

Abstract
The efficacy of programmed cell death–1 (PD‐1) blockade in patients with non–small cell lung cancer (NSCLC) positive for epidermal growth factor receptor (EGFR) gene mutations has been found to be limited, but the underlying mechanisms for this poor response have remained obscure. Given that the recognition by T cells of tumor antigens presented by major histocompatibility complex class I (MHC‐I) molecules is essential for an antitumor immune response, we examined the effects of EGFR tyrosine kinase inhibitors (TKIs) on MHC‐I expression in NSCLC cell lines. Appropriate EGFR‐TKIs increased MHC‐I expression at the mRNA and cell surface protein levels in NSCLC cells positive for EGFR mutations including those with the T790M secondary mutation. Trametinib, an inhibitor of the extracellular signal–regulated kinase (ERK) kinase MEK, also increased MHC‐I expression, whereas the phosphatidylinositol 3‐kinase (PI3K) inhibitor buparlisib did not, suggesting that the MEK‐ERK pathway mediates the down‐regulation of MHC‐I expression in response to EGFR activation. Immunohistochemical analysis of EGFR‐mutated NSCLC specimens obtained before and after EGFR‐TKI treatment also revealed down‐regulation of phosphorylated forms of EGFR and ERK in association with up‐regulation of MHC‐I, an increased number of infiltrating CD8+ T cells, and increased PD‐1 ligand 1 expression after such treatment. Our results thus suggest that mutational activation of EGFR inhibits MHC‐I expression through the MEK‐ERK pathway in NSCLC and thereby contributes to the poor response of such tumors to immunotherapy. Further studies are warranted to evaluate the relation between EGFR‐MEK‐ERK signaling in and the immune response to EGFR‐mutated NSCLC.
Keywords: epidermal growth factor receptor, major histocompatibility complex class I, non–small cell lung cancer, phosphatidylinositol 3‐kinase, T790M mutation
Abbreviations
- EGFR
epidermal growth factor receptor
- ERK
extracellular signal‐regulated kinase
- IFN
interferon
- MHC
major histocompatibility complex
- NSCLC
non‐small cell lung cancer
- PCR
polymerase chain reaction
- PD‐1
programmed cell death‐1
- PI3K
phosphatidylinositol 3‐kinase
- RT
reverse transcription
- TKI
tyrosine kinase inhibitor
1. INTRODUCTION
Lung cancer is the most common cause of cancer deaths worldwide.1 Mutations of the epidermal growth factor receptor (EGFR) gene are common drivers of non–small cell lung cancer (NSCLC),2 and EGFR tyrosine kinase inhibitors (TKIs) are considered the best choice for first‐line treatment of advanced or recurrent NSCLC harboring activating mutations of EGFR. All patients treated with EGFR‐TKIs eventually develop resistance to these drugs, however, with a secondary, gatekeeper mutation (T790M) of EGFR being responsible for the resistance in almost half of such patients. Osimertinib is a third‐generation EGFR‐TKI that has been found to confer a survival benefit compared with cytotoxic chemotherapy in T790M‐positive patients,3 but no specific agents or treatment strategies have been developed to overcome resistance mechanisms other than that mediated by T790M. New treatment options are thus needed for patients whose tumors have developed resistance to EGFR‐TKIs by such mechanisms.
Immune‐checkpoint inhibitors are emerging as an important new mode of treatment for NSCLC. Programmed cell death–1 (PD‐1, CD279) is a cell surface receptor of the immunoglobulin superfamily that is expressed on T cells and pro‐B cells and which binds the ligands PD‐L1 and PD‐L2. PD‐1 and its ligands play an important role as a checkpoint in regulation of the immune system by inhibiting T cell activation, thereby suppressing autoimmunity and promoting autoimmune tolerance.4 Many types of tumor cells express PD‐L1, and inhibition of the binding of PD‐L1 to PD‐1 has been found to enhance T cell responses in vitro as well as to have an antitumor effect in preclinical studies.5 Immune‐checkpoint inhibitors that interfere with the interaction between PD‐1 and PD‐L1 have recently been found to be superior to docetaxel in terms of both overall survival and tolerability for the treatment of NSCLC, although a durable benefit is observed in only ~20% of patients and some individuals may experience unprecedented immune‐related adverse events.6, 7, 8, 9
Subgroup analysis has suggested that inhibition of the PD‐1–PD‐L1 axis is less effective in NSCLC patients positive for EGFR mutations than in those wild type for EGFR.7, 10 Low levels of both PD‐L1 expression and CD8+ tumor‐infiltrating lymphocytes (TILs) within the tumor microenvironment have been suggested to underlie such an unfavorable clinical response to immune‐checkpoint inhibitors in patients with NSCLC positive for activating EGFR mutations.11 However, the mechanisms responsible for the low efficacy of immune therapy in such patients have remained obscure.
Major histocompatibility complex class I (MHC‐I) molecules expressed on the cell surface present peptide fragments from foreign or native intracellular proteins. The induction of a CD8+ TIL response for tumor eradication requires the recognition by these cells of tumor antigens presented by MHC‐I molecules on tumor cells, with limited presentation of such antigens by MHC‐I being a key obstacle to effective immunotherapy.12 We have now examined whether EGFR signaling might inhibit surface MHC‐I expression, resulting in loss of immunogenicity, in EGFR mutation–positive NSCLC. We found that inhibition of a specific EGFR signaling pathway by targeted agents was able to increase MHC‐I expression in such NSCLC cells.
2. MATERIAL AND METHODS
2.1. Human NSCLC cell lines and reagents
The PC9 cell line was kindly provided by Dr. Hayata (Tokyo Medical University). The PC9GR cell line was previously described.13 The cell lines H1944, HCC827, and H1975 were obtained from American Type Culture Collection (Manassas, VA, USA). All cells were maintained under a humidified atmosphere of 5% CO2 at 37°C in RPMI 1640 medium figemented with 10% fetal bovine serum. The cells were routinely tested and found to be negative for mycoplasma contamination with the use of a MycoAlert system (LT07, Lonza, Basel, Switzerland). Erlotinib, osimertinib, trametinib, and buparlisib were obtained from Chemietek (Indianapolis, IN, USA). Recombinant human interferon (IFN) ‐γ was obtained from PeproTech (Rocky Hill, NJ, USA).
