

Abstract
Rationale:
The aim of this study was to analyze the clinical and imaging features of a pediatric patient with mucopolysaccharidosis type IIIA (MPS IIIA) and a novel mutation of the N-sulfoglucosamine sulfohydrolase (SGSH) in 1 pedigree.
Patient concerns:
An 8-year-old female patient presented with developmental regression, seizures, cerebral atrophy, thickened calvarial diploe, apathy, esotropia, slender build, thick hair, prominent eyebrows, hepatomegaly, ankle clonus, muscle and joint contractures, and funnel chest.
Diagnoses:
The patient was diagnosed as autosomal recessive (AR) MPS IIIA with a novel mutation in the SGSH gene.
Interventions:
Genomic DNA was extracted from the peripheral blood and next-generation sequencing (NGS) technology was used to detect pathogenic genes, and the Sanger method was applied to perform pedigree verification for the detected suspicious pathogenic mutations.
Outcomes:
The NGS done for the girl and her family showed 2 variations that were both missense mutations in SGSH. The c.1298G > A (p.Arg433Gln) was a known mutation, and the c.630 G > T (p.Trp210Cys) was a novel variation.
Lessons:
The common clinical manifestations of MPS IIIA were rapid developmental regression, seizures, cerebral atrophy, and thickened calvarial diploe. The results showed that the c.630 G > T was likely pathogenic according to bioinformatics analysis, which probably was a novel mutation. This study reports 1 case of MPS IIIA with some clinical features as determined via clinical and genetic analysis, and found a new mutation in the SGSH gene.
Keywords: mucopolysaccharidosis, novel mutation, SGSH
1. Introduction
Mucopolysaccharidosis (MPS) is a group of lysosomal storage disease. It is caused by lysosomal acidic hydrolytic enzyme deficiency, which could be a genetic defect, resulting in acidic mucopolysaccharide molecules (glycosaminoglycan) not being able to be properly degraded and stored in the lysosome, and disorders of cells, tissues, and organs. This may lead to a series of clinical symptoms and radiographic signs. According to clinical manifestations and different types of enzyme defects, MPS can be divided into 7 types, a total of 12 subtypes. Except for type II as X-linked recessive inheritance, the rest are autosomal recessive hereditary diseases (AR).[1] MPS type III (Sanfilippo syndrome, MPS type III, MPSIII) is divided into 4 subtypes,[2] MPSIIIA, MPSIIIB, MPSIIIC, and MPSIIID. Their clinical manifestations are very similar, mainly for progressive neurodegenerative disease. MPS type IIIA (Sanfilippo syndrome A, MPS IIIA, OMIM 252900) is the most common type with a prevalence of 0.28 to 1.89/0.1 million.[3,4]
MPS IIIA is caused by the genetic defect of N-sulfoglucosamine sulfohydrolase (SGSH, NM_000199) gene, which could cause lysosomal enzyme sulfite enzyme deficiency, leading to accumulation of glycosaminoglycans (GAGs) and heparan sulfate in various organs of the body, followed by a series of rapid progression of lethal diseases characterized by severe neurological symptoms and skeletal deformities. SGSH is located at 7q25.3 with 11 kb spans and contains 8 exons and encodes a protein of 502 amino acids. [5,6] According to OMMIN, 145 SGSH mutations have been reported, including missense mutations, duplication, deletion, insertion, and so on.
