

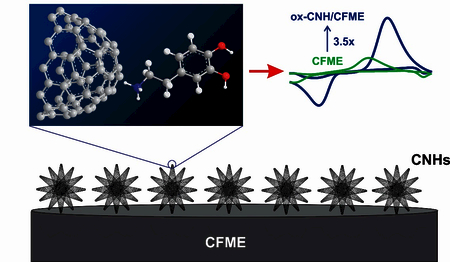
Abstract
Carbon nanohorns (CNHs), closed cone-shaped cages of sp2-hybridized carbons, are a promising nanomaterial to improve carbon-fiber microelectrode (CFME) dues to their high specific surface area and edge planes, but few studies have tested their electrochemical properties. Here, we tested the dopamine detection at electrodeposited CNHs on CFME (CNH/CFME). The optimized concentration of CNHs in the deposition solution is 0.5 mg/mL, and the optimized electrodeposition waveform is 10 cycles of triangular waveform scanned from –1.0 V and +1.0 V at 50 mV/s. Using fast-scan cyclic voltammetry, the optimized CNH/CFME enhances dopamine peak current to 2.3 ± 0.2 times that of the CFME. To further increase the current, CNH/CFMEs were oxidized in NaOH (ox-CNH/CFME), which creates more defects and surface oxide groups to adsorb dopamine. The oxidative etching further increases the peak current to 3.5 ± 0.2 times of the CFME, and ox-CNH/CFME had a limit of detection of 6 ± 2 nM. The dopamine anodic current at ox-CNH/CFME was stable for 8 h of continuous scanning. The ox-CNH/CFME enhanced the anodic peak current for other cationic neurotransmitters including epinephrine, norepinephrine, and serotonin, but less enhancement was found for ascorbic acid, showing higher selectivity for cationic molecules. CNHs also decreased tissue biofouling at CFME. Thus, electrodeposited CNHs are a promising new method for increasing the surface area and current of CFMEs for dopamine detection.
Keywords: carbon nanohorn, cyclic voltammetry, dopamine, electrodeposition, microelectrode
1. Introduction
The carbon nanohorn (CNH) is a cone-shaped cage of graphene consisting of mainly sp2-hybridized carbons. Each individual cone has a diameter of 2–5 nm and a length of 40–50 nm, and has a 5 pentagon-shaped closed cap [1,2]. CNH cones can form dahlia-like aggregates, like petals sticking out from a center, with an overall diameter of 100 nm [2]. The small sizes, combined with conjugated π-bonds, result in high specific surface area and high electronic conductivity [2]. CNHs have similar properties to the ends of carbon nanotubes (CNTs); thus, they can be easily opened and oxidized to provide edge-plane carbon sites and oxide-containing functional groups [2–4]. In addition, the conical structure causes high porosity, ring strain, and high electric field at the cone tip, which is significant for high adsorption capability, electron transfer kinetics, and chemical reactivity [1,2,5]. One key feature distinguishing CNHs from other carbon nanomaterials is high purity because CNHs can be synthesized via laser ablation of graphite at room temperature without any metal catalyst [2,6]. Because of these unique properties, CNHs have been used in various applications including gas adsorptions [7], drug carriers [8], and electrochemical energy devices [9]. However, there have been relatively few reports of using CNHs for electrochemical sensors.
Many carbon nanomaterials have been used to modify and improve the performance of electrochemical sensors, particularly to coat glassy carbon or carbon-fiber microelectrodes (CFMEs) to improve sensitivity [10,11]. Graphene itself has a large specific surface area, high conductivity, and good electrocatalytic properties [10,12] , and has been drop-casted, as reduced graphene oxide, onto glassy carbon electrodes to enhance the signal for dopamine.[13]. Another popular nanomaterial is the CNT, which is a rolled graphene sheet existing as a hollow tube [10,12]. When the end of the tube is opened, it generates defect sites or edge plane sites, which promote electron transfer, causing electrocatalytic effects and enhancing adsorption of cationic neurotransmitters such as dopamine and epinephrine [10,14,15]. CNTs can be made into electrodes in a variety of ways: they can be dip-coated or drop-casted onto an electrode [16–19], grown on a substrate to make an electrode [20,21], or spun as a fiber [22–24] or yarn [25–27] which is then fabricated into a microelectrode. CNT electrodes work best with the ends aligned and directly exposed to the solution, leading to the theory that the greatest electroactive sites are on the exposed ends [20,21]. CNHs have only been sparsely used as electrochemical sensors, but the hypothesis is that they would provide good electrochemical enhancement because their properties are similar to those of CNT ends [1,2,7]. The cone-shaped CNH ends can be oxidized and opened and the dahlia-like aggregates expose the ends like petals, leading to strong adsorption of cationic molecules [2,3,11,14]. CNHs have been used to fabricate electrodes; for example, drop-casting CNHs improved the limit of detection (LOD) of screen-printed electrode [28]. A CNH-modified glassy carbon electrode resolved dopamine, uric acid, and ascorbic acid peaks [29] and resolved dihydroxybenzene isomer peaks [30]. However, smaller CNH-modified electrodes that are suitable for in vivo measurements of neurotransmitters have not been fabricated, and drop-casting is not an effective method for coating cylindrical microelectrodes [10]. Thus, new methods for depositing CNHs must also be developed.
The purpose of this study was to optimize fabrication of CNH-modified microelectrodes and explore their properties for the fast-scan cyclic voltammetric (FSCV) determination of dopamine. Here, we develop electrodeposition as a simple procedure to prepare the carbon nanohorn-modified carbon-fiber microelectrode (CNH/CFME) from a CNH dispersion. Parameters for electrodeposition were optimized and electrodeposition deposits more CNHs than dip-coating alone. CNHs increase the Faradaic current of dopamine redox reaction, and oxidative etching of CNH/CFME (ox-CNH/CFME) increased the current for dopamine even more because it opens the CNH tips. Overall, CNHs are a promising nanomaterial for use in electrochemical neurotransmitter sensors and can easily be electrodeposited to improve dopamine detection.