2.2. RNA isolation, RT, and real‐time PCR analysis
Total RNA was extracted from cells with the use of a RNeasy Mini Kit (74106, Qiagen, Valencia, CA, USA) and was subjected to RT with a High Capacity RNA‐to‐cDNA Kit (4387406, Applied Biosystems, Carlsbad, CA, USA). The resulting cDNA was subjected to reverse transcription (RT) and real‐time polymerase chain reaction (PCR) analysis with PowerUp SYBR Green Master Mix (A25741, Thermo Fisher Scientific, Waltham, MA, USA) and a StepOnePlus Real‐Time PCR system (Applied Biosystems), and the final results were calculated with the ΔΔCt method and normalized by the amount of glyceraldehyde‐3‐phosphate dehydrogenase (GAPDH) mRNA as an internal control. The primer sequences (forward and reverse, respectively) were 5′‐CCGTGGATAGAGCAGGAG‐3′ and 5′‐CGTCGCAGCCATACATTATC‐3′ for human lymphocyte antigen (HLA)–A, 5′‐GCGGCTACTACAACCAGAGC‐3′ and 5′‐GATGTAATCCTTGCCGTCGT‐3′ for HLA–B, 5′‐ GGACAAGAGCAGAGATACACG‐3′ and 5′‐ CAAGGACAGCTAGGACAACC‐3′ for HLA‐C, and 5′‐GAAGGTGAAGGTCGGAGTCA‐3′ and 5′‐GAAGATGGTAGATGGGATTTCC‐3′ for GAPDH. The primers were obtained from Sigma‐Aldrich (St. Louis, MO, USA).
2.3. Flow cytometry
Cells were dissociated and collected with the use of Accutase cell‐detachment solution (561527, BD Biosciences, San Jose, CA, USA), washed three times with 0.5% bovine serum albumin in phosphate‐buffered saline, and incubated for 30 minutes at room temperature with phycoerythrin‐conjugated antibodies to HLA‐A, ‐B, and ‐C (557349, BD Biosciences) or a similarly conjugated isotype control antibody (556640, BD Biosciences). The cells were then washed three times with Stain Buffer containing fetal bovine serum (554656, BD Biosciences) before suspension in Stain Buffer for analysis with a FACS Canto II instrument (BD Biosciences). Viable and dead cells were discriminated with the use of 7‐aminoactinomycin D (559925, BD Biosciences).
2.4. Immunoblot analysis
Cells were washed twice with ice‐cold phosphate‐buffered saline and then lysed with 1× Cell Lysis Buffer (Cell Signaling Technology, Danvers, MA, USA). The protein concentration of the lysates was determined with a bicinchoninic acid assay kit (Thermo Fisher Scientific), and equal amounts of protein were subjected to SDS‐polyacrylamide gel electrophoresis on a 7.5% gel (Bio‐Rad, Hercules, CA, USA). The separated proteins were transferred to a nitrocellulose membrane, which was then incubated for 20 minutes at room temperature with Blocking One or (for phosphorylated proteins) Blocking One‐P solution (both from Nakalai Tesque, Kyoto, Japan) before incubation overnight at 4°C with primary antibodies including those to EGFR (#4405), to phosphorylated EGFR (Tyr1068, #2234), to AKT (#9272), to phosphorylated AKT (Ser473, #9271), or to extracellular signal–regulated kinase (ERK) (#9102) from Cell Signaling Technology; with those to phosphorylated ERK (Thr202/Tyr204, sc‐16982) from Santa Cruz Biotechnology (Santa Cruz, CA, USA); or with those to β‐actin (#10021) from Sigma‐Aldrich. The membrane was washed with phosphate‐buffered saline containing 0.05% Tween 20 before incubation for 2 hours at room temperature with horseradish peroxidase–conjugated secondary antibodies (NA934, GE Healthcare, Indianapolis, IL, USA). Immune complexes were detected with enhanced chemiluminescence reagents (RPN3244, GE Healthcare).
2.5. Immunohistochemistry
Immunohistochemical staining was performed as previously described.14 The primary antibodies included those to HLA class I (ab70328, Abcam, Cambridge, MA, USA), to CD8 (GA623, Agilent Technologies, Palo Alto, CA, USA), to PD‐L1 (ab205921, Abcam), to phosphorylated EGFR (Tyr1068, #3777, Cell Signaling Technology), and to phosphorylated ERK (Thr202/Tyr204, #4370, Cell Signaling Technology).
2.6. Patients and tumor specimens
The use of human tissue was in accordance with the tenets of the Declaration of Helsinki and was approved by the Institutional Review Board of Kindai University. Tumor samples were obtained from NSCLC patients with EGFR mutations both before initiation of EGFR‐TKI therapy as well as after disease progression during the treatment.
2.7. Statistical analysis
Quantitative data are presented as means + SE and were compared with the two‐sided unpaired Student's t test. The analysis was performed with the use of GraphPad Prism v.7 software (GraphPad Inc., La Jolla, CA, USA). A P value < 0.05 was considered statistically significant.
3. RESULTS
3.1. EGFR mutated NSCLC cells tended to show lower expression level of HLA‐B compared to EGFR wild type NSCLC cells
Epidermal growth factor receptor is a transmembrane protein that is activated by binding of its ligands including epidermal growth factor and transforming growth factor–α. Mutations in the kinase domain of EGFR have been identified in a subset of NSCLC patients, the growth of whose tumors is dependent on the associated activation of EGFR signaling. In vitro studies have shown that EGFR signaling suppresses the expression of immunoregulatory genes including those for MHC‐I molecules.15 We therefore hypothesized that mutational activation of EGFR in NSCLC might reduce MHC‐I expression and thereby lower the efficacy of PD‐1–targeted therapy. To examine if mutational activation of EGFR inhibits MHC‐I expression, we compared basal expression level of MHC‐I gene in the presence of IFN‐γ in EGFR mutation positive and negative cells by RT‐PCR (Figure 1). As expected, HLA‐B expression level of EGFR mutated NSCLC cells tended to be lower than those of EGFR wild type positive cells.
Figure 1.
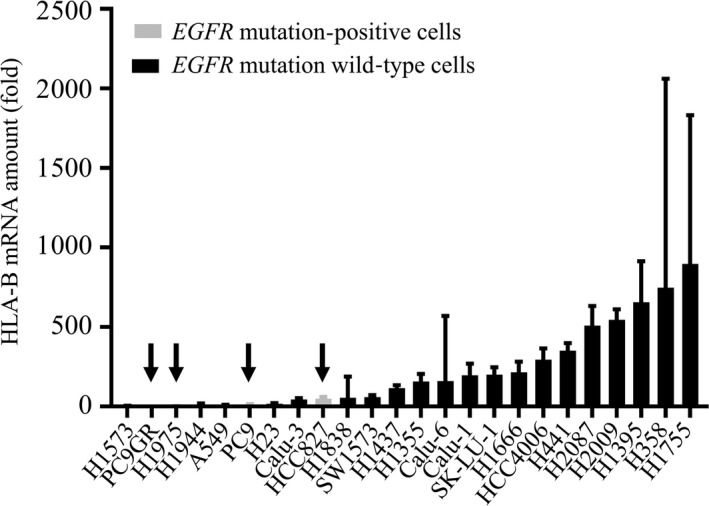
Human lymphocyte antigen (HLA)‐B gene expression in epidermal growth factor receptor (EGFR) mutation positive and EGFR mutation wild type non–small cell lung cancer cells in the presence of interferon (IFN)‐γ. Cells were incubated with IFN‐γ (I, 20 U/mL) for 48 h, after which the amount of HLA‐B mRNA (fold over H1573 control) was determined by reverse transcription and real‐time polymerase chain reaction analysis. Arrow indicates EGFR mutation positive cells
3.2. EGFR inhibition increases MHC‐I expression in the absence or presence of IFN‐γ
We next examined the effects of inhibition of EGFR activity with the EGFR‐TKIs erlotinib and osimertinib on HLA–B gene expression in various NSCLC cell lines (Figure 2A). RT‐PCR analysis revealed that the first‐generation, reversible EGFR‐TKI erlotinib induced only a small increase in the amount of HLA‐B mRNA in the H1944 cell line, which is wild type for EGFR. In the EGFR‐mutated cell lines HCC827, erlotinib significantly increased HLA‐B mRNA abundance. In another EGFR‐mutated cell line, PC9, the increase of HLA‐B expression by erlotinib was small, but was statistically significant. On the other hand, in the T790M‐positive cell line H1975, whereas erlotinib had no significant effect, the third‐generation, T790M‐selective EGFR‐TKI osimertinib significantly increased the amount of HLA‐B mRNA. Neither erlotinib nor osimertinib affected the HLA‐B mRNA level in the T790M‐positive cell line PC9GR.