2. Materials and methods
2.1. Collection of medical history and related auxiliary examination
An 8-year-old girl was admitted to our hospital on June 25, 2015, due to “8 years of global developmental delay, 4 years of retrogression and seizures with dysphagia 1 year.” The girl's development was normal before she was 6 months of age. She could sit without assistance at the 8th month and walk at the 21st month with ataxia. However, she began speaking single, simple words (e.g., sister, brother) at the age of 4 years. Now, she cannot sit down and walk independently with unclear articulation and sleep disturbances, and she was misdiagnosed as “cerebral palsy.” The girl is the first child born to young and healthy parents who are nonconsanguineous. The course of pregnancy and birth was normal. There is no epilepsy in her family history. She had a younger and healthy sister. The physical examination showed that the girl had impaired pursuit initiation and maintenance, apathy, esotropia, slender build, thick hair, prominent eyebrows (Fig. 1A) without speaking and communication. She had visual deterioration, sensitivity to light, and apathetic facial expressions. She had funnel chest and hepatomegaly. Her neurologic examination revealed hypertonia of the extremities and increased tendon reflexes of the lower and upper limbs. She also had ankle clonus, muscle, and joint contractures (Fig. 1B, C). Furthermore, the girl underwent cerebrospinal fluid examination for protein, glucose, lactate levels, and cell count yielding normal results. Metabolic screening indicated normal results. The chest X-ray showed mild broadening of ribs (Fig. 1D). Brain MRI indicated cerebral atrophy and thickening of calvarial diploe with increased signal (Fig. 2).
Figure 1.
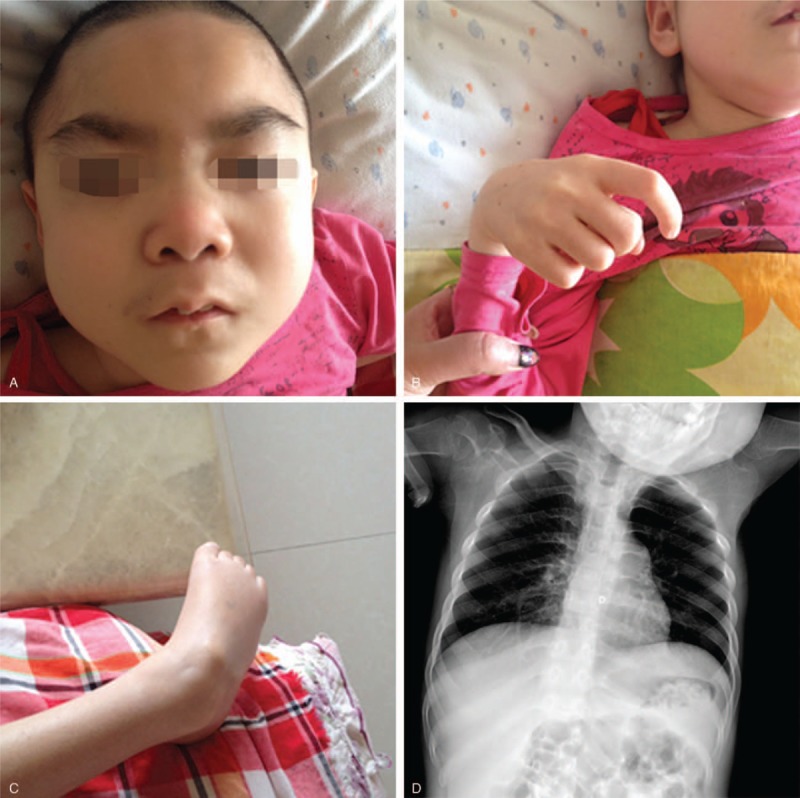
(A) Heavy hair, word brow; (B, C) Fingers and elbow flexion, straightening difficulties; (D) Mild broadening of the ribs.
Figure 2.
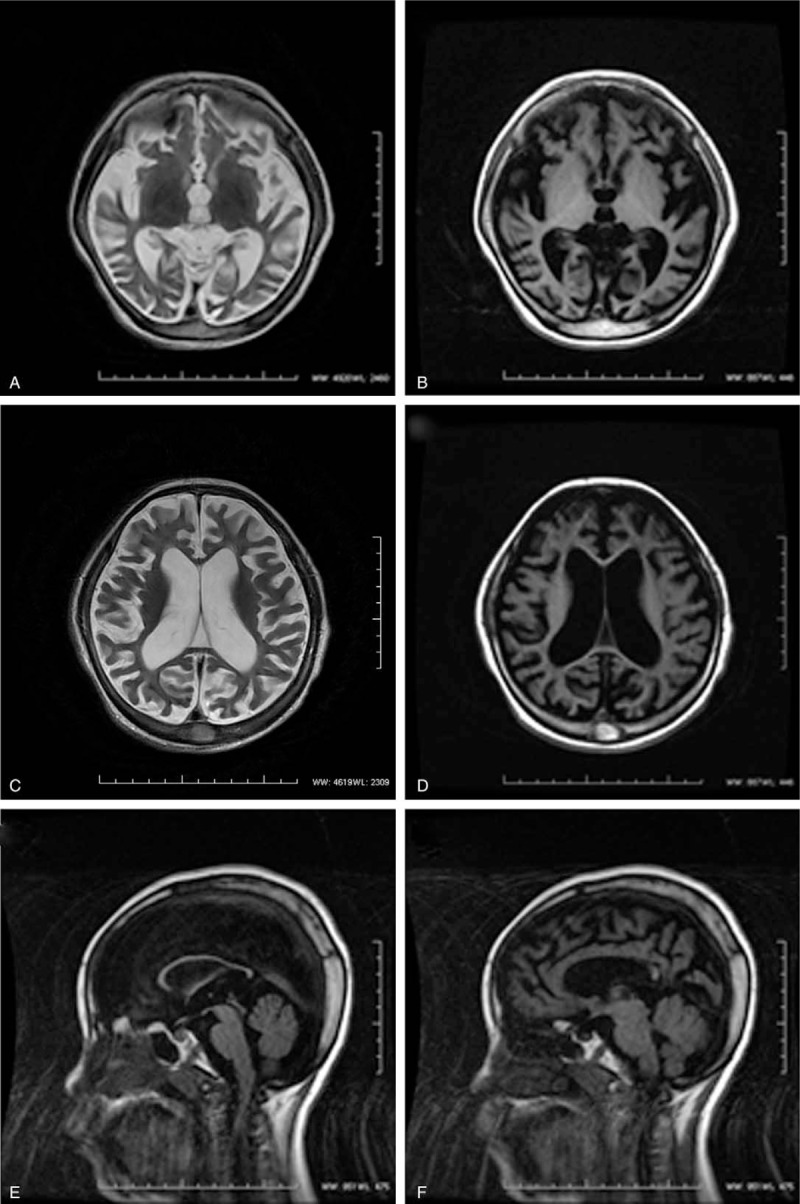
(A–D) Diffuse brain atrophy. (E, F) Corpus callosum atrophy, skull plate thicken.
2.2. Detection of pathogenic genes
2.2.1. Capture of target sequence and high-throughput sequencing
To identify the pathogenic gene, the genomic DNA isolated from blood samples of the family was amplified and sequenced by next-generation sequencing (NGS) in Kangso Medical Inspection (Beijing, China). The panel test includes 270 genes related to 251 types of hereditary metabolic diseases such as sticky lipid storage disease, Sanfilippo syndrome, glutaric acidemia, and familial hypercholesterolemia with an average sequencing depth of 100 × . Samples of the pediatric patient, the parents, and the younger sister were validated using first-generation sequencing.
2.2.2. Validation not a single nucleotide polymorphism (SNP)
In order to verify whether the mutation c.630 G > T was a single nucleotide polymorphism (SNP), 100 healthy children were randomly selected as the control group and 2 mL of peripheral venous blood was collected from each of these children. DNA was extracted by the molecular pathology laboratory of our hospital. Specific primers were designed and the sequences of primers were as follows: The forward primer was CGGAACCTTCTGTGAGAAG, and the reverse primer was GAGCCAGGCTTAGAACAGAC. Then, the region of variability was amplified by PCR. The product was sequenced by Sanger sequencing and the corresponding sequences were compared with GenBank. The results showed that c.630 G > T did not mean SNP.
2.2.3. Bioinformatics analysis
The SIFT (http://sift.jcvi.org/), PolyPhen-2 (Polymorphism Phenotyping v2) software8 (http:// genetics.bwh.harvard.edu/pph2/), and MutationTaster online tool9 (http:// www. Mutationtaster. org/) were used to predict the damaging effects of these 2 mutations.
All subjects involved provided written informed consent, and the study protocol was approved by the medical ethics committee of the Inner Mongolia Medical University.
3. Results
Compound heterozygous missense mutations at nucleotide position c.1298 (c.1298G > A) and c.630 (c.630G > T) were identified in the proband, which could induce amino acid changes from Arg to Gln (p.Arg433Gln) and Trp to Cys (p.Trp210Cys), respectively (Table 1). According to OMIM, the c.1298G > A mutation was reported to cause Sanfilippo syndrome previously [7] (Fig. 3 A), and the c.630 G > T mutation has not been reported yet (Fig. 4E). The girl's parents (Figs. 3B, 3C and 4F, 4G) and sister were heterozygous (Figs. 3D and 4H).
Table 1.
N-sulfoglucosamine sulfohydrolase (SGSH) gene mutations identified in the proband's family.

Figure 3.
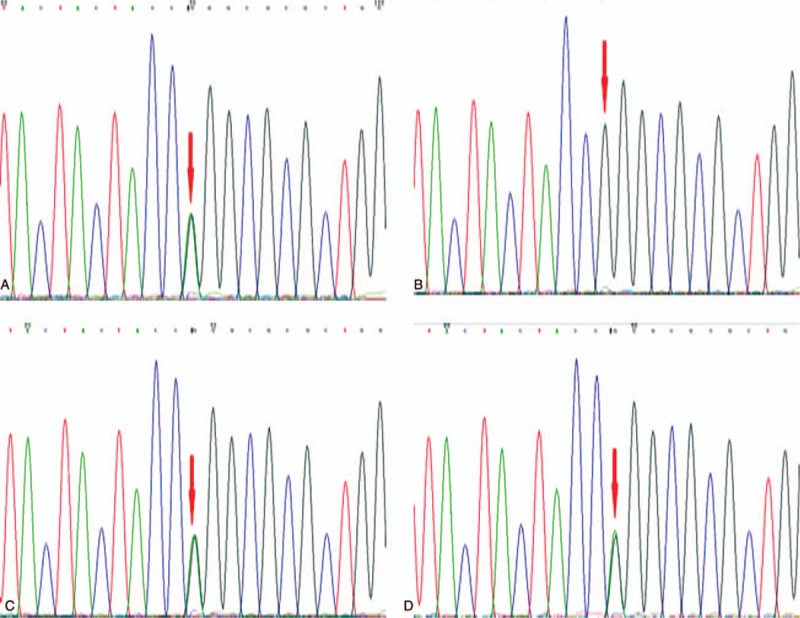
(A, C) SGSH mutations c. 1298G > A, p.Arg433Gln, for the proband and her mother, respectively. (B, D) Normal sequencing maps for her father and sister, respectively.
Figure 4.
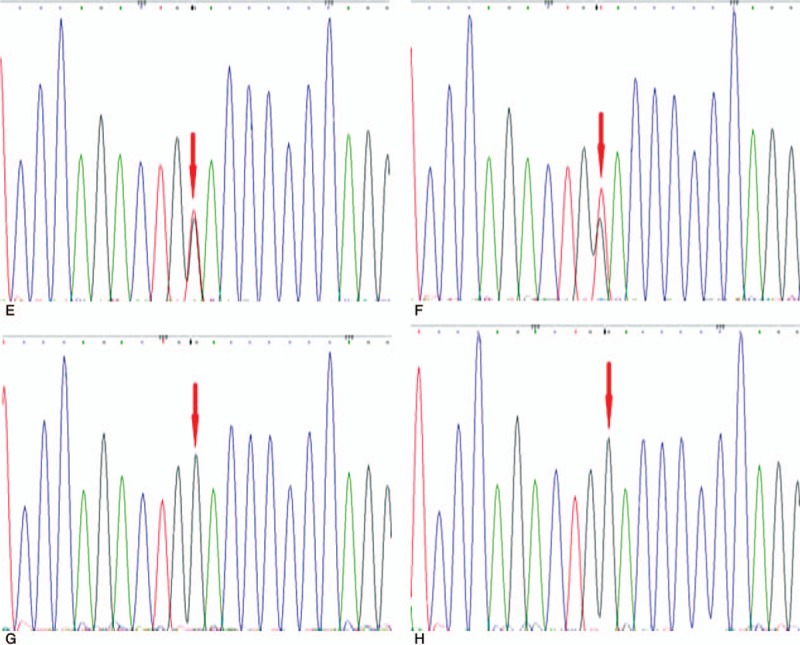
(E, F) SGSH mutations c.630G > T,p.Trp210Cys for the proband and her father, respectively; (G, H) Normal sequencing maps for her mother and sister, respectively.
Three mutation effect predication software were used. The SIFT predicted that the c.1298G > A and c.630 G > T mutations were “deleterious” and “tolerated,” respectively. The MutationTaster software showed that both mutations were “disease-causing.” The Polyphen2 prediction found that both mutations were “probably damaging” (Table 1).
Mutation c.630 G > T did not mean SNP. According to OMMIN, there were 145 mutations except for c.630 G > T, so c.630 G > T was considered as a novel probably pathogenic mutation.
4. Discussion
In general, the MPS IIIA patients develop normally until the age of 4 to 5 years followed by seizures and deaths usually occurring between 20 and 30 years of age.[8] But there are data suggesting that the clinical course of MPS IIIA is more severe, with an earlier onset, more rapid progression of symptoms, and shorter survival than the other subtypes.[9] Our patient's clinical manifestations and developmental regression were similar with them. In addition, many children are misdiagnosed as idiopathic development/language delay, attention deficit/hyperactivity disorder, or autism spectrum disorder.[10] The patient had been misdiagnosed as "cerebral palsy.” The early diagnosis is very important. The MPS IIIA patients have thick hair, special face, but short stature and skeletal anomalies are not serious. In this case, the patient had an ugly face, depressed nasal bridge, abnormality of the eyebrow, and thick hair. The patient had hepatomegaly without splenomegaly, and her ankle and wrist flexion were contracture (Fig. 1B, C). The cranial magnetic resonance imaging (MRI) showed a special phenomenon of calvarial diploe thickening and extensive cortical atrophy and more prominent white matter such as corpus callosum atrophy, a finding consistent with what Rajech et al[11] reported. X-ray examination showed mild broadening of the ribs. The chest radiograph shown in Fig. 1 had no ribbon-like rib and vertebral body deformity. We speculated that it may be related to the type of genetic mutations in children, which needs to be confirmed by further increasing the sample size.
MPS IIIA is an autosomal recessive hereditary disease, and SGSH gene is the disease-causing gene. For such a mode of inheritance, compound heterozygous mutations may cause disease. The pediatric gene diagnosis was completed by NGS and verified by PCR. In the SGSH gene of the patient, there were compound heterozygous nucleotide variations of c.1298G > A (nucleotide position c.1298 changed from G to A) and c.630G > T (the nucleotide position c.630 changed from G to T), which led to the changes of amino acids from Arg to Gln (p.Arg433Gln) and from Trp to Cys (p.Trp210Cys), respectively. The mutations were both missense mutations. They might affect protein functions as the above 3 software predicted. Genetic analysis showed that both of the mutations were from his parents who were both heterozygotes. The mutations are in accordance with autosomal recessive inheritance. Combined with clinical manifestations of the patient and the predicted results of 3 software, and according to the ACMG 2015 guidelines, the mutations were very likely to be pathogenic. The MPS-IIIA diagnosis was established. In pedigree analysis, the patient's sister had a normal phenotype, a heterozygous mutation c.1298G > A was found, and there was no variation at c.630 G > T. The pathogenicity of c.630G > T has not been reported yet, while the pathogenicity of mutation c.1298G > A has been reported to be related to MPS-IIIA.[7]
After retrieving databases (1000 Genomes and dbSNP), we found that the mutations were not at polymorphic sites and the frequency of occurrence in the population was extremely low. In order to verify whether the mutation c.630G > T was a pathogenic mutation, PCR was performed on the 100 healthy control children to prove that the site was not a gene polymorphism site. Considering the pathogenicity predicted by 3 software, the site may be judged as disease-causing mutation. So far, there are 145 mutations reported in the gene SGSH, while the mutation c.630G > T was not included. So, it may be a novel mutation and a functional verification study is needed, which is one of the main research contents in the future. Through this case, we have found a novel mutation site of SGSH gene, which provides a new research material for the genetic background of MPSIIIA children.
In summary, we conclude that the mutation c.630G > T was a novel mutation of SGSH gene, and cerebral atrophy and calvarial diploe thickening may be associated with the mutation of c.630G > T. The compound heterozygous mutations of SGSH identified in the child with MPSIIIA are likely cause for the disease. Genetic testing allows identification of mutations even in early life of the suspected cases and will help to confirm the diagnosis and clinician will be able to counsel the parents regarding prognosis and can guide for antenatal diagnosis during next pregnancy.
Acknowledgment
The authors thank the Foundation of Millions of project of science and technology of Inner Mongolia Medical University [YKD2016KJBW (LH) 011] for the support.
Author contributions
Conceptualization: Xiaohua Li, Xiaomeng Zhang, Junxian Fu.
Data curation: Baiyu Chen, Mengli Zhuang, Xiaohua Li.
Formal analysis: Xiaomeng Zhang, Xiaohua Li.
Methodology: Rui Xiao, Junxian Fu, Xiaohua Li, Yinglong Huang.
Resources: Guanglu Yang.
Validation: Zhuo Fu.
Writing – original draft: Xiaohua Li, Yinglong Huang.
Writing – review & editing: Yinglong Huang.
Footnotes
Abbreviations: AR= autosomal recessive, MPS IIIA= mucopolysaccharidosis type IIIA, NGS = next-generation sequencing, SGSH= N-sulfoglucosamine sulfohydrolase, SNP= single nucleotide polymorphism.
XL and RX contributed equally to this article.
The authors have nothing to disclose and no conflicts of interest.
References
- [1].Parenti G, Andria G, Ballabio A. Lysosomal storage diseases: from pathophysiology to therapy. Annu Rev Med 2015;66:471–86. [DOI] [PubMed] [Google Scholar]
- [2].Andrade F, Aldamiz-Echevarria L, Llarena M, et al. Sanfilippo syndrome: overall review. Pediatr Int 2015;573:331–8. [DOI] [PubMed] [Google Scholar]
- [3].Anthony O. Sanfilippo syndrome: causes, consequences, and treatments. Appl Clin Genet 2015;8:269–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Heron B, Mikaeloff Y, Froissart R, et al. Incidence and natural history of mucopolysaccharidosis type III in France and comparison with United Kingdom and Greece. Am J Med Genet A 2011;155A:58–68. [DOI] [PubMed] [Google Scholar]
- [5].Suzan J, Knottnerus, Stephanie C, et al. Prediction of phenotypic severity in mucopolysaccharidosis type IIIA. Ann Neurol 2017;82:686–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Karageorgos LE, Guo XH, Blanch L, et al. Structure and sequence of the human sulphamidase gene. DNA Res 1996;3:269–71. [DOI] [PubMed] [Google Scholar]
- [7].Chabás A, Montfort M, Martínezcampos M, et al. Mutation and haplotype analyses in 26 Spanish Sanfilippo syndrome type A patients: possible single origin for 1091delC mutation. Am J Med Genet 2001;100:223–8. [DOI] [PubMed] [Google Scholar]
- [8].Christine L, Chris J, Simon A. Mortality in patients with Sanfilippo syndrome. Orphan J Rare Dis 2017;12:168–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Elsa G, Shapiro, Igor N, et al. A prospective natural history study of mucopolysaccharidosis type IIIA. J Pediatr 2016;170:278–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Robin K, Kyle R, Kathleen D, et al. Acquired autistic behaviors in children with mucopolysaccharidosis type IIIA. J Pediatr 2014;164:1147–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Rajech S, Muhammad M, Abdelnaser Z, et al. Sanfilippo type A: new clinical manifestations and neuro-imaging findings in patients from the same family in Israel: a case report. J Med Case Rep 2014;8:78–83. [DOI] [PMC free article] [PubMed] [Google Scholar]