Research Network. (2012)Comprehensive genomic characterization of squamous cell lung cancers. Nature, 489, 519–525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Cancer Genome Atlas Research Network. (2014)Comprehensive molecular profiling of lung adenocarcinoma. Nature, 511, 543–550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Irizarry R.A., et al. (2003)Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics, 4, 249–264. [DOI] [PubMed] [Google Scholar]
- 14. Johnson W.E., et al. (2007)Adjusting batch effects in microarray expression data using empirical Bayes methods. Biostatistics, 8, 118–127. [DOI] [PubMed] [Google Scholar]
- 15. Wang K., et al. (2010)MapSplice: accurate mapping of RNA-seq reads for splice junction discovery. Nucleic Acids Res., 38, e178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Li B., et al. (2010)RNA-Seq gene expression estimation with read mapping uncertainty. Bioinformatics, 26, 493–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Fabregat A., et al. (2018)The reactome pathway knowledgebase. Nucleic Acids Res., 46, D649–D655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Spitz M.R., et al. (2012)Multistage analysis of variants in the inflammation pathway and lung cancer risk in smokers. Cancer Epidemiol. Biomarkers Prev., 21, 1213–1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Subramanian A., et al. (2005)Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA, 102, 15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Bossel Ben-Moshe N., et al. (2018)mRNA-seq whole transcriptome profiling of fresh frozen versus archived fixed tissues. BMC Genomics, 19, 419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Zhao W., et al. (2014)Comparison of RNA-Seq by poly (A) capture, ribosomal RNA depletion, and DNA microarray for expression profiling. BMC Genomics, 15, 419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Rosenberg S.A., et al. (1994)Treatment of 283 consecutive patients with metastatic melanoma or renal cell cancer using high-dose bolus interleukin 2. JAMA, 271, 907–913. [PubMed] [Google Scholar]
- 23. Pine S.R., et al. (2011)Increased levels of circulating interleukin 6, interleukin 8, C-reactive protein, and risk of lung cancer. J. Natl. Cancer Inst., 103, 1112–1122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Pine S.R., et al. (2016)Differential serum cytokine levels and risk of lung cancer between African and European Americans. Cancer Epidemiol. Biomarkers Prev., 25, 488–497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Brenner D.R., et al. (2017)Inflammatory cytokines and lung cancer risk in 3 prospective studies. Am. J. Epidemiol., 185, 86–95. [DOI] [PubMed] [Google Scholar]
- 26. Putoczki T., et al. (2010)More than a sidekick: the IL-6 family cytokine IL-11 links inflammation to cancer. J. Leukoc. Biol., 88, 1109–1117. [DOI] [PubMed] [Google Scholar]
- 27. Putoczki T.L., et al. (2013)Interleukin-11 is the dominant IL-6 family cytokine during gastrointestinal tumorigenesis and can be targeted therapeutically. Cancer Cell, 24, 257–271. [DOI] [PubMed] [Google Scholar]
- 28. Johnson R.W., et al. (2016)Induction of LIFR confers a dormancy phenotype in breast cancer cells disseminated to the bone marrow. Nat. Cell Biol., 18, 1078–1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Metz S., et al. (2008)Novel inhibitors for murine and human leukemia inhibitory factor based on fused soluble receptors. J. Biol. Chem., 283, 5985–5995. [DOI] [PubMed] [Google Scholar]
- 30. Whittington H.A., et al. (2004)Interleukin-22: a potential immunomodulatory molecule in the lung. Am. J. Respir. Cell Mol. Biol., 31, 220–226. [DOI] [PubMed] [Google Scholar]
- 31. Sheppard P., et al. (2003)IL-28, IL-29 and their class II cytokine receptor IL-28R. Nat. Immunol., 4, 63–68. [DOI] [PubMed] [Google Scholar]
- 32. Dennis K.L., et al. (2013)Current status of interleukin-10 and regulatory T-cells in cancer. Curr. Opin. Oncol., 25, 637–645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Compérat E., et al. (2010)Tissue expression of IL16 in prostate cancer and its association with recurrence after radical prostatectomy. Prostate, 70, 1622–1627. [DOI] [PubMed] [Google Scholar]
- 34. Donati K., et al. (2017)Neutrophil-derived interleukin 16 in premetastatic lungs promotes breast tumor cell seeding. Cancer Growth Metastasis, 10. doi:10.1177/1179064417738513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Yellapa A., et al. (2014)Interleukin 16 expression changes in association with ovarian malignant transformation. Am. J. Obstet. Gynecol., 210, 272.e1–272.10. [DOI] [PubMed] [Google Scholar]
- 36. Labelle M., et al. (2014)Platelets guide the formation of early metastatic niches. Proc. Natl. Acad. Sci. USA, 111, E3053–E3061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Nash G.F., et al. (2002)Platelets and cancer. Lancet Oncol., 3, 425–430. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.