2. Experimental Section
2.1. Chemicals
Dopamine, epinephrine, norepinephrine, serotonin, and ascorbic acid were purchased from Acros Organics (Morris Plains, NJ). A 10 mM stock solution of each analyte was prepared in 0.1 M HClO4. The final working solutions were daily prepared by diluting the stock solution in a phosphate buffer saline (PBS) (131.25 mM NaCl, 3.00 mM KCl, 10 mM NaH2PO4, 1.2 mM MgCl2, 2.0 mM Na2SO4, and 1.2 mM CaCl2 with pH adjusted to 7.4) to the desired concentration.
2.2. Preparation of CNH/CFME and ox-CNH/CFME
Prior to the electrode modification, a cylindrical CFME was prepared by the same procedure as the previous work [31]. Briefly, a T-650 carbon fiber (7-μm diameter, Cytec Engineering Materials, West Patterson, NJ) was pulled into a glass capillary (1.28 mm inner diameter × 0.68 mm outer diameter, A-M Systems, Sequim, WA) by an aspirating pump. Then, the capillary was pulled by an electrode puller (model PE-21, Narishige, Tokyo, Japan) to get two electrodes. The extended fiber was cut to a length of 100 μm. After that, each electrode was epoxied by dipping in an 80°C-solution of Epon Resin 828 (Miller-Stephenson, Danbury, CT) and 14% m-phenylenediamine hardener (Acros Organics, Morris Plains, NH) for 30 s to seal the fiber with the glass capillary. Finally, the epoxied electrode was left overnight at room temperature, cured in an oven at 100°C for 2 h, and 150°C overnight. An electrical connection between the fiber and a connecting wire was made by filling 1 M KCl in the capillary before using.
To prepare the modified electrodes, single-walled CNHs synthesized at Oak Ridge National Laboratory (ORNL), Oak Ridge, TN (Tunneling electron microscopy (TEM) image of CNHs is shown in Fig. S1A) were dispersed in water with 0.1 M NaCl and 0.01 M sodium dodecyl sulfate (SDS) and homogenized with an ultrasonic tissue homogenizer (model 150VT, Biologics, Manassas, VA) for 10 min. After that, the electrodeposition was performed in a batch electrochemical cell containing Ag/AgCl reference electrode, Pt counter electrode, and CFME working electrode in the CNH dispersion. Using a potentiostat (Gamry Instruments, Warminster, PA), a repeated cyclic voltammetric waveform between –1.0 to +1.0 V at a scan rate of 50 mV/s for 5–25 cycles was applied to the CFME to obtain the CNH/CFME. Finally, the oxidative etching was done by applying a constant potential of +1.5 V for 1–4 min to the CNH/CFME to obtain the ox-CNH/CFME. All modified electrodes were left overnight before they were tested.
2.3. Surface Characterization
Scanning electron microscopy (SEM) images were taken using Quanta 650 (FEI Company, Hillsboro, OR) at the Nanoscale Materials Characterization Facility, Department of Materials Science and Engineering, University of Virginia. The secondary electron detector images were recorded with an accelerating voltage of 2 kV and a working distance of approximately 10 mm. Raman spectra were obtained from a Renishaw 100 confocal micro-Raman system (Renishaw, Hoffman Estates, IL) at ORNL, using 532-nm laser.
2.4. FSCV Instrumentation
FSCV experiments were performed with a ChemClamp potentiostat and headstage (Dagan, Minneapolis, MN). The triangular waveform with a holding potential of –0.4 V, a switching potential of +1.3 V, a scan rate of 400 V/s, and a repetition rate of 10 Hz was applied to a working microelectrode versus Ag/AgCl reference electrode. The buffer and test solutions were injected through the flow cell at 2 mL/min by a syringe pump (Harvard Apparatus, Holliston, MA) and a flow-injection system consisting of a six-port loop injector with an air actuator (VIVI Valco Instruments, Houston, TX). The data were collected with HDCV Analysis software (Department of Chemistry, University of North Carolina at Chapel Hill).
2.5. Statistics
All values are given as the mean ± standard error of the mean (SEM) for n number of electrodes. All statistical tests were performed in GraphPad Prism 7.0 (GraphPad Software, La Jolla, CA), and significance was defined at p < 0.05.
3. Results and Discussion
The goal of this study is to investigate the extent to which CNHs and oxidative etching improve FSCV electrochemical detection of dopamine. CNHs were prepared as a conductive, homogeneous dispersion in order to be compatible with the electrodeposition technique and to prevent inter-dahlia agglomeration of CNHs [32]. The solvent contained 0.1 M NaCl as a supporting electrolyte and 0.01 M SDS as a surfactant to disperse CNHs. Fig. S1A shows a TEM image of the CNH dahlias. Electrodeposition was performed by using CV waveform which can attract CNH particles from the dispersion onto the CFME surface by electrostatic interaction.[33] Each dahlia CNHs particle is surrounded by SDS molecules as a negatively charged micelle. The positively scanned CV waveform attracts the negative micelles to bring and attach CNHs on the CFME surface; then the SDS molecules are dissolved back into the aqueous solution.