Figure 2.
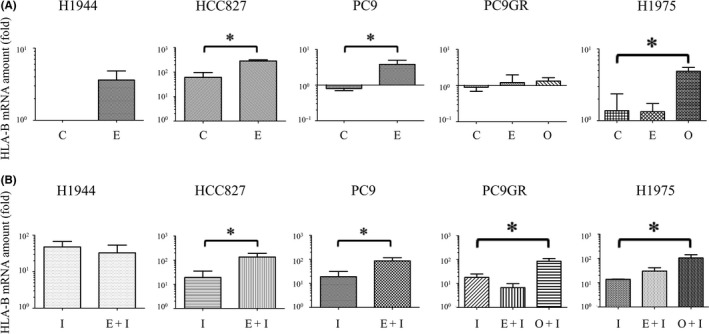
Effects of epidermal growth factor receptor (EGFR)‐tyrosine kinase inhibitors on human lymphocyte antigen (HLA)‐B gene expression in non–small cell lung cancer cell lines in the absence or presence of interferon (IFN)‐γ. A, The indicated cell lines were incubated in the absence (C, control) or presence of erlotinib (E, 50 nmol/L) or osimertinib (O, 50 nmol/L) for 48 h, after which the amount of HLA‐B mRNA (fold over H1944 control) in the cells was determined by reverse transcription and real‐time polymerase chain reaction analysis. B, Cells were incubated with IFN‐γ (I, 20 U/mL) in the absence or presence of erlotinib (E, 50 nmol/L) or osimertinib (O, 50 nmol/L) for 48 h, after which the amount of HLA‐B mRNA (fold over H1944 control) was determined as in (A). All data are means + SE from three independent experiments. *P < 0.05 (unpaired Student's t test)
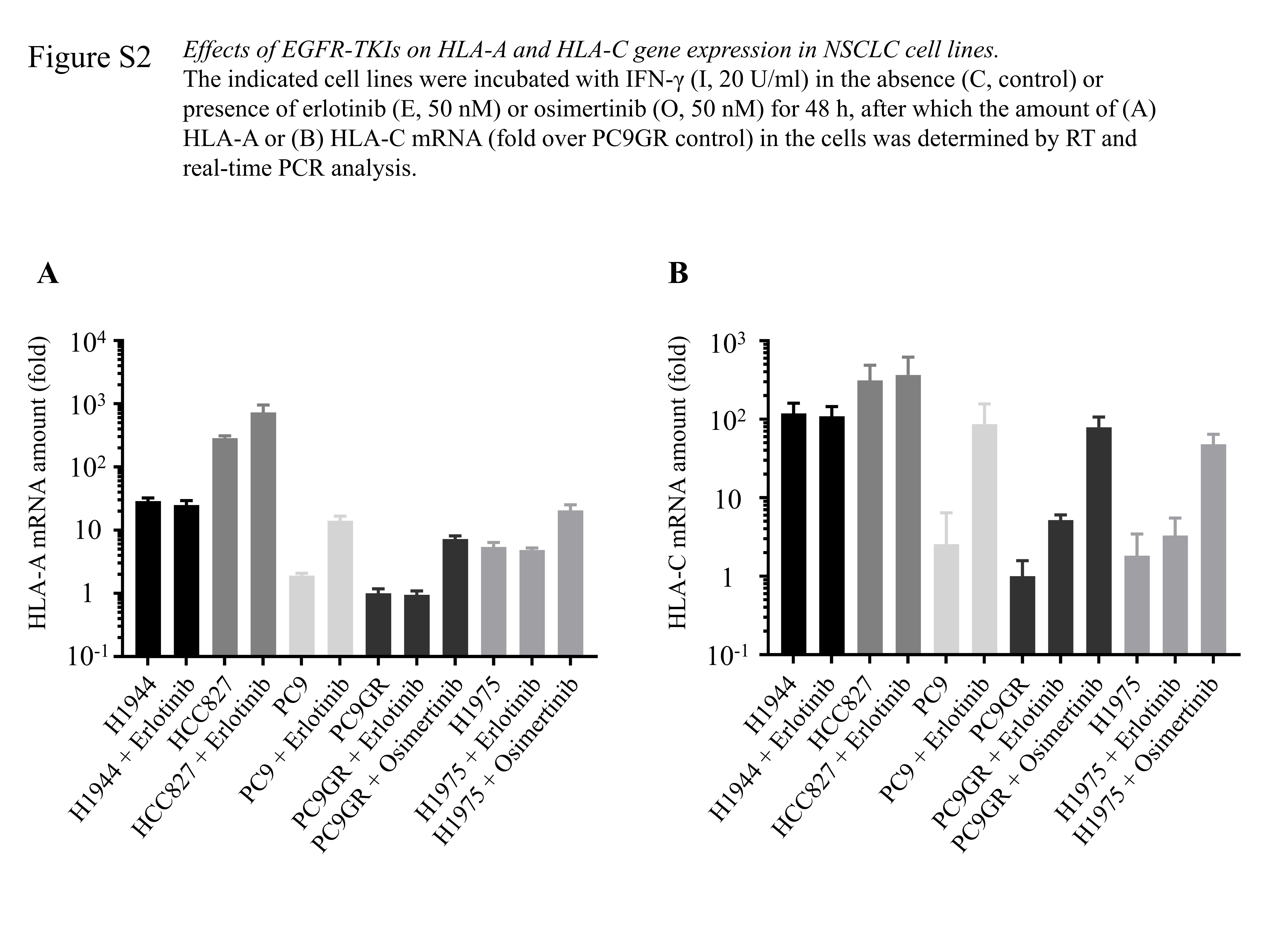
Given that IFN–γ up‐regulates cell surface expression of MHC‐I in vivo,16, 17 we next exposed NSCLC cells to EGFR‐TKIs in the presence of IFN‐γ (Figure 2B). Under this condition, erlotinib did not affect HLA‐B mRNA abundance in H1944 cells but significantly increased it in HCC827 and PC9 cells. Osimertinib significantly increased the HLA‐B mRNA level in both H1975 and PC9GR cells, whereas erlotinib had no such effect. Treatment of osimertinib resulted in no increase of HLA‐B in H1944 cells, and only small increase compared to erlotinib in HCC827 and PC9 cells (Figure S1). The effect of EGFR‐TKIs on MHC‐I gene expression was also seen in HLA‐A and HLA‐C, which was determined by RT‐PCR (Figure S2). In consistent with HLA‐B expression, appropriate EGFR‐TKI increased HLA‐A or C expression in EGFR‐mutated cells, whereas, there was no effect of EGFR‐TKIs on EGFR wild type H1944 cells.
To determine whether these effects of EGFR‐TKIs on MHC‐I gene (HLA‐A, B, and C) expression were accompanied by up‐regulation of MHC‐I protein at the cell surface, we treated PC9 and PC9GR cells with these drugs in the absence or presence of IFN‐γ and then subjected them to flow cytometric analysis with antibodies to HLA‐A, ‐B, and ‐C (Figure 3). In EGFR‐mutated PC9 cells, erlotinib increased the expression of MHC‐I in the absence or presence of IFN‐γ, although these effects were not statistically significant. In T790M‐positive PC9GR cells, erlotinib and osimertinib each increased the expression of HLA‐B in the absence or presence of IFN‐γ, with the effect of osimertinib in the presence of IFN‐γ being the most pronounced and achieving statistical significance. Overall, these findings were thus similar to those obtained by RT and real‐time PCR analysis of HLA‐B mRNA and suggested that EGFR inhibition by appropriate EGFR‐TKIs increases both MHC‐I gene expression and cell surface protein abundance.
Figure 3.
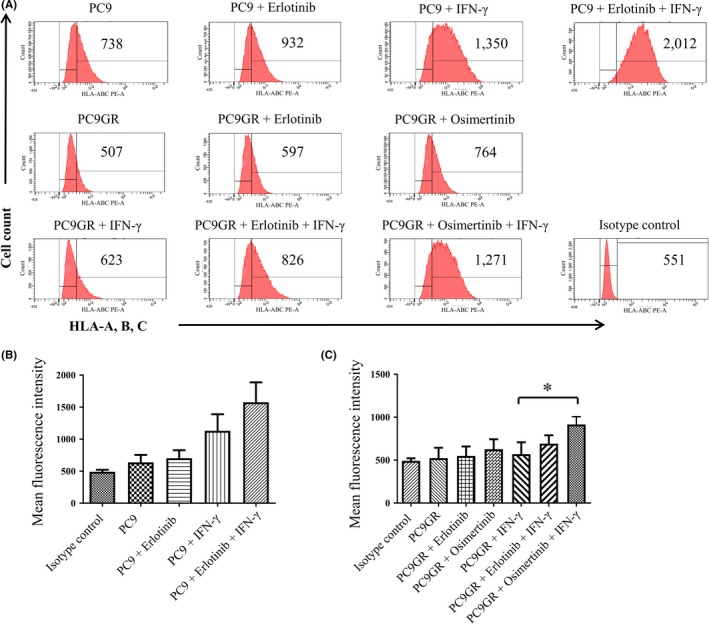
Effects of epidermal growth factor receptor (EGFR)‐tyrosine kinase inhibitors on major histocompatibility complex class I (MHC‐I) protein expression at the surface of non–small cell lung cancer cells in the absence or presence of interferon (IFN)‐γ. A, PC9 or PC9GR cells were incubated in the absence or presence of erlotinib (50 nmol/L), osimertinib (50 nmol/L), or IFN‐γ (20 U/mL) for 48 h, after which cell surface MHC‐I expression was analyzed by flow cytometry with antibodies to a shared epitope of human lymphocyte antigen (HLA)‐A, ‐B, and ‐C. Representative flow cytometric profiles and the corresponding mean fluorescence intensity (MFI) of MHC‐I–positive cells are shown. B,C, The MFI of MHC‐I–positive cells is shown as the mean + SE from three independent experiments. *P < 0.05 (unpaired Student's t test)
3.3. Role of the MEK‐ERK pathway in induction of MHC‐I expression by EGFR inhibition
The phosphatidylinositol 3‐kinase (PI3K)–AKT and ERK signaling pathways are the major mediators of EGFR signal transduction. Immunoblot analysis revealed that, in T790M‐positive PC9GR cells, osimertinib (which increased MHC‐I expression) inhibited the phosphorylation of AKT and ERK, whereas erlotinib (which did not significantly induce MHC‐I expression) had minimal effects on AKT and ERK phosphorylation (Figure 4A), suggesting that inhibition of either PI3K‐AKT or ERK pathways may play a role in up‐regulation of MHC‐I expression. To investigate further the role of these two pathways, we examined the effects of trametinib, which inhibits the upstream ERK kinase MEK, and of the pan‐PI3K inhibitor buparlisib on MHC‐I expression in PC9GR cells. We first confirmed that trametinib and buparlisib inhibited the activation of ERK and AKT, respectively, in PC9GR cells (Figure 4B). Flow cytometric analysis then revealed that trametinib significantly increased MHC‐I expression in PC9GR cells in the absence or presence of IFN‐γ, whereas buparlisib had no such effect (Figure 3C,D). These results thus suggested that the inhibition of MHC‐I expression by EGFR activity is mediated by MEK‐ERK signaling.
Figure 4.
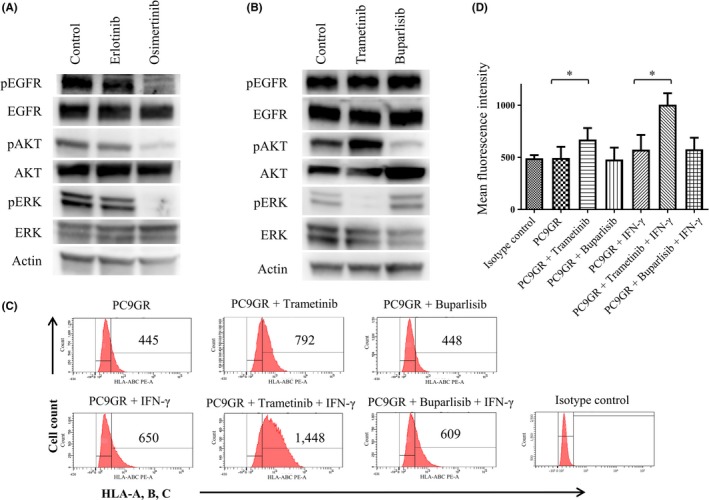
Effects of MEK and PI3K inhibitors on major histocompatibility complex class I (MHC‐I) expression in PC9GR cells. A,B, Cells were incubated for 6 h in the absence or presence of erlotinib (50 nmol/L) or osimertinib (50 nmol/L) (A) or of trametinib (100 nmol/L) or buparlisib (500 nmol/L) (B), after which cell lysates were subjected to immunoblot analysis with antibodies to phosphorylated (p) or total forms of epidermal growth factor receptor, AKT, or extracellular signal‐regulated kinase or with those to β‐actin (loading control). C,D, Cells were incubated with or without trametinib (100 nmol/L) or buparlisib (500 nmol/L) and in the absence or presence of interferon‐γ (20 U/mL) for 48 h, after which cell surface expression of MHC‐I was analyzed by flow cytometry with antibodies to a shared epitope of HLA‐A, ‐B, and ‐C. Representative flow cytometric profiles (C) and the mean fluorescence intensity (MFI) of MHC‐I–positive cells determined as means + SE from three independent experiments (D) are shown. *P < 0.05 (unpaired Student's t test)
3.4. Down‐regulation of phospho‐EGFR and phospho‐ERK associated with up‐regulation of MHC‐I in tumor tissue
To investigate the possible role of EGFR‐MEK‐ERK signaling in MHC‐I expression in vivo, we examined the abundance of MHC‐I and phosphorylated forms of EGFR and ERK as well as of CD8 and PD‐L1 in specimens of an EGFR‐mutated NSCLC tumor obtained before and after EGFR‐TKI treatment (Figure 5). Immunohistochemical analysis revealed that the amounts of phosphorylated EGFR and ERK were markedly decreased after the onset of EGFR‐TKI treatment, possibly as a result of the inhibition of EGFR activation. In contrast, expression of MHC‐I at the cell surface and in the cytoplasm was substantially increased after treatment onset. Furthermore, the number of CD8+ TILs and the expression of PD‐L1 were also increased after EGFR‐TKI exposure. These results are thus consistent with the notion that increased signaling by the EGFR‐MEK‐ERK pathway in EGFR‐mutated NSCLC results in down‐regulation of MHC‐I expression, and that inhibition of EGFR activity with EGFR‐TKIs attenuates MEK‐ERK signaling and thereby increases MHC‐I expression, possibly leading to activation of CD8+ TILs.
Figure 5.
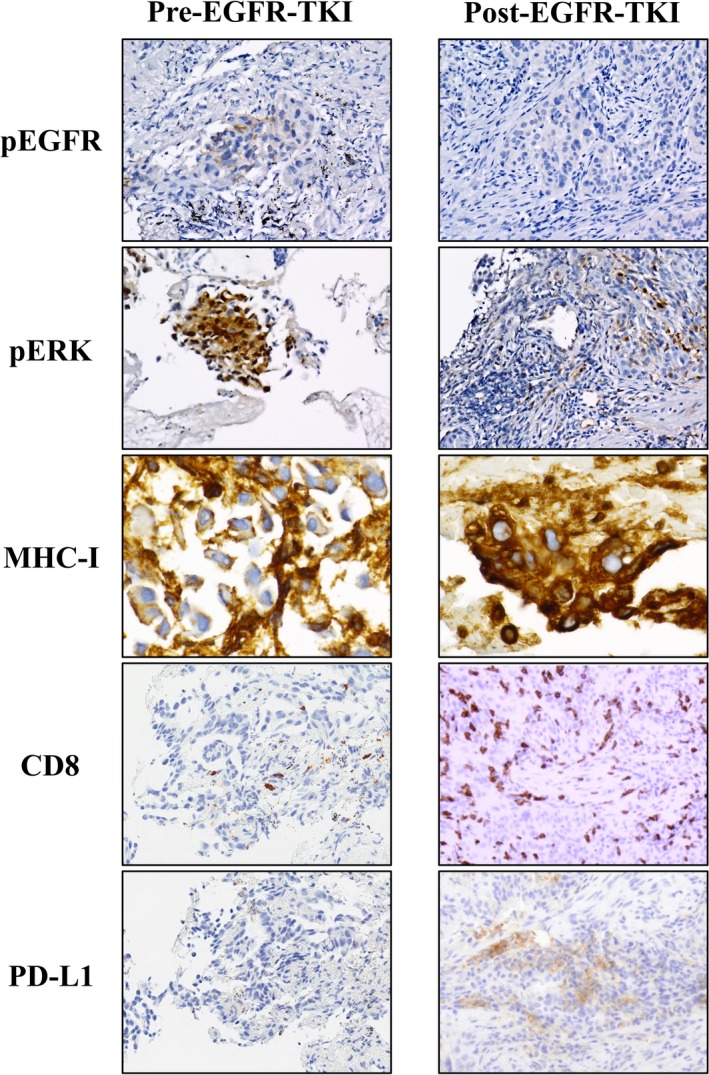
Immunohistochemical analysis of tumor biopsy specimens obtained before and after epidermal growth factor receptor (EGFR)‐ tyrosine kinase inhibitor treatment in a patient with EGFR‐mutated non–small cell lung cancer. The tumor samples were stained with antibodies to phosphorylated (p) forms of EGFR and extracellular signal‐regulated kinase as well as with those to human lymphocyte antigen class I, to CD8, and to PD‐L1
4. DISCUSSION
Epidermal growth factor receptor is a receptor tyrosine kinase that is commonly activated or overexpressed in numerous types of cancer. Up‐regulation of EGFR activity is mediated by various mechanisms including kinase domain mutations that confer ligand‐independent signaling. MEK‐ERK and PI3K‐AKT signaling pathways mediate the biological effects of EGFR activation and are well‐characterized targets of cancer therapy. With the use of EGFR‐mutated NSCLC cell lines, we have now shown that pharmacological inhibition of the MEK‐ERK pathway, but not that of the PI3K‐AKT pathway, restored the expression of MHC‐I molecules at the cell surface that had been attenuated as a result of EGFR activation. Our results thus support previous findings that inhibition of EGFR or MEK increases the expression of MHC‐I in primary and malignant human keratinocytes or NSCLC cell lines.15, 18
Although the development of PD‐1–PD‐L1 inhibitors has improved treatment outcome for NSCLC patients, the presence of EGFR mutations, which have been identified in ~40% to 60% of lung adenocarcinomas in East Asia,19 has been associated with a low response rate to such drugs.10, 11 Antibodies to PD‐1 or to PD‐L1 relieve effector T cell suppression, with CD8+ TILs being key determinants of the efficacy of these agents. Tumor infiltration by CD8+ T cells and their distribution at tumor invasive margins that precede PD‐1 blockade appear to predict the efficacy of PD‐1–PD‐L1 inhibitors in advanced melanoma.20, 21 Expression of MHC‐I and antigen presentation at the surface of target cells are essential for the adaptive immune response. Our present histopathologic analysis of EGFR‐mutated NSCLC tumor samples suggested that EGFR‐TKI treatment induced CD8+ T cell infiltration and PD‐L1 expression in the tumor as well as increased MHC‐I expression both at the cell surface and in the cytoplasm and down‐regulated the amounts of phosphorylated EGFR and ERK in tumor cells. Although the precise nature of the relation between MHC‐I expression and CD8+ TIL infiltration in EGFR‐mutated tumors remains unclear, our results suggest the possibility that a low level of MHC‐I expression due to EGFR‐MEK‐ERK pathway activation might contribute to the poor response to PD‐1–PD‐L1 inhibitors in EGFR‐mutated NSCLC. However, the current in vivo analysis has a limitation due to lack of sample size, since we only showed one representative case in this study. Analysis of a larger number of clinical specimens will be necessary to validate this possibility.
Epidermal growth factor receptor‐TKIs are considered the best choice for first‐line treatment of advanced EGFR‐mutated NSCLC,22 but treated individuals eventually develop resistance to these drugs, most often as a result of the emergence of a secondary T790M mutation of EGFR. Osimertinib was designed to overcome such resistance conferred by the T790M mutation and has been approved and now widely adopted for treatment of NSCLC positive for this mutation.23 On the other hand, we previously found that, among EGFR‐TKI–resistant patients, those negative for T790M tended to show a more favorable response to the PD‐1–targeted antibody nivolumab compared with those positive for T790M.24 The results of the present and previous studies suggest that EGFR pathway activation is maintained in T790M‐positive tumors that generally show a durable response to osimertinib and may lead to suppression of MHC‐I expression.18 We found that osimertinib overcomes such suppression of MHC‐I in T790M‐positive PC9GR and H1975 cells. Therefore, even in NSCLC patients with EGFR‐TKI resistance due to T790M, appropriate EGFR‐TKI therapy may increase MHC‐I expression through inhibition of EGFR signaling and thereby improve the efficacy of immune‐checkpoint inhibitors.
Our flow cytometric analysis revealed that the down‐regulation of MHC‐I expression by EGFR activation was dependent on the MEK‐ERK pathway, but not on the PI3K‐AKT pathway. Consistent with our in vitro findings, activation of the MEK‐ERK pathway or of its upstream activator RAS has previously been associated with low MHC expression in several other types of malignancy.25, 26, 27, 28, 29 MEK inhibition was also shown to increase MHC‐I expression in tumor cell lines.30 MHC‐I expression has been found to be induced by tumor necrosis factor–α, interleukin‐1, IFN‐β, and IFN‐γ, all of which up‐regulate HLA‐A through activation of the JAK‐STAT pathway.31 Furthermore, the combination of kinase inhibitors (an irreversible EGFR‐TKI, afatinib, or trametinib) and IFN‐γ had additive effects on HLA expression as well as on that of the antigenic peptide transporter TAP1 and β2‐microglobulin.18 The MHC‐II transcription factor CIITA was also found to increase MHC‐I gene expression,32 and our RT and real‐time PCR analysis showed that EGFR inhibition induced CIITA gene expression in EGFR‐mutated NSCLC cell lines (unpublished data). Together, these various observations suggest that further investigations of the combination of immune‐checkpoint inhibitors and agents that target the EGFR‐MEK‐ERK pathway for cancer treatment are warranted. A phase Ib study of the combination of the MEK inhibitor cobimetinib and the PD‐L1 inhibitor atezolizumab in patients with metastatic colorectal cancer demonstrated a promising tumor response associated with increased T cell infiltration.33 In addition, several early‐phase studies of the concurrent combination of an EGFR‐TKI and immunotherapy have been undertaken with NSCLC patients,34, 35, 36, 37 although the data have been disappointing as a result of high toxicity as manifested by adverse events such as interstitial lung disease and elevation of liver enzymes.35, 36 Further studies will thus be required to identify optimal treatment strategies based on concurrent or sequential administration of an EGFR‐TKI or MEK inhibitor in combination with immunotherapy in patients with NSCLC positive for EGFR mutations.
In summary, our results have suggested that mutational activation of EGFR signaling inhibits MHC‐I expression through the MEK‐ERK pathway in NSCLC cells. An appropriate EGFR‐TKI or MEK inhibitor can inhibit EGFR‐MEK‐ERK signaling and thereby increase MHC‐I expression and possibly activate an immune response to the tumor (Figure 6). Down‐regulation of MHC‐I expression may be responsible, at least in part, for the low response rate of immune‐checkpoint blockade in patients with EGFR‐mutated NSCLC. Further study is thus warranted to evaluate the relation between EGFR‐MEK‐ERK signaling in EGFR‐mutated NSCLC and the antitumor immune response.
Figure 6.
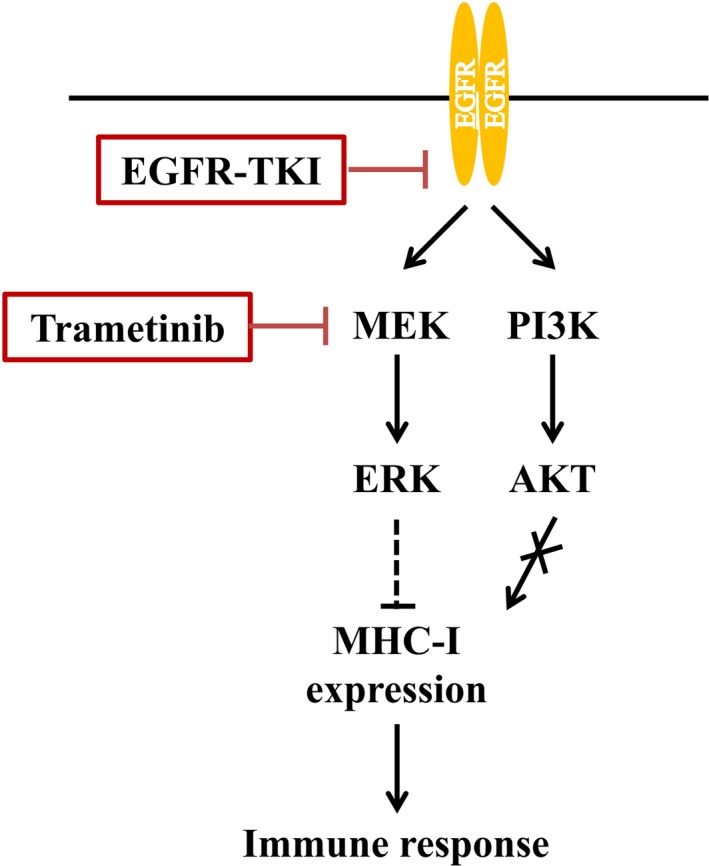
Model for the regulation of major histocompatibility complex class I (MHC‐I) expression in non–small cell lung cancer cells by epidermal growth factor receptor (EGFR)‐MEK‐extracellular signal‐regulated kinase (ERK) signaling. Mutational activation of EGFR leads to down‐regulation of MHC‐I expression through the MEK‐ERK signaling pathway. EGFR‐tyrosine kinase inhibitors and the MEK inhibitor trametinib increase MHC‐I expression via inhibition of ERK activation. The increase in MHC‐I expression facilitates recognition of the cancer cells by intratumoral T cells and thereby promotes an antitumor immune response
CONFLICT OF INTERESTS
Hidetoshi Hayashi reports grants from Japan Society for the Promotion of Science, during the conduct of the study; personal fees from AstraZeneca K.K., personal fees from Boehringer Ingelheim Japan Inc, personal fees from Bristol‐Myers Squibb Co. Ltd, personal fees from Eli Lilly Japan K.K., personal fees from Ono Pharmaceutical Co. Ltd., personal fees from Taiho Pharmaceutical Co. Ltd, personal fees from Chugai Pharmaceutical Co. Ltd., personal fees from Kyowa Hakko Kirin Co., Ltd., personal fees from MSD K.K., personal fees from Pfizer Japan Inc., outside the submitted work; Koji Haratani reports personal fees from Ono Pharmaceutical Co. Ltd., personal fees from Bristol‐Myers Squibb Co. Ltd., personal fees from Pfizer Japan Inc., grants from AstraZeneca K.K., outside the submitted work; Junji Tsurutani reports grants from Daiichisankyo Inc., grants and other from Chugai Pharmaceutical Co Ltd., grants and other from Eisai Co., Ltd., other from Taiho pharmaceutical Co., Ltd., other from Novartis Pharma K.K, other from Kyowa‐Hakko Co., Ltd., other from Astra Zeneca K.K., outside the submitted work; Dr. Togashi reports grants and personal fees from Ono Pharmaceutical Co Ltd, grants and personal fees from Bristol‐Myers Squibb Co Ltd, personal fees from MSD K.K., personal fees from Chugai Pharmaceutical Co Ltd, personal fees from AstraZeneca K.K., personal fees from Boehringer Ingelheim Japan Inc, personal fees from Novartis Pharma K.K., personal fees from Eli Lilly Japan KK, outside the submitted work; Kazuhiko Nakagawa reports grants and personal fees from MSD K.K., grants from A2 Healthcare Corp, grants from inVentiv Health Japan, grants and personal fees from Astellas Pharma Inc, grants from Daiichi Sankyo Co., Ltd., grants and personal fees from Novartis Pharma K.K., grants from AbbVie Inc., grants from Quintiles Inc., grants from ICON Japan K.K., grants from Chugai Pharmaceutical Co.,Ltd., grants from Takeda Pharmaceutical Co.,Ltd., grants from EP‐CRSU CO., LTD., grants from GRITSONE ONCOLOGY.INC, grants from Linical Co.,Ltd., grants and personal fees from Eli Lilly Japan K.K., grants from Eisai Co., Ltd., grants and personal fees from Taiho Pharmaceutical Co.,Ltd, grants from PAREXEL International Corp., grants and personal fees from Ono Pharmaceutical Co.,Ltd., personal fees from Clinical Trial Co.,Ltd., personal fees from Nippon Boehringer Ingelheim Co.,Ltd., personal fees from SymBio Pharmaceuticals Limited., personal fees from Pfizer Japan Inc., grants and personal fees from Bristol Myers Squibb Company, outside the submitted work. No potential conflicts of interest were disclosed by the other authors.
Supporting information
{kind=link}
{kind=link}
ACKNOWLEDGMENTS
This work was supported by Japan Society for the Promotion of Science under KAKENHI Grant number 16K21506. We thank Yuka Kitamura and Yuki Imaoka of N Lab Co., Ltd. for performing immunohistochemistry as well as Shinji Kurashimo, Eiko Honda, Haruka Sakamoto, Yume Shinkai, Michiko Kitano, and Mami Kitano of Kindai University for technical support.
Watanabe S, Hayashi H, Haratani K, et al. Mutational activation of the epidermal growth factor receptor down‐regulates major histocompatibility complex class I expression via the extracellular signal‐regulated kinase in non–small cell lung cancer. Cancer Sci. 2019;110:52‐60. 10.1111/cas.13860
REFERENCES
- 1. Ferlay J, Soerjomataram I, Dikshit R, et al. Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. Int J Cancer. 2015;136:E359‐E386. [DOI] [PubMed] [Google Scholar]
- 2. Mitsudomi T, Yatabe Y. Mutations of the epidermal growth factor receptor gene and related genes as determinants of epidermal growth factor receptor tyrosine kinase inhibitors sensitivity in lung cancer. Cancer Sci. 2007;98:1817‐1824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Mok TS, Wu YL, Ahn MJ, et al. Osimertinib or platinum‐pemetrexed in EGFR T790M‐positive lung cancer. N Engl J Med. 2016;376:629‐640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Okazaki T, Honjo T. PD‐1 and PD‐1 ligands: from discovery to clinical application. Int Immunol. 2007;19:813‐824. [DOI] [PubMed] [Google Scholar]
- 5. Johnson DB, Rioth MJ, Horn L. Immune checkpoint inhibitors in NSCLC. Curr Treat Options Oncol. 2014;15:658‐669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Brahmer J, Reckamp KL, Baas P, et al. Nivolumab versus docetaxel in advanced squamous‐cell non‐small‐cell lung cancer. N Engl J Med. 2015;373:123‐135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Borghaei H, Paz‐Ares L, Horn L, et al. Nivolumab versus docetaxel in advanced nonsquamous non‐small‐cell lung cancer. N Engl J Med. 2015;373:1627‐1639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Herbst RS, Baas P, Kim DW, et al. Pembrolizumab versus docetaxel for previously treated, PD‐L1‐positive, advanced non‐small‐cell lung cancer (KEYNOTE‐010): a randomised controlled trial. Lancet. 2016;387:1540‐1550. [DOI] [PubMed] [Google Scholar]
- 9. Rittmeyer A, Barlesi F, Waterkamp D, et al. Atezolizumab versus docetaxel in patients with previously treated non‐small‐cell lung cancer (OAK): a phase 3, open‐label, multicentre randomised controlled trial. Lancet. 2017;389:255‐265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Lee CK, Man J, Lord S, et al. Checkpoint inhibitors in metastatic EGFR‐mutated non‐small cell lung cancer‐a meta‐analysis. J Thorac Oncol. 2017;12:403‐407. [DOI] [PubMed] [Google Scholar]
- 11. Gainor JF, Shaw AT, Sequist LV, et al. EGFR mutations and ALK rearrangements are associated with low response rates to PD‐1 pathway blockade in non‐small cell lung cancer (NSCLC): a retrospective analysis. Clin Cancer Res. 2016;22:4585‐4593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Garrido F, Aptsiauri N, Doorduijn EM, Garcia Lora AM, van Hall T. The urgent need to recover MHC class I in cancers for effective immunotherapy. Curr Opin Immunol. 2016;39:44‐51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Yoshida T, Zhang G, Smith MA, et al. Tyrosine phosphoproteomics identifies both codrivers and cotargeting strategies for T790M‐related EGFR‐TKI resistance in non‐small cell lung cancer. Clin Cancer Res. 2014;20:4059‐4074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Tanaka K, Arao T, Maegawa M, et al. SRPX2 is overexpressed in gastric cancer and promotes cellular migration and adhesion. Int J Cancer. 2009;124:1072‐1080. [DOI] [PubMed] [Google Scholar]
- 15. Pollack BP, Sapkota B, Cartee TV. Epidermal growth factor receptor inhibition augments the expression of MHC class I and II genes. Clin Cancer Res. 2011;17:4400‐4413. [DOI] [PubMed] [Google Scholar]
- 16. Restifo NP, Esquivel F, Kawakami Y, et al. Identification of human cancers deficient in antigen processing. J Exp Med. 1993;177:265‐272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Schoenborn JR, Wilson CB. Regulation of interferon‐gamma during innate and adaptive immune responses. Adv Immunol. 2007;96:41‐101. [DOI] [PubMed] [Google Scholar]
- 18. Brea EJ, Oh CY, Manchado E, et al. Kinase regulation of human MHC Class I molecule expression on cancer cells. Cancer Immunol Res. 2016;4:936‐947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Sun Y, Ren Y, Fang Z, et al. Lung adenocarcinoma from East Asian never‐smokers is a disease largely defined by targetable oncogenic mutant kinases. J Clin Oncol. 2010;28:4616‐4620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Tumeh PC, Harview CL, Yearley JH, et al. PD‐1 blockade induces responses by inhibiting adaptive immune resistance. Nature. 2014;515:568‐571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Taube JM, Anders RA, Young GD, et al. Colocalization of inflammatory response with B7‐h1 expression in human melanocytic lesions supports an adaptive resistance mechanism of immune escape. Sci Transl Med. 2012;4:127ra37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Hanna N, Johnson D, Temin S, et al. Systemic therapy for stage IV non‐small‐cell lung cancer: American Society of Clinical Oncology Clinical Practice Guideline Update. J Clin Oncol. 2017;35:3484‐3515. [DOI] [PubMed] [Google Scholar]
- 23. Mok TS, Wu YL, Ahn MJ, et al. Osimertinib or platinum‐pemetrexed in EGFR T790M‐positive lung cancer. N Engl J Med. 2017;376:629‐640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Haratani K, Hayashi H, Tanaka T, et al. Tumor immune microenvironment and nivolumab efficacy in EGFR mutation‐positive non‐small‐cell lung cancer based on T790M status after disease progression during EGFR‐TKI treatment. Ann Oncol. 2017;28:1532‐1539. [DOI] [PubMed] [Google Scholar]
- 25. Sers C, Kuner R, Falk CS, et al. Down‐regulation of HLA Class I and NKG2D ligands through a concerted action of MAPK and DNA methyltransferases in colorectal cancer cells. Int J Cancer. 2009;125:1626‐1639. [DOI] [PubMed] [Google Scholar]
- 26. Frederick DT, Piris A, Cogdill AP, et al. BRAF inhibition is associated with enhanced melanoma antigen expression and a more favorable tumor microenvironment in patients with metastatic melanoma. Clin Cancer Res. 2013;19:1225‐1231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Mimura K, Shiraishi K, Mueller A, et al. The MAPK pathway is a predominant regulator of HLA‐A expression in esophageal and gastric cancer. J Immunol. 2013;191:6261‐6272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. El‐Jawhari JJ, El‐Sherbiny YM, Scott GB, et al. Blocking oncogenic RAS enhances tumour cell surface MHC class I expression but does not alter susceptibility to cytotoxic lymphocytes. Mol Immunol. 2014;58:160‐168. [DOI] [PubMed] [Google Scholar]
- 29. Loi S, Dushyanthen S, Beavis PA, et al. RAS/MAPK activation is associated with reduced tumor‐infiltrating lymphocytes in triple‐negative breast cancer: therapeutic cooperation between MEK and PD‐1/PD‐L1 immune checkpoint inhibitors. Clin Cancer Res. 2016;22:1499‐1509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Ebert PJR, Cheung J, Yang Y, et al. MAP kinase inhibition promotes T cell and anti‐tumor activity in combination with PD‐L1 checkpoint blockade. Immunity. 2016;44:609‐621. [DOI] [PubMed] [Google Scholar]
- 31. Girdlestone J, Isamat M, Gewert D, Milstein C. Transcriptional regulation of HLA‐A and ‐B: differential binding of members of the Rel and IRF families of transcription factors. Proc Natl Acad Sci USA. 1993;90:11568‐11572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Gobin SJ, Peijnenburg A, van Eggermond M, van Zutphen M, van den Berg R, van den Elsen PJ. The RFX complex is crucial for the constitutive and CIITA‐mediated transactivation of MHC class I and beta2‐microglobulin genes. Immunity. 1998;9:531‐541. [DOI] [PubMed] [Google Scholar]
- 33. Bendell JC, Kim TW, Goh BC, et al. Clinical activity and safety of cobimetinib (cobi) and atezolizumab in colorectal cancer (CRC). J Clin Oncol. 2016;34:3502.27458302 [Google Scholar]
- 34. Gettinger S, Rizvi N, Chow LQ, et al. 1054PDNIVOLUMAB (ANTI‐PD‐1; BMS‐936558, ONO‐4538) in combination with platinum‐based doublet chemotherapy (PT‐DC) or erlotinib (ERL) in advanced non‐small cell lung cancer (NSCLC). Ann Oncol. 2014;25:iv363. [Google Scholar]
- 35. Ahn MJ, Yang J, Yu H, et al. 136O: Osimertinib combined with durvalumab in EGFR‐mutant non‐small cell lung cancer: results from the TATTON phase Ib trial. J Thorac Oncol. 2016;11:S115. [Google Scholar]
- 36. Gibbons DL, Chow LQ, Kim DW, et al. 57O Efficacy, safety and tolerability of MEDI4736 (durvalumab [D]), a human IgG1 anti‐programmed cell death‐ligand‐1 (PD‐L1) antibody, combined with gefitinib (G): a phase I expansion in TKI‐naïve patients (pts) with EGFR mutant NSCLC. J Thorac Oncol. 2016;11:S79. [Google Scholar]
- 37. Ma BBY, Rudin CM, Cervantes A, et al. 441O Preliminary safety and clinical activity of erlotinib plus atezolizumab from a Phase Ib study in advanced NSCLC. Ann Oncol. 2016;27:mdw594.005. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials