

Abstract
Metabolic reprogramming has been proposed to be a hallmark of cancer. Aside from the glycolytic pathway, the metabolic changes of cancer cells primarily involve amino acid metabolism. However, in glioma, the characteristics of the amino acid metabolism‐related gene set have not been systematically profiled. In the present study, RNA sequencing expression data from 309 patients in the Chinese Glioma Genome Atlas database were included as a training set, while another 550 patients within The Cancer Genome Atlas database were used to validate. Consensus clustering of the 309 samples yielded two robust groups. Compared with Cluster1, Cluster2 correlated with a better clinical outcome. We then developed an amino acid metabolism‐related risk signature for glioma. Our results showed that patients in the high‐risk group had dramatically shorter overall survival than low‐risk counterparts in any subgroup, stratified by isocitrate dehydrogenase and 1p/19q status based on the 2016 World Health Organization classification guidelines. The 30‐gene signature showed better prognostic value than the traditional factors “age” and “grade” by analyzing the receiver operating characteristic curve with areas under curve of 0.966, 0.692, 0.898 and 0.975, 0.677, 0.885 for 3‐ and 5‐year survival, respectively. Moreover, univariate and multivariate analysis showed that the 30‐gene signature was an independent prognostic factor for glioma. Furthermore, Gene Ontology analysis and Gene Set Enrichment Analysis showed that tumors with a high risk score correlated with various aspects of the malignancy of glioma. In summary, we demonstrated a novel amino acid metabolism‐related risk signature for predicting prognosis for glioma.
Keywords: amino acid metabolism, CGGA, glioma, prognosis, risk signature
Abbreviations
- AUC
area under curve
- CGGA
Chinese Glioma Genome Atlas
- CI
confidence interval
- GBM
glioblastoma
- GO
gene ontology
- GSEA
gene set enrichment analysis
- HR
hazard ratio
- IDH
isocitrate dehydrogenase
- KEGG
Kyoto Encyclopedia of Genes and Genomes
- LGG
lower‐grade glioma
- OS
overall survival
- ROC
receiver operating characteristic
- TCGA
The Cancer Genome Atlas
- WHO
World Health Organization
1. INTRODUCTION
Metabolic reprogramming, as both direct and indirect consequences of oncogenic mutations, has been proposed to be a hallmark of cancer.1, 2 Amino acid metabolism might represent an “Achilles heel” in cancer as a number of tumors acquire an altered dependency on some of these metabolic pathways.3, 4, 5 Amino acid metabolism involving serine, glycine and threonine and the carbon units they provide satisfies cell growth and proliferation, as well as the maintenance of cellular redox, genetic and epigenetic status.6, 7, 8 Also, glutamine, as a super nutrient, plays surprising roles in supporting the biological hallmarks of malignancy.9, 10 Additionally, several lines of evidences have shown that an individual amino acid metabolism‐related gene plays a pivotal role in tumor progression. For instance, inhibition of glutaminase (GLS) with siRNA or small molecule inhibitor preferentially slows growth of glioma cells with mutant IDH 1.11 Yue et al12 found that oncogenic MYC selectively activates SLC7A5/SLC43A1 transcription and the MYC‐SLC7A5/SLC43A1 signaling circuit promotes essential amino acid transport and tumorigenesis. ASCT2 (encoded by SLC1A5) is a sodium‐dependent neutral amino acid transporter, and pharmacological blockade of ASCT2 with V‐9302 led to attenuated cancer cell growth, increased cell death and raised oxidative stress, which collectively contributed to antitumor responses in vitro and in mouse models in vivo.13 Nevertheless, currently, the characteristic of the amino acid metabolism‐related gene set has not been systematically profiled.
In our study, we focused on gliomas, the most common form of primary malignant brain tumor, which can be subdivided into grades II‐IV in light of WHO classification. Compared with WHO Grades II‐III, which comprise LGG, GBM WHO IV bears a dismal prognosis with median survival rates of 14.6 months.14, 15, 16 The 2016 WHO classification of central nervous system (CNS) tumors combines molecular parameters and histology to define diffuse gliomas.17 Based on traditional histopathology but enriched with IDH and 1p/19q codeletion status, gliomas could be classified into five subtypes (three LGG and two GBM), as follows: (i) LGG with wild‐type IDH (LGG‐IDHwt); (ii) LGG with IDH mutation and 1p/19q non‐codeletion (LGG‐IDHmut‐noncodel); (iii) LGG with IDH mutation and 1p/19q codeletion (LGG‐IDHmut‐codel); (iv) GBM with wild‐type IDH (GBM‐IDHwt); and (v) GBM with IDH mutation (GBM‐IDHmut).18, 19 These five subtypes of glioma show distinct tumor characteristics and OS outcomes.
In the present study, we conducted systematic and comprehensive research on the characteristics of the amino acid metabolism‐related gene set in glioma. First, we demonstrated that amino acid metabolism‐related gene sets could stratify the clinical and molecular characteristics of gliomas, highlighting their significance in the malignancy of glioma. Then, we developed an amino acid metabolism‐related signature for glioma patients in the CGGA RNA sequencing (RNAseq) dataset, and validated in TCGA RNAseq dataset. Furthermore, the 30‐gene‐based risk signature was verified as an independent prognostic factor for gliomas, indicating an association between amino acid metabolism‐related signature and prognosis. Finally, GO analysis and GSEA identified that a tumor with a higher risk score of amino acid metabolism‐related signature was involved in many aspects of tumor progression, including cell division, angiogenesis, cell adhesion and immune response. These results might provide a new insight into the research of glioma malignancy and individual therapy.
2. MATERIALS AND METHODS
2.1. Samples and data collection
We retrospectively collected whole‐genome RNA‐seq expression data and corresponding clinical and molecular information from 309 patients (gender, age, IDH mutational status, status of loss of 1p/19q and methylguanine methyltransferase [MGMT] promoter methylation and survival information) from the CGGA database (http://www.cgga.org.cn 6) as the training set.20, 21 Tumor tissue samples were obtained from patients with newly diagnosed glioma who were treated by the CGGA group. All tissues were independently diagnosed histologically by two or more neuropathologists. Only samples containing above 80% tumor cells were selected for whole‐genome expression profiling. OS was calculated from the date of diagnosis until death or the end of follow up. The study protocol was approved by the ethics committee of the Beijing Tiantan Hospital. We selected the TCGA‐RNAseq cohort as the validation set, which contains 683 samples (http://cancergenome.nih.gov/),22, 23 and after eliminating cases in which clinical information was incomplete and lacked prognostic information, 550 samples were retained.