3.1. Surface and Electrochemical Characterization of CNH/CFME
CFMEs were initially subjected to the electrodeposition from a 0.5 mg/mL CNH dispersion with a slow-scan CV waveform (–1.0 V to +1.0 V, 50 mV/s, 10 cycles). The SEM images in Fig. 1A show distributed spherical particles on the carbon fiber which are not present on an unmodified CFME (Fig. S2). From Fig. 1B, which is the enlarged image of CNH/CFME, these CNHs are between 80–120 nm, which is consistent with the reported diameter of the dahlia-aggregated CNHs [2]. This spherical shape and size is also similar to other reported literature on the CNH-modified electrodes [34,35] and CNH assemblies [36]. Therefore, these SEM images confirm that CV electrodeposition technique is an appropriate technique to prepare CNH/CFME. CNH particles did not completely cover the carbon-fiber surface, even when the CNH concentration or electrodeposition cycles were increased. Instead, using higher CNH concentrations led to agglomeration of the dahlia-CNHs, and resulted in thicker, but uneven coatings which are undesirable for our electroanalytical application [10].
Fig. 1.

SEM images show the electrodeposited CNHs on the CFME surface. (A-B) CNH/CFME prepared from electrodeposition of 0.5 mg/mL CNH using 10 cycles of CV scanned between –1.0 V to +1.0 V at a scan rate of 50 mV/s. (C-D) ox-CNH/CFME prepared from the oxidative etching of CNH/CFME in 1 M NaOH at a constant potential of +1.5 V vs. Ag/AgCl for 1.5 min.
Fig. 2 shows FSCV detection of 1 μM dopamine before and after electrodeposition of CNHs. From the representative cyclic voltammogram (Fig. 2A), CNHs enhanced the dopamine anodic peak current from 20 nA to about 50 nA, an increase of 2.5-fold. The cathodic current also increased by the same magnitude. The background current, or charging current, is proportional to the electrode surface area and specific capacitance of the electrode material [37]. From Fig. 2B, the background current increased from about 500 nA to 750 nA, about 1.5-fold, at this particular electrode. The magnitude of background current increase is less than that of the peak current. The other features of the CV, including ΔEp, and ratio of anodic to cathodic peak did not obviously change, which implies that there is no electrocatalytic effect of CNHs.
Fig. 2.
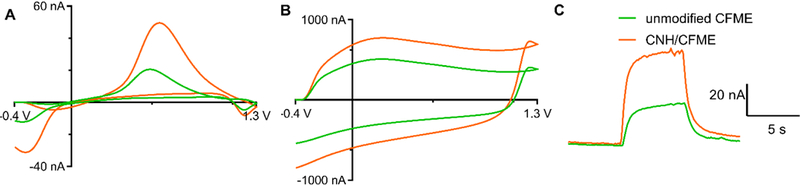
Example data before and after CNH deposition. FSCV response from unmodified CFME (green line) and CNH/CFME (orange line) prepared from the electrodeposition of 0.5 mg/mL CNH using 10 cycles of CV scanned between –1.0 V to +1.0 V at a scan rate of 50 mV/s. (A) background-subtracted CV of 1 μM dopamine, (B) background current in PBS pH 7.4, and (C) current vs. time trace of bolus injection of 1 μM dopamine.
3.2. Optimization of CNH Electrodeposition
We optimized the CNH electrodeposition process by varying the concentration of CNHs in the dispersion and the number of electrodeposition cycles. In Fig. 3A, electrodeposition was performed in CNH dispersions at concentrations from 0.1 to 1.5 mg/mL and there was a significant main effect of CNH concentration on the anodic peak current for 1 μM dopamine (one-way ANOVA, p < 0.001, n = 4–6). The control here is blank electrodeposition, where the electrodeposition waveform was applied to the CFME in the solvent without CNHs. All CNH concentrations above 0.1 mg/mL significantly increased the anodic peak current of 1 μM dopamine compared to control (Bonferonni post-test, n = 4–6). The anodic peak current increased with increasing CNH concentration from 0.1 to 0.5 mg/mL, but then plateaued or even decreased slightly after that. The background current also significantly increased with CNH concentration (one-way ANOVA, p < 0.05, n = 4–6, Fig. S4A), thus the cause of the plateau is likely a thicker, multilayer CNH deposition with higher concentrations. Here, our result indicates 0.5 mg/mL as the optimum concentration of CNH and this was used in all further experiments.
Fig. 3.
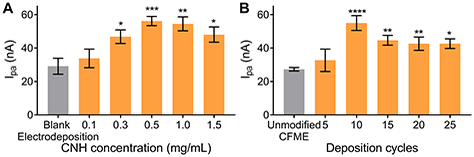
Optimization of CNH deposition parameters. Average 1 μM dopamine anodic peak currents for CNH/CFME preparation. (A) Effect of CNH concentration while using 10 cycles of electrodeposition waveform. Blank electrodeposition was used as a control; the electrodeposition waveform was applied to a CFME in the solvent without CNHs. (B) Effect of number of deposition cycles while using 0.5 mg/mL CNH dispersion. Unmodified CFME was used as a control. (n = 4–6, error bars represent SEM, one-way ANOVA with Bonferroni post-test compared to the control (gray bars). *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001)
Fig. 3B shows the effect of number of deposition cycles, from 5 cycles to 25 cycles. The control is an unmodified CFME. There is a significant main effect of number of deposition cycles (one-way ANOVA, p < 0.0001, n = 4–6). Increasing from 5 to 10 cycles enhances the anodic peak current of the CNH/CFME because using higher number of deposition cycles provides more time for negatively-charged CNHs to be electrodeposited on the CFME. However, the response plateaus after 10 cycles, despite the fact that the background keeps increasing (Fig. S4B), likely because the thicker coating layer would not all be accessible for electrochemical reactions. CVs are noisier when CNH/CFMEs are prepared with higher number of electrodeposition cycles or CNH concentrations. This noise may come from the agglomeration of CNH particles on the electrode surface or in the dispersion and noise is proportional to background current and electrode surface area [38]. Therefore, the optimized number of electrodeposition cycles is 10 cycles.
In addition, a dip coating control was performed by dipping the CFME in the 0.5 mg/mL without applying a potential. Immersing the CFME in CNH dispersion for 800 s (the same length of time as 10 cycles of electrodeposition) caused no effect as the anodic peak currents for dopamine before (26 ± 4 nA) and after dip coating (27 ± 4 nA) were not significantly different (paired t-test, p = 0.898, n = 5). Thus, electrodeposition is crucial to coat the CNHs on the carbon-fiber surface.
The optimized CNH/CFME was electrochemically characterized and average data from five electrodes is shown in Table 1. CNH/CFME has a 2.3 ± 0.2 times higher anodic peak current than CFME and a 1.5 ± 0.1 times higher background current. The LOD was found by determining the concentration of dopamine giving signal-to-noise ratio (S/N) of 3. Because CNH/CFME gives higher noise than CFME due to greater surface area, the LOD (11 ± 1 nM) is only slightly smaller and not significantly different than that of CFME (15 ± 1 nM) (unpaired t-test, p = 0.095, n = 4). Other electrochemical parameters, including anodic-cathodic peak separation (ΔEp), anodic-cathodic peak current ratio (ipa/ipc), and rise time are not different between CNH/CFME and unmodified CFME. The LOD of CNH/CFME is compared with previous literature in Table 2. Our electrode has better LOD than previous CNH-modified screen-printed [28] and glassy carbon electrodes [29] because of less agglomeration from electrodeposition compared to drop-casting. The LOD is also better than other CNT or graphene-modified electrodes prepared from other techniques [16,39,40].
Table 1.
Average electrochemical response to 1 μM dopamine and limit of detection for dopamine at unmodified CFME, CNH/CFME, and ox-CNH/CFME (n = 5).
| Electrode | Peak current enhancementa | Background enhancement | ΔEp (mV) | ipa/ipc | rise timeb (s) | LOD (nM) |
|---|---|---|---|---|---|---|
| CFME | 1 (defined) | 1 (defined) | 832 ± 2 | 1.65 ± 0.05 | 1.1 ± 0.1 | 15 ± 1 |
| CNH/CFME | 2.3 ± 0.2 | 1.5 ± 0.1 | 835 ± 2 | 1.68 ± 0.05 | 1.1 ± 0.2 | 11 ± 1 |
| ox-CNH/CFME | 3.5 ± 0.1 | 1.6 ± 0.3 | 825 ± 10 | 1.63 ± 0.05 | 1.1 ± 0.1 | 6 ± 2 |
ratio of the anodic peak current obtained from modified electrode to that obtained from unmodified CFME.
time from 10% to 90% of the peak current
Table 2.
LOD for dopamine detection compared at several carbon nanohorn-electrochemical sensors.
| Electrode material | Techniquea | LOD | Ref. |
|---|---|---|---|
| CNH/CFME | FSCV | 15 nM | This work |
| ox-CNH/CFME | FSCV | 6 nM | This work |
| CNH/glassy carbon electrode | LSV | 60 nM | [29] |
| CNH/screen-printed carbon electrode | DPV | 100 nM | [28] |
| oxidized CNH/ screen-printed carbon electrode | DPV | 400 nM | [28] |
FSCV = fast-scan cyclic voltammetry, LSV = linear seep voltammetry, DPV = differential pulse voltammetry
CNHs form a dahlia-like aggregate on the carbon-fiber surface. Each dahlia particle has a spherical shape with rough, porous surface, which provide interstitial surface area to adsorb dopamine on the electrode surface [1,7]. The sparse, monolayer of CNHs on the carbon-fiber surface is similar to that of CNTs grown on metal electrode [21]. This morphology provides a more accessible surface area for electroactive species, and leads to increases in redox current. Because the dopamine redox reaction at carbon-based microelectrode is adsorption-controlled,[41] the peak current increase should be proportional to the surface area increase, which can be estimated from the background charging current, assuming the carbon nanohorns and fibers have the same capacitance [37]. As shown in Fig. 2 and Table 1, CNH/CFMEs have a higher increase in Faradaic current than background current. Thus, CNHs enhance the specific adsorption of dopamine on the CNH/CFME surface in addition to increasing the surface area. Previous computer simulation and adsorption modeling suggest that CNHs have a high electric field at their cone tips, which also have defect sites containing surface oxide groups [1,2,7]. Hence, the CNH tips, which point radially outward from the aggregate center, enhance the dopamine adsorption. However, CNHs do not show electrocatalytic effect for dopamine redox reaction at CNH/CFME and thus peak potentials are not changed.
3.3. Surface Characterization and Optimization of Oxidative Etching
The CNH/CFME was further modified to improve the sensitivity toward dopamine detection by oxidative etching. This process generates defects in carbon microstructure and increases the amount of surface oxides [21,42]. The procedure was adapted from a study which oxidatively etched a vertically-aligned CNT-sheathed CFME for neurotransmitter detection [43]. The optimized CNH/CFME was oxidized by applying a constant potential of +1.5 V vs. Ag/AgCl in 1 M NaOH. Fig. 1C and 1D shows the SEM images of ox-CNH/CFME. No difference is observed in the morphology of the carbon-fiber or deposited CNHs between CNH/CFME and ox-CNH/CFME, which is expected because the oxidative etching should be on a smaller scale. Raman spectra were compared (Fig. S3) before and after etching to evaluate the intensity ratio of the D (defect) band around 1350 cm–1 and the G (graphitic) band around 1580 cm–1. The D/G ratio indicates proportion of the sp3-hybridized carbons compared to that of sp2-hybridized carbons [12,21]. The D/G ratio of ox-CNH/CFME (3.1 ± 0.1) is significantly higher than that ratio of CNH/CFME (2.7 ± 0.1) (unpaired t-test, p < 0.05, n = 4), so the ox-CNH/CFME has more defect sites or edge planes than CNH/CFME and can lead to increased dopamine adsorption [11,14].
Fig. 4 shows the electrochemical response of ox-CNH/CFME compared to that of CNH/CFME. In Fig. 4A, the anodic peak current of 1 μM dopamine at ox-CNH/CFME is increased from 50 nA to 75 nA, or about 1.5 times that of the CNH/CFME. The cathodic peak current also increases by the same magnitude. The peaks are shifted about 200 mV in the positive direction after oxidation. The background current of the ox-CNH/CFME is almost unchanged, from 960 nA to 980 nA after etching (Fig. 4B), and the background peaks are also positively shifted. The time response trace for ox-CNH/CFME and CNH/CFME to an injection of dopamine are not different from each other, showing that a fast time response is maintained after oxidation (Fig. 4C). As a control, unmodified CFMEs were also applied the +1.5 V constant potential for 90 s in 1 M NaOH. The anodic peak current for dopamine after the oxidation (34 ± 3 nA) is not significantly changed from the unmodified CFME (30 ± 2 nA) (paired t-test, p = 0.071, n = 5), indicating that the current increase of ox-CNH/CFME is due to oxidation of the CNHs.
Fig. 4.
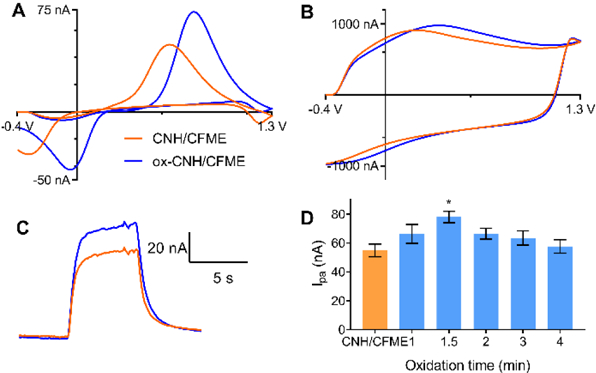
Oxidized CNH electrodes. (A-C). FSCV response of ox-CNH/CFME (blue line) prepared from 1.5-min oxidative etching of the optimized CNH/CFME (orange line). (A) background-subtracted CV of 1 μM dopamine, (B) background current in PBS pH 7.4, and (C) current vs. time trace in response to a bolus of 1 μM dopamine. (D) Optimization of oxidative etching time, response is anodic current for 1 μM dopamine. (n = 4–6, error bars represent SEM, one-way ANOVA with Bonferroni post-test compared to CNH/CFME (an orange bar), *p < 0.05)
Fig. 4D shows the effect of varying the oxidative etching time from 1 to 4 min on the anodic peak current of 1 μM dopamine. There is a significant main effect of oxidation time on the anodic current (one-way ANOVA, p < 0.05, n = 4–6). The peak current of 1 μM dopamine at ox-CNH/CFME was maximal at 1.5 min and significantly higher than the non-oxidized CNH/CFME (Bonferonni post-test, p < 0.05, n = 4–6). The background current of ox-CNH/CFME is not affected by the oxidation time (Fig. S5) (one-way ANOVA, p = 0.891, n = 4–6), implying there are no changes in electrode surface area from oxidative etching.
The average electrochemical characterization data from the optimized ox-CNH/CFME with 1 μM dopamine are shown in Table 1. The dopamine peak current is increased from CNH/CFME by 1.5 times, or a total of 3.5 ± 0.2 times compared to unmodified CFME. The background current enhancement is 1.6 ± 0.3 times of the unmodified CFME, as the oxidation does not affect the background current significantly. The ΔEp and ipa/ipc ratio of ox-CNH/CFME are also unchanged after the oxidative etching. The dopamine LOD for ox-CNH/CFME is 6 ± 2 nM, which is significantly improved from the CNH/CFME (unpaired t-test, p < 0.01, n = 5) while maintaining the same rise time (unpaired t-test, p = 0.831, n = 5).
The oxidation of CNHs has been performed by various methods including O2 [4], H2O2 [3], or HNO3 [36]. These studies found the oxidation process opening the tips of each CNH creates sp3-hybridized carbons and surface oxide groups [3,4,36]. The CNH tips have ring strain caused by pentagon nonplanar structure, so they are easier oxidized than the cone wall [1]. The oxidized CNHs, as shown in the TEM image in Fig. S1B, had looser and more opened dahlia-aggregated structure than the pristine CNHs. Our results indicate the increasing of D/G ratio of ox-CNH/CFME from that of CNH/CFME from Raman spectroscopy, so the edge planes and surface oxide groups are created while the same surface area is maintained because of the unchanged background current. The increased oxide groups without a change in background improves the S/N ratio and the LOD, making the ox-CNH/CFME a better probe for FSCV detection of dopamine. The whole CV of the dopamine obtained from ox-CNH/CFME is positively shifted in potential from that of CNH/CFME by 200 mV (Fig. 4A). Because the etching process introduces oxide groups, which are negatively charged at pH 7.4, more positive charge is required to overcome the surface charge. This shifting is similar to a previous study on oxygen-plasma etched CNT yarn microelectrodes that also had shifted CVs [27].
3.4. Analytical Performance of the ox-CNH/CFME
The electrochemical properties of ox-CNH/CFME were studied and analytical figures of merit obtained for dopamine. Fig. 5A and 5B illustrate the relationship between anodic peak current of dopamine with varied scan rate from 50 to 1000 V/s at the ox-CNH/CFME. The linearity of the peak current with scan rate (R2 = 0.998) and not square root of scan rate (R2 = 0.982) indicates that the dopamine redox reaction at ox-CNH/CFME is adsorption-controlled [37]. The result indicates the important role of ox-CNH/CFME surface, i.e. surface oxide groups, in dopamine adsorption [11].
Fig. 5.
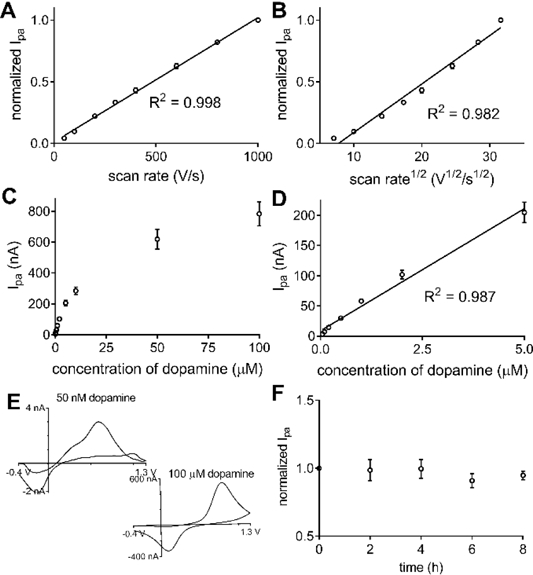
Analytical performance of ox-CNH/CFME. (A-B) Scan rate dependence. Anodic peak current of 1 μM dopamine is plotted vs (A) scan rate or (B) square root of scan rate (n = 4). The plot is more linear vs scan rate indicating adsorption controlled kinetics. (C-D) Concentration dependence. (C) Plot of anodic peak current vs. dopamine concentration from 50 nM to 100 μM (n = 4). (D) The current is linear from 50 nM to 5 μM (n = 4). (E) Example CVs of 50 nM and 100 μM dopamine in PBS pH 7.4. (F) Stability. Peak current was stable when dopamine was measured every 2 h for 8 h (n = 3). FSCV waveform was continuously applied. Error bars represent SEM.
Fig. 5C shows the concentration dependence of dopamine anodic peak current at ox-CNH/CFME. The electrode was tested with 50 nM to 100 μM dopamine in PBS pH 7.4. The peak current was linear with dopamine concentration up to 5 μM (R2 = 0.987), or two orders of magnitudes from 50 nM, as shown in Fig. 5D. At higher concentration, the peak current deviates from linearity because all the surface sites are occupied and the dopamine redox reaction could become diffusion-controlled, as supported by the shape of CV of 100 μM dopamine (Fig. 5E). The CV of adsorption-controlled electron transfer has symmetrical peak shapes because the process is similar to the thin-layer electrochemical cell condition, while the CV peak of diffusion-controlled process is unsymmetrical, and has the more traditional “duck-like” shape [37]. This saturation is similar to the previous work, [41] which found that redox reaction at CFMES is diffusion-controlled at higher dopamine concentrations. In addition, the CV of 50 nM is shown (Fig. 5E) to demonstrate the potential of the ox-CNH/CFME to detect dopamine at a very low concentration near the LOD.
In biological experiments, neurotransmitters are usually monitored for several hours. To investigate the stability and reproducibility of ox-CNH/CFME in dopamine detection, the FSCV waveform was continuously applied to the electrode, and the anodic peak current of dopamine was measured every 2 h for 8 h. Fig. 5F shows that the peak current does not dramatically change throughout the experiment. The relative standard deviation (RSD) of the peak current at the same electrode is 7.4 + 1.0% (n = 3). These results confirm good stability of the ox-CNH/CFME and indicate that the deposited CNH on the carbon-fiber surface does not fall off the electrode, even when it is scanned for a long period of time.
We also examined the FSCV response from ox-CNH/CFME toward other neurochemicals including epinephrine, norepinephrine, serotonin, and ascorbic acid (Fig. 6). In general, the whole CV of all neurochemicals (Fig. 6A to 6D) shifts positively from CV obtained at the unmodified CFME, similar to that of dopamine (Fig. 4A), due to the surface charge. The cationic neurotransmitters epinephrine, norepinephrine, and serotonin had similar peak current enhancements (2.8 ± 0.1 times for epinephrine (Fig. 6A), 2.1 ± 0.2 times for norepinephrine (Fig. 6B), and 2.2 ± 0.2 times for serotonin (Fig. 6C)). Ascorbic acid is an anion with a high concentration in biological matrix [44] that can interfere electrochemical analysis. Fig. 6D shows that the current enhancement of ascorbic acid (1.4 ± 0.1 times) is significantly less than that of dopamine (unpaired t-test, p < 0.0001, n = 4) because of the electrostatic repulsion between negatively-charged ascorbic acid and surface oxide groups on ox-CNH/CFME. The current enhancement of ascorbic acid is likely only caused by electrode surface area increase. These differences in the current enhancements show the enhanced selectivity of ox-CNH/CFME towards cations.
Fig. 6.
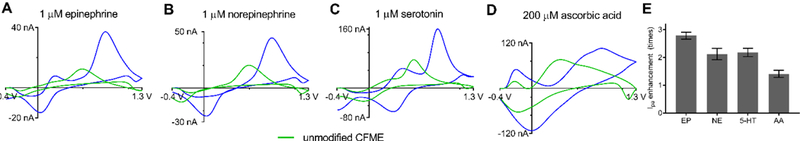
Response to other neurochemicals. Response of ox-CNH/CFME (blue line) compared with unmodified CFME (green line). (A) 1 μM epinephrine (EP), (B) 1 μM norepinephrine (NE), (C) 1 μM serotonin (5-HT), and (D) 200 μM ascorbic acid (AA) in PBS pH 7.4. (E) anodic peak current enhancement at ox-CNH/CFME (compared to unmodified CFME (n = 4)). Error bars represent SEM.
A smaller peak was observed at about +0.4 V in the CV of epinephrine and serotonin ox-CNH/CFME. This peak corresponds to a cyclization of their quinone form via 1,4-Michael addition yielding leucoaminochromes [45,46]. The peak is observed at ox-CNH/CFME but not CFME because the quinone form, which is the oxidation products of those catecholamines, are adsorbed in the interstitial pore and tips of CNHs. Therefore, the products have longer time at the electrode surface and can undergo another electron transfer causing such cyclization. However, dopamine and norepinephrine have lower cyclization rate constant [47], so their cyclization peaks were not observed.
One concern for possible use of microelectrodes in vivo is the fouling of the surface from proteins or from oxidation products of compounds, such as serotonin which can polymerize after oxidation. [48] The resistance of ox-CNH/CFME toward biofouling was determined by comparing the anodic peak current of 1 μM dopamine from the electrode before and after placing the electrode in the brain slice tissue for 2 h. The current after fouling (39 ± 5 nA) significantly decreases from original current (64 ± 3 nA) (paired t-test, p < 0.005, n = 4). However, the decrease is only about 40%, which is less than the decrease observed at unmodified CFMEs, where the dopamine anodic current decreased by 70% [49]. The background current at ox-CNH/CFME before (1080 ± 240 nA) and after biofouling (960 ± 140 nA) is not significantly different (paired t-test, p = 0.500, n = 4), consistent with our previous studies of oxygen plasma etched-CNT yarn microelectrode [27]. Edge planes and surface oxide groups from CNHs and oxidative etching increase the hydrophilicity of the electrode surface, which decreases the irreversible protein adsorption generally caused by hydrophobic interactions [50]. Higher hydrophilicity also increases wetting at the electrode surface, which helps to maintain the reproducibility of the response and for CNHs to stay on the CFME surface. Future studies can test the performance of these CNH electrodes in vivo, but the resistance to biofouling will help maintain the signal enhancements in tissue.
4. Conclusions
We have optimized electrodeposition of carbon nanohorns to enhance the signal for dopamine using FSCV. The signal is further improved by oxidative etching that introduces more defect sites and surface oxide groups to enhance dopamine adsorption. Overall, the current for dopamine improves 3.5-time higher than unmodified CFME and the LOD is 6 nM, while the rapid time response is maintained, and the measurements are stable for 8 h, longer than a typical biological experiment. In addition, ox-CNH/CFME increased the signal from other cationic neurotransmitters including epinephrine, norepinephrine, and serotonin but not the anionic interferent ascorbic acid, demonstrating improved selectivity. Oxidized CNHs also improved the biofouling resistance of the microelectrode. Overall, CNHs are a promising nanomaterial to enhance sensitivity and selectivity for cationic neurotransmitters.
Supplementary Material
Acknowledgments
This work was supported by the National Institute of Health (NIH) grants R21 DA037584 and R01 NS076875. Characterization of single-walled CNHs and electrodes were conducted at the Center for Nanophase Materials Sciences, ORNL, which is a DOE Office of Science User Facility (User grant CNMS2017–076). We thanked Jihua Chen for obtaining TEM images of CNH. Travel aid to ORNL was supported by ORNL-UVA Travel Award (University of Virginia). We also thanked Scott T. Lee for preparing the brain slices for the biofouling experiment.
References
- [1].Berber S, Kwon Y-K, Tománek D, Phys. Rev. B 2000, 62, R2291–R2294. [DOI] [PubMed] [Google Scholar]
- [2].Karousis N, Suarez-Martinez I, Ewels CP, Tagmatarchis N, Chem. Rev. 2016, 116, 4850–4883. [DOI] [PubMed] [Google Scholar]
- [3].Xu J, Zhang M, Nakamura M, Iijima S, Yudasaka M, Appl. Phys. A Mater. Sci. Process. 2010, 100, 379–383. [Google Scholar]
- [4].Yang CM, Kim YJ, Miyawaki J, Kim YA, Yudasaka M, Iijima S, Kaneko K, J. Phys. Chem. C 2015, 119, 2935–2940. [Google Scholar]
- [5].Wang X, Lou M, Yuan X, Dong W, Dong C, Bi H, Huang F, Carbon 2017, 118, 511–516. [Google Scholar]
- [6].Iijima S, Yudasaka M, Chem. Phys. Lett. 1999, 309, 165–170. [Google Scholar]
- [7].Russell BA, Migone AD, Petucci J, Calbi MM, Phys. Chem. Chem. Phys. 2016, 18, 15436–15446. [DOI] [PubMed] [Google Scholar]
- [8].Ajima K, Murakami T, Mizoguchi Y, Tsuchida K, Ichihashi T, Iijima S, Yudasaka M, ACS Nano 2008, 2, 2057–2064. [DOI] [PubMed] [Google Scholar]
- [9].Zhang Z, Han S, Wang C, Li J, Xu G, Nanomaterials 2015, 5, 1732–1755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Yang C, Denno ME, Pyakurel P, Venton BJ, Anal. Chim. Acta 2015, 887, 17–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].McCreery RL, Chem. Rev. 2008, 108, 2646–2687. [DOI] [PubMed] [Google Scholar]
- [12].Georgakilas V, Perman JA, Tucek J, Zboril R, Chem. Rev. 2015, 115, 4744–4822. [DOI] [PubMed] [Google Scholar]
- [13].Yang L, Liu D, Huang J, You T, Sensors Actuators B Chem. 2014, 193, 166–172. [Google Scholar]
- [14].Banks CE, Compton RG, Analyst 2006, 131, 15–21. [DOI] [PubMed] [Google Scholar]
- [15].E SP, Miller TS, Macpherson JV, Unwin PR, Phys. Chem. Chem. Phys. 2015, 17, 26394–26402. [DOI] [PubMed] [Google Scholar]
- [16].Jacobs CB, Vickrey TL, Venton BJ, Analyst 2011, 136, 3557–3565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Swamy BEK, Venton BJ, Analyst 2007, 132, 876–884. [DOI] [PubMed] [Google Scholar]
- [18].Ross AE, Venton BJ, Analyst 2012, 137, 3045–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Peairs MJ, Ross AE, Venton BJ, Anal. Methods 2011, 3, 2379. [Google Scholar]
- [20].Xiao N, Venton BJ, Anal. Chem. 2012, 84, 7816–7822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Yang C, Jacobs CB, Nguyen MD, Ganesana M, Zestos AG, Ivanov IN, Puretzky AA, Rouleau CM, Geohegan DB, Venton BJ, Anal. Chem. 2016, 88, 645–652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Harreither W, Trouillon R, Poulin P, Neri W, Ewing AG, Safina G, Anal. Chem. 2013, 85, 7447–7453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Zestos AG, Nguyen MD, Poe BL, Jacobs CB, Venton BJ, Sensors Actuators B Chem. 2013, 182, 652–658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Yang C, Trikantzopoulos E, Jacobs CB, Venton BJ, Anal. Chim. Acta 2017, 965, 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Jacobs CB, Ivanov IN, Nguyen MD, Zestos AG, Venton BJ, Anal. Chem. 2014, 86, 5721–5727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Yang C, Trikantzopoulos E, Nguyen MD, Jacobs CB, Wang Y, Mahjouri-Samani M, Ivanov IN, Venton BJ, ACS Sensors 2016, 508–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Yang C, Wang Y, Jacobs CB, Ivanov I, Venton BJ, Anal. Chem. 2017, 5605–5611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Valentini F, Ciambella E, Boaretto A, Rizzitelli G, Carbone M, Conte V, Cataldo F, Russo V, Casari CS, Chillura-Martino DF, et al. , Electroanalysis 2016, 28, 2489–2499. [Google Scholar]
- [29].Zhu S, Li H, Niu W, Xu G, Biosens. Bioelectron. 2009, 25, 940–943. [DOI] [PubMed] [Google Scholar]
- [30].Zhu S, Gao W, Zhang L, Zhao J, Xu G, Sensors Actuators B Chem. 2014, 198, 388–394. [Google Scholar]
- [31].Huffman ML, Venton BJ, Electroanalysis 2008, 20, 2422–2428. [Google Scholar]
- [32].Zhang Y, Ji Y, Wang Z, Liu S, Zhang T, RSC Adv. 2015, 5, 106307–106314. [Google Scholar]
- [33].Oakes L, Westover A, Mahjouri-Samani M, Chatterjee S, Puretzky AA, Rouleau C, Geohegan DB, Pint CL, ACS Appl. Mater. Interfaces 2013, 5, 13153–13160. [DOI] [PubMed] [Google Scholar]
- [34].Zhang L, Lei J, Zhang J, Ding L, Ju H, Analyst 2012, 137, 3126. [DOI] [PubMed] [Google Scholar]
- [35].Zhu S, Zhao X, Chen G, Wang H, Xu G, You J, Ionics 2015, 21, 2911–2917. [Google Scholar]
- [36].Yang CM, Noguchi H, Murata K, Yudasaka M, Hashimoto A, Iijima S, Kaneko K, Adv. Mater. 2005, 17, 866–870. [Google Scholar]
- [37].Bard AJ, Faulkner LR, Electrochemical Methods: Fundamentals and Applications, John Wiley And Sons, New York, 2001. [Google Scholar]
- [38].Morgan DM, Weber SG, Anal. Chem. 1984, 56, 2560–2567. [DOI] [PubMed] [Google Scholar]
- [39].Rodthongkum N, Ruecha N, Rangkupan R, Vachet RW, Chailapakul O, Anal. Chim. Acta 2013, 804, 84–91. [DOI] [PubMed] [Google Scholar]
- [40].Ding X, Bai J, Xu T, Li C, Zhang H-M, Qu L, Electrochem. Commun. 2016, 72, 122–125. [Google Scholar]
- [41].Bath BD, Michael DJ, Trafton BJ, Joseph JD, Runnels PL, Wightman RM, Anal. Chem. 2000, 72, 5994–6002. [DOI] [PubMed] [Google Scholar]
- [42].Takmakov P, Zachek MK, Keithley RB, Walsh PL, Donley C, McCarty GS, Wightman RM, Anal. Chem. 2010, 82, 2020–2028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Xiang L, Yu P, Hao J, Zhang M, Zhu L, Dai L, Mao L, Anal. Chem. 2014, 86, 3909–14. [DOI] [PubMed] [Google Scholar]
- [44].Robinson DL, Venton BJ, V Heien MLA, Wightman RM, Clin. Chem. 2003, 49, 1763–1773. [DOI] [PubMed] [Google Scholar]
- [45].Palomäki T, Chumillas S, Sainio S, Protopopova V, Kauppila M, Koskinen J, Climent V, Feliu JM, Laurila T, Diam. Relat. Mater. 2015, 59, 30–39. [Google Scholar]
- [46].Chen SM, Chen JY, Vasantha VS, Electrochim. Acta 2006, 52, 455–465. [Google Scholar]
- [47].Hu M, Fritsch I, Anal. Chem. 2016, 88, 5574–5578. [DOI] [PubMed] [Google Scholar]
- [48].Harreither W, Trouillon R, Poulin P, Neri W, Ewing AG, Safina G, Electrochim. Acta 2016, 210, 622–629. [Google Scholar]
- [49].Singh YS, Sawarynski LE, Dabiri PD, Choi WR, Andrews AM, Anal. Chem. 2011, 83, 6658–6666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Downard AJ, Roddick AD, Electroanalysis 1995, 7, 376–378. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.