2.2. Bioinformatics analysis
We carried out consensus clustering with the R programming language (http://cran.r-project.org)24, 25 to access expression patterns of amino acid metabolism‐related genes from the CGGA and TCGA datasets. GO analysis and KEGG pathway analysis were carried out in DAVID (http://david.abcc.ncifcrf.gov/home.jsp) for functional annotation of the genes positively and negatively correlated with risk score in the two cohorts.26, 27 GSEA (http://www.broadinstitute.org/gsea/index.jsp) was carried out to determine whether confirmed gene sets were significantly distinct between the two groups (high risk score vs low risk score).26, 28 We evaluated tumor purity of each sample using the ESTIMATE algorithm, which reflects the enrichment of stromal and immune cell gene signatures in a transcriptional profile.29 Protein‐protein interactions among 30 amino acid metabolism‐related proteins were analyzed using the STRING database (http://www.string-db.org/).
2.3. Statistical analysis
Amino acid metabolism‐related gene sets (REACTOME_METABOLISM_OF_AMINO_ACIDS_AND_DERIVATIVES) were first extracted from the Molecular Signatures Database v5.1 (MSigDB) (http://www.broad.mit.edu/gsea/msigdb/),20 which contained a total of 200 genes. After overlapping with genes in CGGA and TCGA RNA‐seq datasets, 194 and 196 genes related to amino acid metabolism, respectively, remained.
Univariate Cox regression analysis was carried out to assess the prognostic value of genes associated with amino acid metabolism and 121 genes correlating with survival (P < 0.05) were selected to achieve further gene signature selection and risk‐based classification in the training datasets. A risk signature was formulated according to the Least Absolute Shrinkage and Selection Operator (LASSO) regression algorithm.30, 31, 32 The penalty parameter λ was chosen based on 10‐fold cross‐validation within the training set, which produced the minimum mean cross‐validated error for the Cox model. Based on this, 30 genes and their regression coefficients were obtained. We then computed the risk score according to the formula followed in the training and validation datasets.
On the basis of the median risk value, patients were separated into high‐ and low‐risk groups in both CGGA and TCGA databases. Kaplan‐Meier survival curves and the log rank test were exploited to evaluate the prognostic significance.33 Differences in clinicopathological features between groups were tested by Student's t or chi‐squared tests. Multivariate Cox regression analyses were carried out to determine independent prognostic factors, and the statistical analyses were conducted using SPSS version 16.0 software (SPSS Inc., Chicago, IL, USA). P value <.05 was regarded as statistically significant.
3. RESULTS
3.1. Stratification of gliomas based on amino acid metabolism‐related gene sets
Amino acid metabolism‐related gene expression profiling of 309 samples was obtained from the CGGA RNAseq datasets, and we analyzed the genes identified as having highly variable expression among the samples. Consensus clustering of the 309 samples determined two robust clusters with clustering stability increasing between k = 2 and k = 10 (Figure 1A‐D and Figure S1). We observed that consensus clustering determined striking differences in the clinical and molecular features of the two glioma subclasses (Figure 1E, Table S1). In the training cohort, Cluster1 was strongly linked with older age at diagnosis (median age = 46, P < .001), classical or mesenchymal subtypes (72.3%, P < .001), GBM phenotype (71.8%, P < .001), IDH wild type (72.9%, P < .001) and 1p/19q non‐codeletion (96.3%, P < .001). Cluster2 cluster mainly represented the proneural or neural subtypes (92.0%, P < .001), lower grade (88.5%, P < .001), and IDH mutational status (81.3%, P < .001). These findings were validated in the TCGA datasets (Figure S2). Furthermore, OS analysis showed that glioma patients with the Cluster1 subgroup had a better prognosis compared with the Cluster2 subgroup (P < .001, log‐rank; Figure 1F). These results indicated that amino acid metabolism‐related gene sets were involved in the malignancy of gliomas and closely related to prognosis of patients. According to the CGGA cohort, TCGA samples were also clearly stratified into two different prognostic subgroups (Figure S2F).
Figure 1.
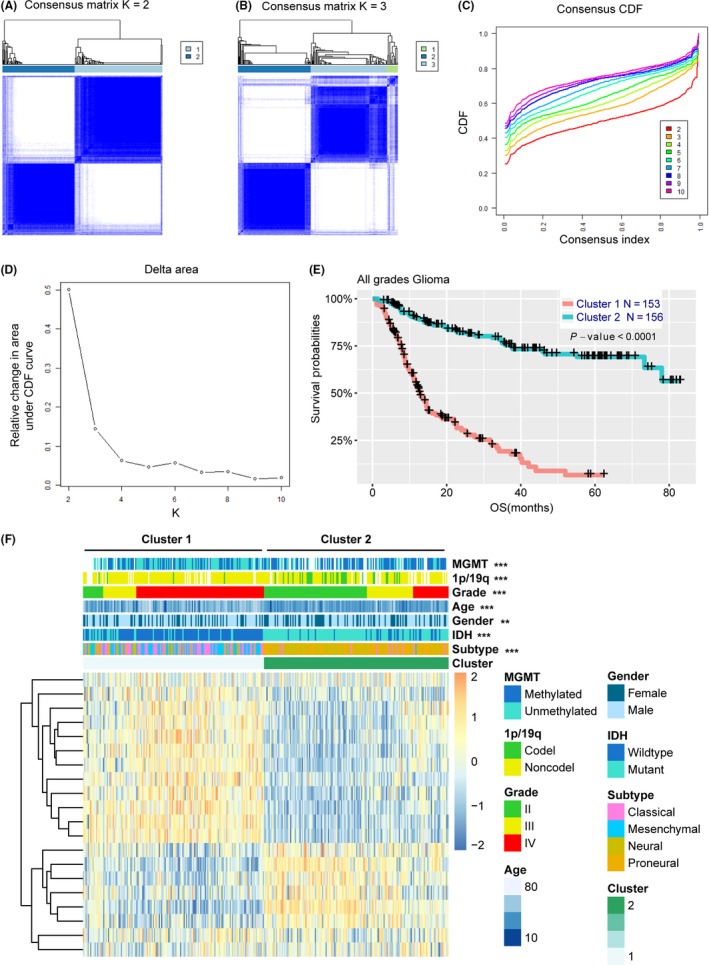
Amino acid‐related gene sets could classify the clinical and molecular features of gliomas. A,B, Consensus clustering matrix of 309 CGGA samples for k = 2 and k = 3. C, Consensus clustering CDF for k = 2 to k = 10. D, Relative change in area under CDF curve according to various k values. E, Heat map and clinicopathological features of the two clusters defined by the amino acid‐related gene sets. F, Survival analysis of Cluster 1 and Cluster 2 subgroups in CGGA samples. CDF, cumulative distribution function; CGGA, Chinese Glioma Genome Atlas; Codel, codeletion; IDH, isocitrate dehydrogenase; MGMT, methylguanine methyltransferase; Noncodel, noncodeletion; OS, overall survival
3.2. Identification of a 30‐gene risk signature associated with amino acid metabolism
To identify an amino acid metabolism‐related gene signature, first, we selected 121 genes associated with OS (P < .05) by univariate Cox regression analysis in the training cohort. Then, by LASSO regression algorithm, 30 genes were selected as active covariates to evaluate the prognostic value, and the risk scores for the patients in the training cohort were obtained (Figure 2A,B). To assess performance of the signature genes as classifier, we distinguished the training dataset into high‐risk and low‐risk groups by using the median risk score as the cutoff value, and found a significant difference in the clinical and molecular features between the two groups (Figure 2C and Table 1). In comparison with the low‐risk group, patients in the high‐risk group tended to be older (P < .001). As shown in Table 1, classical and mesenchymal subtypes were found in 12.9% and 73.5% of low‐risk and high‐risk groups, respectively (P < .001). Moreover, we found that GBM accounted for a large proportion, 72.9% of the total, in the high‐risk group, whereas GBM was 12.9% in the low‐risk group (P < .001). We found that 78.6% and 24.5% of samples in the low‐risk and high‐risk groups, respectively, were found to carry IDH mutations (P < .001). Loss of chromosome 1p/19q was found in 24.6% and 3.1% of low‐risk and high‐risk groups, respectively (P < .001). Our results also showed that MGMT promoter methylation was found in 70.6% and 42.8% of low‐risk and high‐risk groups, respectively (P < .001).
Figure 2.
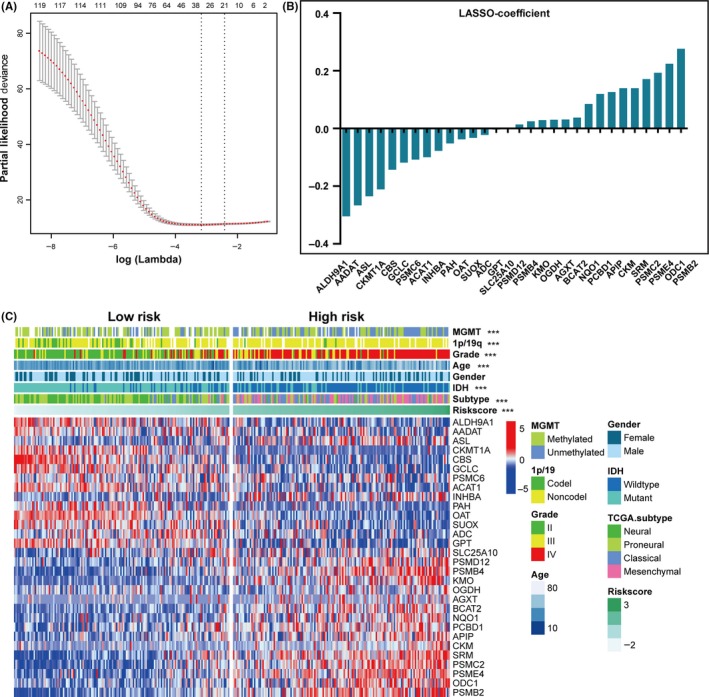
Identification of 30‐gene risk signature for OS by LASSO regression analysis in CGGA datasets. A, Partial likelihood deviance as function of regularization parameter λ in the training dataset. Each red point marks a λ value along regularization paths, and gray error bars represent confidence intervals for the cross‐validated error rate. Left vertical dotted line marks the minimum error, whereas the right vertical dotted line marks the largest λ value, the error of which is within 1 SD of the minimum. Horizontal row of numbers above the plot marks the gene number in each condition upon shrinkage and selection based on linear regression. Results of 30 genes selected and their regression coefficients by LASSO are shown in (B). C, Heat map shows the association of risk scores and clinicopathological features based on the 30‐gene risk signature. CGGA, Chinese Glioma Genome Atlas; Codel, codeletion; IDH, isocitrate dehydrogenase; LASSO, Least Absolute Shrinkage and Selection Operator; MGMT, methylguanine methyltransferase; Noncodel, noncodeletion; OS, overall survival; TCGA, The Cancer Genome Atlas
Table 1.
Correlation between 30‐gene‐based risk scores and clinicopathological factors of glioma patients in the two cohorts
| Features | Training set CGGA RNA‐seq cohort (n = 309) | Validation set TCGA RNA‐seq cohort (n = 550) | ||||
|---|---|---|---|---|---|---|
| Low‐risk score (n = 154) | High‐risk score (n = 155) | P‐value | Low‐risk score (n = 275) | High‐risk score (n = 275) | P‐value | |
| Age | ||||||
| Mean (range) | 40 (10‐75) | 47 (8‐81) | <.001 | 40 (14‐87) | 56 (21‐89) | <.001 |
| Gender | ||||||
| Female | 62 | 53 | .113 | 119 | 112 | .390 |
| Male | 92 | 102 | 156 | 163 | ||
| TCGA subtype | ||||||
| Pro | 65 | 34 | <.001 | 237 | 108 | <.001 |
| Neural | 69 | 7 | 28 | 5 | ||
| Classical | 17 | 52 | 9 | 132 | ||
| Mes | 3 | 62 | 1 | 30 | ||
| WHO grade | ||||||
| II | 95 | 9 | <.001 | 160 | 31 | <.001 |
| III | 34 | 33 | 115 | 96 | ||
| IV | 25 | 113 | 0 | 148 | ||
| IDH status | ||||||
| WT | 33 | 117 | <.001 | 17 | 195 | <.001 |
| Mut | 121 | 38 | 258 | 80 | ||
| 1p/19q status | ||||||
| Codel | 32 | 4 | <.001 | 134 | 3 | <.001 |
| Noncodel | 98 | 124 | 141 | 266 | ||
| NA | 24 | 27 | 0 | 6 | ||
| MGMT promoter status | ||||||
| Unmethy | 32 | 79 | <.001 | 30 | 105 | <.001 |
| Methy | 77 | 59 | 245 | 138 | ||
| NA | 45 | 17 | 0 | 32 | ||
Bold type indicates a statistically significant difference ( P value < .05).
CGGA, Chinese Glioma Genome Atlas; Codel, codeletion; IDH, isocitrate dehydrogenase; Mes, mesenchymal; Methy, methylated; MGMT, methylguanine methyltransferase; Mut, mutation; NA, not applicable; Noncodel, noncodeletion; Pro, proneural; TCGA, The Cancer Genome Atlas; Unmethy, unmethylated; WHO, World Health Organization; WT, wildtype.
To validate the 30 amino acid metabolism‐related risk signature in other populations, we formulated the risk scores for each patient in TCGA database based on the 30‐gene coefficients derived from the training dataset. Consistent with the above results, we also found that there was significant difference between the two groups in an independent validation cohort (Figure S3 and Table 1). In brief, compared to the low‐risk group, the high‐risk group tended to comprise the patients with poor prognostic features.
3.3. Identification of 30‐gene signature for prognostication in glioma
In view of the close correlation between risk groups and clinicopathological features, we sought to assess the prognostic value of the risk score. In all gliomas, patients were assigned to two groups according to the median risk score. Our results showed that patients in the high‐risk group (n = 155) had dramatically shorter OS than their low‐risk counterparts (n = 154) in the training cohort (median OS = 9.0 vs 37.9 months; P < .0001; Figure 3A). Moreover, we explored the prognostic value of risk score in gliomas of different grades and found that OS differed significantly between high‐risk and low‐risk groups in WHO grade II (median OS = 26.5 vs 56.8 months; P = .0024), grade III (median OS = 11.4 vs 33.6 months; P < .0001) and GBM (median OS = 7.2 vs 12.7 months; P < .0001; Figure 3B‐D).
Figure 3.
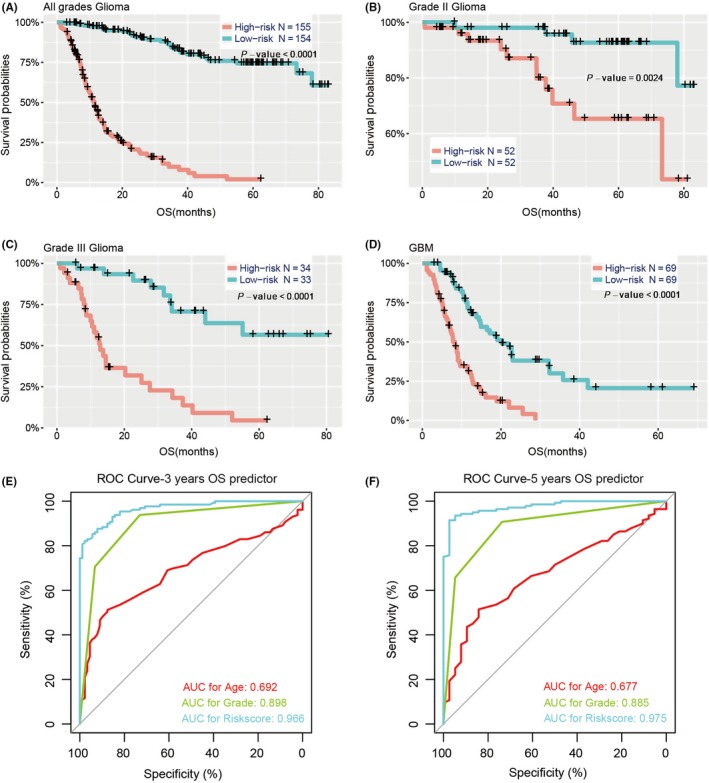
Prognostic significance of the 30‐gene signature‐derived risk scores in different WHO grades. Prognosis efficiency of the 30‐gene risk signature in all grades (A), grade II (B), grade III (C) and GBM (D) from the CGGA datasets. P‐value shown in each panel is determined by a log‐rank test between the two groups. E,F, ROC curves indicating the sensitivity and specificity of predicting 3‐ and 5‐y survival with the amino acid metabolism‐related signature in the CGGA datasets. CGGA, Chinese Glioma Genome Atlas; GBM, glioblastoma; OS, overall survival; ROC, receiver operating characteristic; WHO, World Health Organization
The 2016 update to the WHO proposed a classification strategy and, thus, gliomas were classified into five subtypes based on traditional histopathology and the status of IDH and 1p/19q codeletion. Given that these five glioma subtypes showed distinct tumor characteristics and OS outcomes, we determined whether the risk score had prognostic value in the five various populations. For LGG, survival time of the high‐risk group was remarkably shorter than that of the low‐risk group in LGG‐IDHmut‐noncodel (P < .0001; Figure 4A) and LGG‐IDHwt (P < .0001; Figure 4B), whereas there was no significant difference in LGG‐IDHmut‐codel (P = .1175; Figure 4C). For both GBM‐IDHwt and GBM‐IDHmut, there were significant differences in OS between the two risk groups (P < .0001; P = .0015, respectively; Figure 4D,E).
Figure 4.
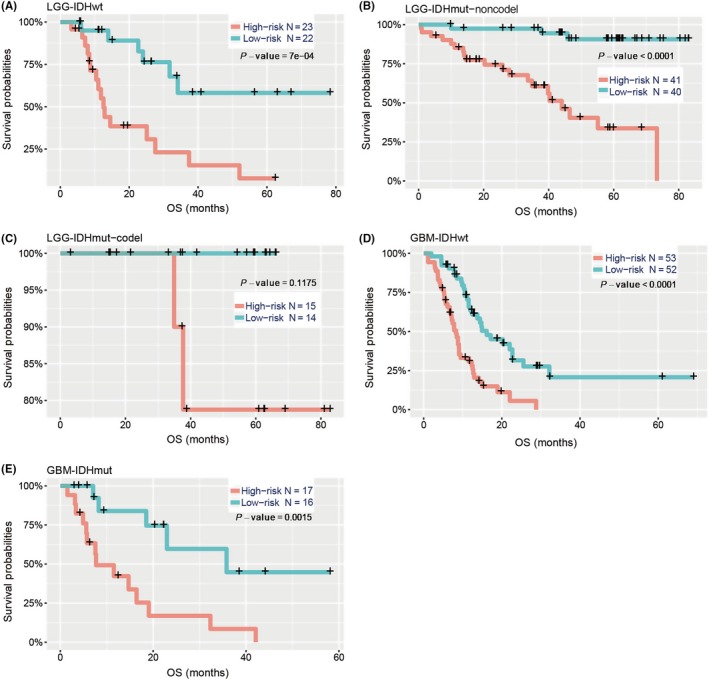
Prediction of outcome in diverse cohorts stratified by IDH mutation and 1p/19q codeletion status. Kaplan‐Meier survival curves for LGG patients with IDH‐wild type (A), IDH‐mutation but not the 1p/19q codeletion (B) and IDH‐mutation with 1p/19q codeletion (C), classified into two groups based on 30‐gene signature‐derived risk scores. Kaplan‐Meier survival curves also show the prognostic value of GBM patients with IDH‐wild type (D) and IDH‐mutation (E) in the CGGA cohort. P‐value is the result of a log‐rank test between the two groups shown in each panel. CGGA, Chinese Glioma Genome Atlas; Codel, codeletion; GBM, glioblastoma; IDH, isocitrate dehydrogenase; LGG, lower‐grade glioma; OS, overall survival
Meanwhile, the signature value showed significant differences between samples stratified by WHO grade in the CGGA and TCGA cohorts (Figure 5A and Figure S5A). Such being the case, gliomas were classified into five principal groups on the basis of IDH status and 1p/19q codeletion status. Based on the critical molecular markers IDH and 1p/19q, we investigated the distribution of the 30‐gene signature in patients stratified by IDH status among distinct WHO grades (Figure 5B‐D and Figure S5B‐D) and 1p/19q codeletion status in LGG‐IDH mutation patients (Figure 5E and Figure S5E). Verhaak et al34 have identified four clinically relevant subtypes (neural, proneural, classical, mesenchymal) of GBM characterized by abnormalities in platelet derived growth factor receptor alpha (PDGFRA), IDH1, epidermal growth factor receptor (EGFR) and neurofibromin 1 by an integrated genomic analysis. Therefore, we explored the distribution of TCGA subtypes for GBM in the CGGA and TCGA cohorts (Figure 5F and Figure S5F).
Figure 5.
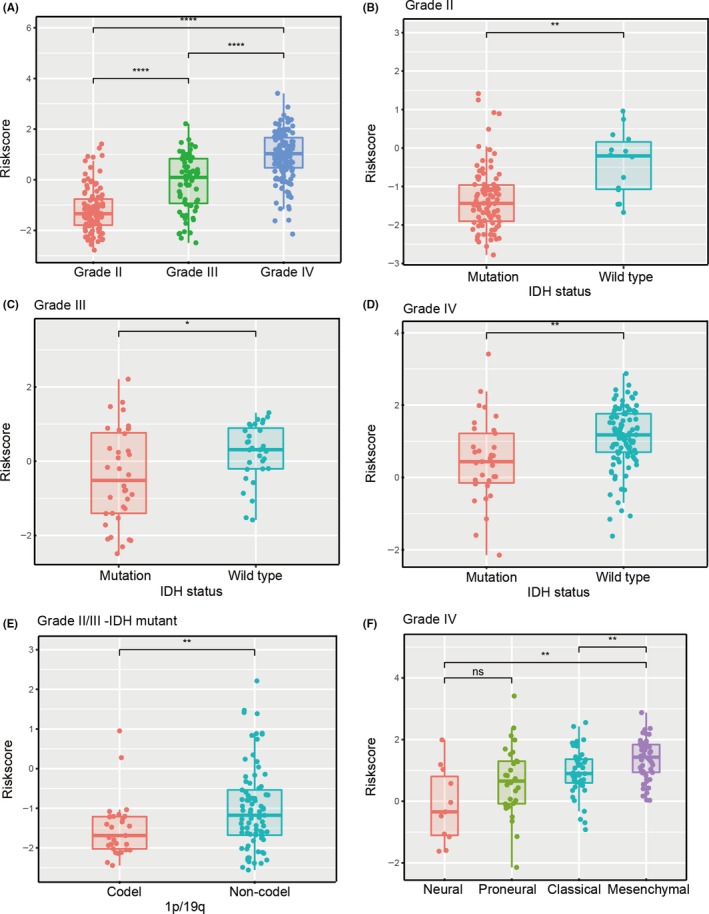
Associations between the amino acid‐related signature and other features in CGGA datasets. Distribution of the amino acid‐related gene signature in patients stratified by WHO grade (A), IDH1 status in each grade (B‐D), 1p/19q status in IDH mutation‐LGG (E) and TCGA subtypes in GBM (F). *P < .05; **P < .01; ****P < .0001; ns, not significant. CGGA, Chinese Glioma Genome Atlas; Codel, codeletion; GBM, glioblastoma; IDH, isocitrate dehydrogenase; LGG, lower‐grade glioma; TCGA, The Cancer Genome Atlas; WHO, World Health Organization
3.4. Prognostic validity of the 30‐gene signature for glioma
Subsequently, we investigated the specificity and sensitivity of risk score in the prediction of 3‐ and 5‐year survival by analyzing the ROC curve, and compared the 30‐gene signature with traditional “age” and “grade”. The 30‐gene signature showed striking prognostic validity, with AUC of 0.966 and 0.975 for 3‐ and 5‐year survival, respectively, which were higher than for the traditional factors (Figure 3E,F), underscoring the superior predictive value of the 30‐gene signature.
3.5. Univariate and multivariate analysis shows prognostic value of 30‐gene signature
To further explore whether the risk score was an independent prognostic factor of prognosis in glioma, we carried out univariate and multivariate Cox regression analyses in the CGGA cohort. Results showed that the 30‐gene signature was independently correlated with OS by adjusting for clinicopathological factors (age, gender, WHO grade, TCGA subtype, IDH status, MGMT promoter status and 1p/19q status; Table 2). Consistently, the local immune‐related risk signature was validated as an independent factor after Cox regression analyses in TCGA cohort (Table S2).
Table 2.
Uni‐ and multivariate Cox regression analysis of the clinical features and 30‐gene‐based risk score for OS in CGGA datasets
| Variables | Univariate analysis | Multivariate analysis | ||||
|---|---|---|---|---|---|---|
| HR | 95% CI | P‐value | HR | 95% CI | P‐value | |
| Age | 1.038 | 1.022‐1.053 | <.001 | 0.995 | 0.979‐1.012 | .593 |
| Gender | 1.187 | 0.841‐1.675 | .330 | NA | NA | NA |
| WHO grade | 3.469 | 2.709‐4.443 | <.001 | 1.090 | 0.738‐1.610 | .666 |
| TCGA subtype | 1.936 | 1.642‐2.282 | <.001 | 0.880 | 0.687‐1.127 | .310 |
| IDH status | 0.229 | 0.159‐0.331 | <.001 | 0.770 | 0.391‐1.514 | .448 |
| MGMT promoter status | 0.529 | 0.374‐0.750 | <.001 | 0.989 | 0.644‐1.517 | .959 |
| 1p/19q status | 0.165 | 0.067‐0.404 | <.001 | 0.970 | 0.362‐2.596 | .951 |
| Risk score | 4.077 | 3.326‐4.999 | <.001 | 3.825 | 2.830‐5.171 | <.001 |
Bold type indicates a statistically significant difference ( P value < .05).
Variables with prognostic significance in univariate Cox regression analysis were included in further multivariate Cox analysis.
Gender (female and male); WHO grade (II, III and IV); TCGA subtype (neural, proneural, mesenchymal and classical); IDH status (mutant and wildtype); MGMT promoter status (methylated and unmethylated); 1p/19q status (codeletion and non‐codeletion); Risk score (low and high). CI, confidence interval; CGGA, Chinese Glioma Genome Atlas; HR, hazard ratio; IDH, isocitrate dehydrogenase; MGMT, methylguanine methyltransferase; NA, not applicable; OS, overall survival.
3.6. Functional annotation of 30‐gene signature
To explore the potentially altered functional characteristics associated with the 30‐gene signature, GO analysis was carried out to study differences in biological processes between the two risk groups. First, we demonstrated 1346 high‐risk score positively related genes (P < .05) and 922 negatively related genes (P < .05) using Pearson correlation analysis. Genes upregulated in the high‐risk group were primarily involved in tumor progression, including “extracellular matrix organization”, “cell division”, “angiogenesis”, “cell adhesion”, “apoptotic process” and “immune response”. In contrast, downregulated genes in the high‐risk group were closely related to neurogenesis, such as “chemical synaptic transmission”, “learning”, “neurotransmitter secretion” and “nervous system development” (Figure 6A).
Figure 6.
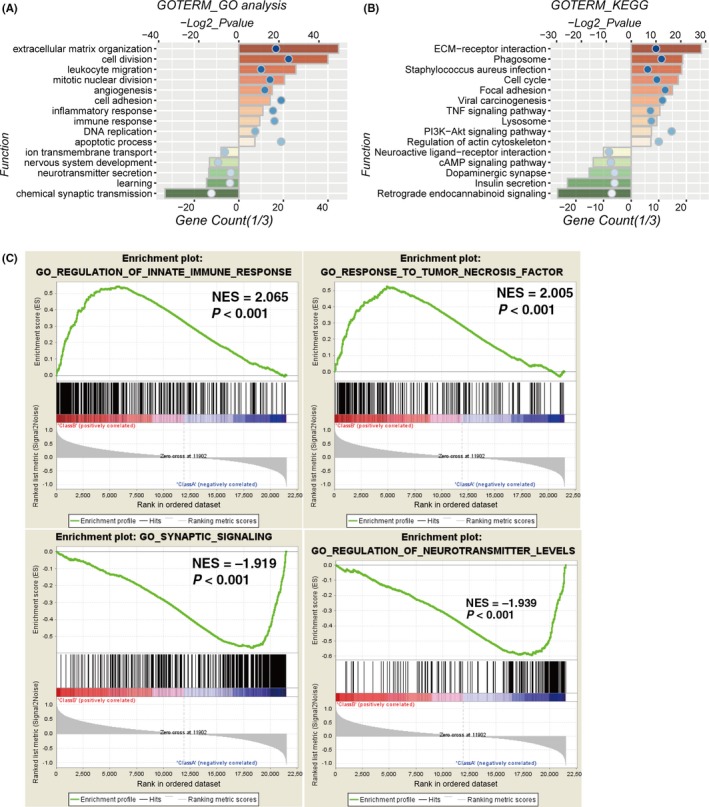
Altered functional characteristics related to the 30‐gene signature. A,B, Functional annotation of genes positively (red bar chart) or negatively (green bar chart) correlated with the risk score using GO terms of biological processes (A) and KEGG pathway (B). C, Gene set enrichment analysis (GSEA) shows that higher risk score was positively associated with immune response and negatively correlated with synaptic signaling and neurotransmitter levels. Codel, codeletion; NES, normalized enrichment score; Noncodel, noncodeletion. Orange and green bars represent P‐value, and the blue dots represent the 1/3 gene count
Moreover, GO analysis was implemented to explore the differences in KEGG pathway between the high‐ and low‐risk score groups. We found that positively related genes were mainly enriched in KEGG terms including “ECM‐receptor interaction”, “cell cycle”, “focal adhesion” and “TNF signaling pathway”, whereas the negatively correlated genes were enriched in terms including “retrograde endocannabinoid signaling”, “insulin secretion” and “dopaminergic synapse” (Figure 6B). These results were validated in TCGA cohort (Figure S6).
Next, GSEA analyses were carried out for validation, showing that the high‐risk groups were positively associated with regulation of innate immune response (P < .001) and response to tumor necrosis factor (P < .001), negatively with synaptic signaling (P < .001) and regulation of neurotransmitter levels (P < .001; Figure 6C).
4. DISCUSSION
Fast‐growing tumor cells largely draw energy out of typically increasing aerobic glycolysis, a phenomenon known as the Warburg effect.1, 12 Aside from the glycolytic pathway, the metabolic changes of cancer cells primarily involve amino acid metabolism.3 A previous study has identified a glucose‐related risk signature for the malignancy of glioma and the survival of patients through bioinformatic profiling.20 Also, metabolomic investigations have provided novel biomolecular insights into the aggressive phenotype of the malignancy of brain tumors.24, 33, 34 However, there continues to be a gap in systematically understanding the characteristics of the amino acid metabolism‐related gene set in glioma.
In the present study, for the first time, we built an amino acid metabolism‐related risk signature to predict the prognosis of glioma. RNAseq expression data from 309 patients in the CGGA database were included as the training set, whereas another 550 patients with TCGA database were used to validate. First, the 309 samples were apparently clustered into two distinct subclasses (k = 2), and the two subclasses showed significant differences in clinical and molecular features in both the CGGA and TCGA cohort. However, for k = 3, the area under the cumulative distribution function (CDF) curve was increased by more than 0.1‐fold that of k = 2 (Figure 1C,D), and we found the ratio of samples in the third subclass was very small (Figure 1B). It also meant that for k > 2, clustering stability did not improve significantly.
Next, we developed a 30‐gene‐based risk signature to determine the status of amino acid metabolism in glioma patients. We observed that the high‐risk group was closely associated with IDH wildtype, 1p/19q noncodeletion, higher WHO grades and worse TCGA subtypes (classical and mesenchymal) (Figures 2C and 5, Figure S2C and S5), which implies that the amino acid metabolism‐related risk signature may, to some extent, result in the poor prognosis of patients with IDH wildtype, 1p/19q noncodeletion, higher WHO grades and worse TCGA subtypes.
We further showed that the 30‐gene signature could predict the prognosis of glioma regardless of WHO grade and the five subgroups of WHO 2016 classification based on the stratification of IDH and 1p/19q status in the CGGA cohort (Figures 3A‐D and 4). Then, ROC curves were carried out to compare the prognostic values between the 30‐gene signature and traditional factors “age” and “grade”, with AUC of 0.966, 0.692, 0.898 and 0.975, 0.677, 0.885 for 3‐ and 5‐year survival, respectively (Figure 3E,F). These results suggested that the 30‐gene signature could better predict the prognosis of glioma.
In the validation cohort, in contrast with WHO grade II and III, the 30‐gene signature predicted poor overall prognosis for GBM (Figure S4A‐D). One possible reason is that there were distinct differences in the distribution of grades and subtypes between CGGA and TCGA cohorts. As shown in Table S3, GBM patients in the CGGA cohort accounted for 44.7% of the total, whereas GBM accounted for a proportion of 26.9% in TCGA cohort. LGG were divided into three subgroups based on the status of IDH and 1p/19q codeletion. As for LGG‐IDHwt, glioma patients in the high‐risk group had a poorer prognosis than those with low‐risk score, with a significant difference (P = .001). However, for LGG‐IDHmut‐codel and LGG‐IDHmut‐noncodel, the OS of high‐risk patients tended to be worse, although the difference showed no significance (P > .05) (Figure S4E‐G). We considered that if the sample sizes were increased, there might be a statistical difference in GBM and these subtypes.
Of note, we identified that the amino acid metabolism‐related risk signature remained an independent prognostic factor after adjustment of clinical and molecular features. There is great potential for the status of amino acid metabolism to refine the clinicopathological features of accurate prognostication, so combining the risk signature and other features could better predict the prognosis of glioma.
Functional annotation of the 30‐gene signature showed that biological functions of angiogenesis, cell adhesion and immune response may contribute to patients’ high risk and poor clinical outcome. Low‐purity gliomas were characterized by intensive local immune phenotypes and correlated with a poor prognosis.23 Therefore, we applied the ESTIMATE algorithm to predict tumor purity using gene expression profiles28 and found a significant increase in ESTIMATE scores in the high‐risk group (Figure S7), indicating that a greater presence of inflammatory microenvironment components is associated with progressive tumorigenesis.29
In addition, we analyzed the 30‐amino acid metabolism‐related genes and proteins in detail. DAVID functional annotation was carried out to determine the biological process in which each gene selected as 30‐risk signature is involved (Figure S8A). Our results showed that a group of genes (including PSMC6, PSMD12, PSMB4, PSMC2, PSME4 and PSMB2) engaged in similar biological processes, such as “regulation of cellular amino acid metabolic process”, “NIK/NF‐kappaB signaling”, “TNF/T‐cell receptor mediated signaling pathway” and “protein polyubiquitination” etc. Five genes (CBS, PAH, OAT, GPT and BCAT2) among them participated in “cellular amino acid biosynthetic process”. We still found some genes that played roles in certain amino acid metabolic processes. For instance, ODC1 took part in the “polyamine metabolic process”, and AADAT and GCLC were involved in the “glutamate metabolic process”. Moreover, we analyzed the protein‐protein interaction network for 30‐amino acid metabolism‐related genes/proteins using the STRING database (Figure S8B). Further molecular mechanisms as to how these genes affect the progression of glioma remain to be studied in our follow‐up work.
In conclusion, we identified that the amino acid metabolism‐related gene set could distinguish the clinical and molecular features of gliomas. We then developed a 30‐amino acid metabolism‐related gene expression‐based risk signature, which was strongly related to the OS of glioma patients in the five subgroups of WHO 2016 classification for patients based on the stratification of IDH and 1p/19q status, and confirmed that the 30‐risk signature could better predict OS for glioma than traditional factors. Moreover, we carried out functional annotation of the positive and negative amino acid metabolism‐related gene in glioma. Furthermore, the risk signature could contribute to understanding the carcinogenesis and development of glioma, as well as providing new insight into the therapeutic targets for glioma patients. In short, we identified a novel amino acid metabolism‐related risk signature for predicting the prognosis of glioma.
CONFLICTS OF INTEREST
Authors declare no conflicts of interest for this article.
Supporting information
ACKNOWLEDGMENTS
The authors conducting this work represent the Chinese Glioma Cooperative Group (CGCG). This work was supported by grants from the Capital Medical Development Research Fund (2016‐1‐1072), the National Natural Science Foundation of China (Grant Numbers: 81672479, 81773208, and 81502495) and the Beijing Natural Science Foundation (7182076). The study protocol was approved by the ethics committees of participating hospitals, and all patients signed written, informed consent.
Liu Y‐Q, Chai R‐C, Wang Y‐Z, et al. Amino acid metabolism‐related gene expression‐based risk signature can better predict overall survival for glioma. Cancer Sci. 2019;110:321–333. 10.1111/cas.13878
Liu and Chai contributed equally to this work.
Contributor Information
Fan Wu, Email: wufan0510284@163.com.
Tao Jiang, Email: Taojiang1964@163.com.
REFERENCES
- 1. Pavlova NN, Thompson CB. The emerging hallmarks of cancer metabolism. Cell Metab. 2016;23:27‐47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Jain M, Nilsson R, Sharma S, et al. Metabolite profiling identifies a key role for glycine in rapid cancer cell proliferation. Science. 2012;336:1040‐1044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Karpel‐Massler G, Ramani D, Shu C, et al. Metabolic reprogramming of glioblastoma cells by L‐asparaginase sensitizes for apoptosis in vitro and in vivo. Oncotarget. 2016;7:33512‐33528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Alberghina L, Gaglio D. Redox control of glutamine utilization in cancer. Cell Death Dis. 2014;5:e1561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Seyfried TN, Flores R, Poff AM, D'Agostino DP, Mukherjee P. Metabolic therapy: a new paradigm for managing malignant brain cancer. Cancer Lett. 2015;356:289‐300. [DOI] [PubMed] [Google Scholar]
- 6. Locasale JW. Serine, glycine and one‐carbon units: cancer metabolism in full circle. Nat Rev Cancer. 2013;13:572‐583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Kalhan SC, Hanson RW. Resurgence of serine: an often neglected but indispensable amino acid. J Biol Chem. 2012;287:19786‐19791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. DeBerardinis RJ, Cheng T. Q's next: the diverse functions of glutamine in metabolism, cell biology and cancer. Oncogene. 2010;29:313‐324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Hensley CT, Wasti AT, DeBerardinis RJ. Glutamine and cancer: cell biology, physiology, and clinical opportunities. J Clin Invest. 2013;123:3678‐3684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Zhang J, Pavlova NN, Thompson CB. Cancer cell metabolism: the essential role of the nonessential amino acid, glutamine. EMBO J. 2017;36:1302‐1315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Seltzer MJ, Bennett BD, Joshi AD, et al. Inhibition of glutaminase preferentially slows growth of glioma cells with mutant IDH1. Can Res. 2010;70:8981‐8987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Yue M, Jiang J, Gao P, Liu H, Qing G. Oncogenic MYC activates a feedforward regulatory loop promoting essential amino acid metabolism and tumorigenesis. Cell Rep. 2017;21:3819‐3832. [DOI] [PubMed] [Google Scholar]
- 13. Schulte ML, Fu A, Zhao P, et al. Pharmacological blockade of ASCT2‐dependent glutamine transport leads to antitumor efficacy in preclinical models. Nat Med. 2018;24:194‐202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Strickland M, Stoll EA. Metabolic reprogramming in glioma. Front Cell Dev Biol. 2017;5:43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Jiang T, Mao Y, Ma W, et al. CGCG clinical practice guidelines for the management of adult diffuse gliomas. Cancer Lett. 2016;375:263‐273. [DOI] [PubMed] [Google Scholar]
- 16. Pandey R, Caflisch L, Lodi A, Brenner AJ, Tiziani S. Metabolomic signature of brain cancer. Mol Carcinog. 2017;56:2355‐2371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Louis DN, Perry A, Reifenberger G, et al. The 2016 World Health Organization classification of tumors of the central nervous system: a summary. Acta Neuropathol. 2016;131:803‐820. [DOI] [PubMed] [Google Scholar]
- 18. Lu CF, Hsu FT, Hsieh KL, et al. Machine learning‐based radiomics for molecular subtyping of gliomas. Clin Cancer Res. 2018;24:4429‐4436. [DOI] [PubMed] [Google Scholar]
- 19. Brat DJ, Verhaak RG, Aldape KD, et al. Comprehensive, integrative genomic analysis of diffuse lower‐grade gliomas. N Engl J Med. 2015;372:2481‐2498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Zhao S, Cai J, Li J, et al. Bioinformatic profiling identifies a glucose‐related risk signature for the malignancy of glioma and the survival of patients. Mol Neurobiol. 2017;54:8203‐8210. [DOI] [PubMed] [Google Scholar]
- 21. Wu F, Zhao Z, Chai R, et al. Expression profile analysis of antisense long non‐coding RNA identifies WDFY3‐AS2 as a prognostic biomarker in diffuse glioma. Cancer Cell Int. 2018;18:107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Brennan CW, Verhaak RG, McKenna A, et al. The somatic genomic landscape of glioblastoma. Cell. 2013;155:462‐477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Zhang C, Cheng W, Ren X, et al. Tumor purity as an underlying key factor in glioma. Clin Cancer Res. 2017;23:6279‐6291. [DOI] [PubMed] [Google Scholar]
- 24. Subramanian A, Tamayo P, Mootha VK, et al. Gene set enrichment analysis: a knowledge‐based approach for interpreting genome‐wide expression profiles. Proc Natl Acad Sci USA. 2005;102:15545‐15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Cheng W, Ren X, Zhang C, Cai J, Han S, Wu A. Gene expression profiling stratifies IDH1‐mutant glioma with distinct prognoses. Mol Neurobiol. 2017;54:5996‐6005. [DOI] [PubMed] [Google Scholar]
- 26. Cheng W, Ren X, Zhang C, et al. Bioinformatic profiling identifies an immune‐related risk signature for glioblastoma. Neurology. 2016;86:2226‐2234. [DOI] [PubMed] [Google Scholar]
- 27. Hu X, Martinez‐Ledesma E, Zheng S, et al. Multigene signature for predicting prognosis of patients with 1p19q co‐deletion diffuse glioma. Neuro Oncol. 2017;19:786‐795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Gu J, Zhang X, Miao R, et al. A three‐long non‐coding RNA‐expression‐based risk score system can better predict both overall and recurrence‐free survival in patients with small hepatocellular carcinoma. Aging. 2018;10:1627‐1639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Gao J, Kwan PW, Shi D. Sparse kernel learning with LASSO and Bayesian inference algorithm. Neural Netw. 2010;23:257‐264. [DOI] [PubMed] [Google Scholar]
- 30. Qian Z, Li Y, Fan X, et al. Molecular and clinical characterization of IDH associated immune signature in lower‐grade gliomas. Oncoimmunology. 2018;7:e1434466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Yan W, Zhang W, You G, et al. Molecular classification of gliomas based on whole genome gene expression: a systematic report of 225 samples from the Chinese Glioma Cooperative Group. Neuro Oncol. 2012;14:1432‐1440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Verhaak RG, Hoadley KA, Purdom E, et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell. 2010;17:98‐110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Griffin JL, Kauppinen RA. A metabolomics perspective of human brain tumours. FEBS J. 2007;274:1132‐1139. [DOI] [PubMed] [Google Scholar]
- 34. Ahmed KA, Chinnaiyan P. Applying metabolomics to understand the aggressive phenotype and identify novel therapeutic targets in glioblastoma. Metabolites. 2014;4:740‐750. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials