

Abstract
Serine peptidases of the prolyl oligopeptidase (POP) family are of substantial therapeutic importance because of their involvement in diseases such as diabetes, cancer, neurological diseases, and autoimmune disorders. Proper annotation and knowledge of substrate specificity mechanisms in this family are highly valuable. Although endopeptidase, dipeptidyl peptidase, tripeptidyl peptidase, and acylaminoacyl peptidase activities have been reported previously, here we report the first instance of carboxypeptidase activity in a POP family member. We determined the crystal structures of this carboxypeptidase, an S9C subfamily member from Deinococcus radiodurans, in its active and inactive states at 2.3-Å resolution, providing an unprecedented view of assembly and disassembly of the active site mediated by an arginine residue. We observed that this residue is poised to bind substrate in the active structure and disrupts the catalytic triad in the inactive structure. The assembly of the active site is accompanied by the ordering of gating loops, which reduces the effective size of the oligomeric pore. This prevents the entry of larger peptides and constitutes a novel mechanism for substrate screening. Furthermore, we observed structural adaptations that enable its carboxypeptidase activity, with a unique loop and two arginine residues in the active site cavity orienting the peptide substrate for catalysis. Using these structural features, we identified homologs of this enzyme in the POP family and confirmed the presence of carboxypeptidase activity in one of them. In conclusion, we have identified a new type within POP enzymes that exhibits not only unique activity but also a novel substrate-screening mechanism.
Keywords: oligomerization, substrate specificity, carboxypeptidase, enzyme catalysis, peptidase, active site regulation, catalytic triad, POP family, serine carboxypeptidase, substrate screening, S9 peptidase, oligopeptidase, structure-function, S9C subfamily
Introduction
The peptidases of the prolyl oligopeptidase (POP)5 or S9 family are of considerable pharmacological interest. Several members of this family, including prolyl oligopeptidase (the POP enzyme, namesake of the family), dipeptidyl peptidase IV, and oligopeptidase B (OPB), are important drug targets for diseases like celiac sprue and diabetes (1–4). Another member of this family, acylaminoacyl peptidase (AAP), is associated with Alzheimer's disease, cataract formation, and cancer and has been implicated in the DNA damage response (5–8). Given the immense pharmacological relevance of this family, proper annotation and knowledge of substrate specificity mechanisms used by various members of the family are highly valuable.
Members of the POP family are quite varied in their substrate specificities and are grouped into subfamilies (Fig. 1) (9). POP and OPB are endopeptidases that cleave peptide bonds following proline and basic residues, respectively (10, 11). Dipeptidyl peptidase IV and prolyl tripeptidyl peptidase remove dipeptides and tripeptides from the N terminus of oligopeptides with specificity toward proline at the penultimate position (12, 13). AAP, conversely, is an acylaminoacyl peptidase that cleaves N-acetylated peptides to generate N-acetylated amino acids and peptides with free N termini (14, 15). Despite this variation, the members of this family share a common structural fold comprising two domains, a catalytic α/β-hydrolase domain, which harbors residues of the active site, and a cylindrical β-propeller domain, which buries the active site at the domain interface and forms a large dome-shaped cavity over it (16–19). The peptidases of the family have adopted sophisticated mechanisms to regulate access to the buried active site and permit smaller peptides into the active site cavity while excluding longer or structured peptides to prevent their nontargeted digestion (20–26).
Figure 1.

Substrate specificities of POP family peptidases. Based on sequence similarities and the sequence motif around the nucleophilic serine, the enzymes are categorized into subfamilies in the MEROPS database (S9A, S9B, and S9C) (9). The nomenclature of residue position in peptide substrate is as given by Schechter and Berger (50). *, present study.
In this study, we report the first carboxypeptidase from the POP family. This enzyme is currently annotated as AAP in UniProt database, however, our concrete activity data reveal that it is, in fact, a carboxypeptidase. In contrast to all previously characterized members of this family, this peptidase requires a free C terminus in the substrate and catalyzes the sequential removal of C-terminal amino acids. We have determined 2.3-Å resolution crystal structures of this enzyme in both active (assembled catalytic triad) and inactive (disassembled catalytic triad) states. Our structural and mutagenesis studies reveal adaptations in the enzyme that enable its carboxypeptidase activity. We provide an unprecedented view of the assembly and disassembly of the active site for enzyme activation and inactivation. The active site is disassembled via disruption of the catalytic triad and the substrate-binding site. This is accompanied by the disordering of the gating loops that are involved in the oligomeric pore formation. This constitutes a novel substrate-screening mechanism in this family. Thus, our study adds a new dimension to the POP family of peptidases in terms of both enzyme activity and mechanism.
Results
Enzymatic activity
A putative peptidase of the S9C subfamily, S9Cdr from radiation-resistant bacteria Deinococcus radiodurans, was found to be annotated as AAP in the sequence database (UniProt accession number Q9RXY9). However, our preliminary studies with S9Cdr showed that, unlike what is expected from a typical AAP, activity of the enzyme toward N-Ac-Ala-Ala was lower than that toward the unblocked peptide Met-Ala-Ala (Fig. 2A).
Figure 2.
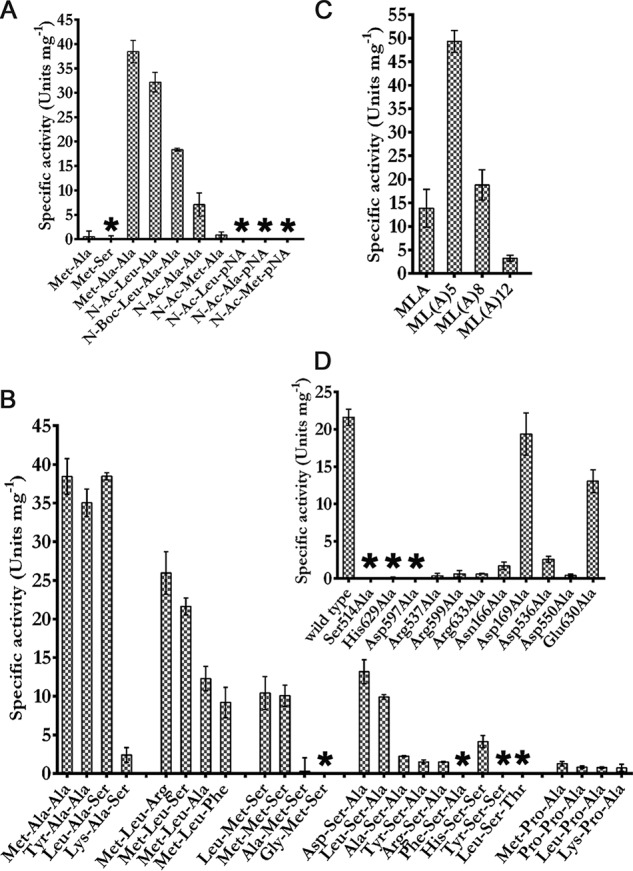
Substrate specificity and selectivity of S9Cdr. The enzyme assay was performed in 50 mm Tris-Cl, pH 8.0, at 37 °C with 2 mm substrate concentration. The specific activity for N- and/or C-blocked peptides (A), tripeptides (B), peptides with different chain length (C), and S9Cdr mutants toward Met-Leu-Ser (D) are shown. The error bars depict S.D. between the replicates (n = 3). Nondetectable activity is depicted by *.
A systematic study was carried out using different substrates to understand the substrate specificity of the enzyme. The ability of the enzyme to degrade various peptide substrates was evaluated by a spectrophotometric assay using ninhydrin reagent. No enzymatic activity was detected toward dipeptide substrates (Fig. 2A). However, tripeptides and longer peptides elicited significant activity. Among the tripeptides tested, the highest activity was observed for Met-Ala-Ala and Leu-Ala-Ser (Fig. 2B). We checked the efficiency of the enzyme in cleaving peptides with different lengths using peptides MLA, ML(A)5, ML(A)8, and ML(A)12. Of these, ML(A)5 was found to be the best substrate, whereas ML(A)12 elicited the least enzymatic activity. This result suggests that the optimum peptide length for the enzyme substrates is in the range of four to nine amino acids (Fig. 2C). The enzymatic activity was also determined for different peptides modified at either the N or C terminus (Fig. 2A). The enzyme exhibited activity toward N-blocked peptides N-Ac-Leu-Ala and N-Ac-Ala-Ala but not toward peptides N-Ac-Leu-pNA and N-Ac-Ala-pNA, which are typical AAP substrates (Fig. 2A). Thus, unlike in the case of AAP, cleavage of N-Ac-Leu-Ala and N-Ac-Ala-Ala by S9Cdr was not due to recognition of a modified N terminus. This suggests that the free C terminus of the modified peptides may be required for the activity. Hence, we suspected that S9Cdr could be a carboxypeptidase. To provide conclusive evidence, the peptide cleavage profile of the enzyme was recorded. Keeping in mind the substrate length preferred by the enzyme, peptides GGLA, MLSAA, and YSSAAAA were chosen for recording the peptide cleavage profile. These substrates were individually treated with the enzyme, and the products of the reactions were monitored using reverse-phase chromatography at different intervals after derivatizing with o-phthalaldehyde. The elution profiles of the products of individual reactions were recorded (Fig. 3). The amino acids at the C termini of the peptides (MLSAA, GGLA, and YSSAAAA) were found to be released sequentially during the reactions (Fig. 3, A, C, and E). Moreover, at no instance was the release of di- or tripeptides from the C terminus of peptides observed. Hence, the dipeptidyl and tripeptidyl activities known to occur in the POP family were ruled out for S9Cdr. Amidation of the free α-carboxylate group of the peptides MLSAA and GGLA made them resistant to hydrolysis by the enzyme (Fig. 3, B and D). This conclusively demonstrates that the S9Cdr enzyme exhibits carboxypeptidase activity.
Figure 3.

HPLC elution profiles showing carboxypeptidase activity of S9Cdr. The enzyme was incubated with different peptides, and the reaction products were monitored at different time intervals by subjecting them to reverse phase C18 column chromatography after derivatizing with o-phthalaldehyde reagent. The elution profiles of the hydrolyzed products for MLSAA (A), MLSAA-NH2 (B), GGLA (C), GGLA-NH2 (D), and YSSAAAA (E) peptides are shown.
Kinetic parameters of the enzyme were evaluated for Met-Leu-Ser, Leu-Ala-Ser, and Met-Ala-Ala substrates (Table 1). The magnitude of the carboxypeptidase activity of the enzyme is significant enough for it to be its biologically relevant activity (27). Residues believed to be critical for enzyme activity based on structural information (details in following sections) were mutated. Eleven mutants (S514A, H629A, D597A, R537A, R599A, R633A, N166A, D169A, D536A, D550A, and E630A) of the enzyme were constructed and assayed for activity toward the substrate Met-Leu-Ser (Fig. 2D). As expected, mutation of the residues of catalytic triad (Ser-514, His-629, and Asp-597) to Ala made the enzyme inactive. Mutagenesis of the substrate-stabilizing arginine residues (Arg-537, Arg-599, and Arg-633) to Ala also practically obliterated the enzyme activity. Even mutating the acidic residues that hold the stabilizing arginine residues in place (Asp-536, Asp-550, and Glu-630) had an adverse effect on activity of the enzyme. Moreover, the importance of the gating loop was highlighted by N166A mutation that led to about 90% loss of activity. Mutation D169A had no significant effect on activity of the enzyme (Fig. 2D).
Table 1.
Kinetic constants of S9Cdr
| Tripeptides | Kma | kcata | kcat/Kma |
|---|---|---|---|
| mm | s−1 | m−1 s−1 | |
| Met-Leu-Ser | 1.0 ± 0.2 | (4.1 ± 0.3) × 101 | (4.1 ± 0.9) × 104 |
| Leu-Ala-Ser | 2.6 ± 0.5 | (7.7 ± 0.7) × 101 | (3.0 ± 0.6) × 104 |
| Met-Ala-Ala | 6.0 ± 1.7 | (1.4 ± 0.3) × 102 | (2.3 ± 0.1) × 104 |
a Enzyme assay was performed in 50 mm Tris-Cl, pH 8.0, at 37 °C at various substrate concentrations. Data at each concentration are from assays performed in triplicate. Kinetic constants were determined by linear regression (Michaelis–Menten) curve fitting using Graph Pad Prism (version 6.0). Errors (S.D.) reported are those from curve fitting.
Crystal structure
The crystal structure of S9Cdr was determined to 2.3-Å resolution (PDB code 5YZM; Table 2) using crystal grown in 50 mm sodium acetate, pH 4.5, 200 mm NaCl, 10 mm MgCl2, 12% PEG 3350 (condition 1). The first seven residues of the construct and the residues of three loops (residues 42–53, 142–175, and 236–241) could not be modeled due to poor electron density. This structure presented a rather accessible active site. Upon close examination, we found that the active site catalytic triad was disassembled (not aligned in a conformation conducive to activity). Hence, we call this the inactive state. The crystal structure of the WT enzyme was solved from crystals obtained in two other crystallization conditions (condition 2: 50 mm Tris-Cl, pH 8.5, 200 mm ammonium citrate, 10 mm CaCl2, 14% PEG 3350; condition 3: 0.17 m ammonium sulfate, 25.5% PEG 4000, 15% glycerol), and in both cases the structure was found to be in the inactive state (PDB codes 6IGP and 6IGQ; Tables S1 and S2). The protein samples used to set up the crystallization trials were buffered at pH 8 using 20 mm Tris-Cl.
Table 2.
Data collection and refinement statistics of S9Cdr proteins
| S9Cdr (inactive) | S9Cdr (active) | S9Cdr S514A mutant (active) | ||
|---|---|---|---|---|
| Data collection statistics | ||||
| Diffraction source | PX-BL21, Indus-2, India | PX-BL21, Indus-2, India | PX-BL21, Indus-2, India | |
| Wavelength (Å) | 0.979 | 0.979 | 0.979 | |
| Space group | P21 | P212121 | P21 | |
| Cell dimensions | ||||
| a, b, c (Å) | 73.7, 195.1, 107.7 | 120.4, 148.2, 190.2 | 77.2, 190.0, 119.5 | |
| α, β, γ (°) | 90, 100, 90 | 90, 90, 90 | 90, 108.5, 90 | |
| Resolution (Å) | 48.77–2.30 (2.34–2.30)a | 39.28–2.30 (2.34–2.30)a | 47.90–1.70 (1.73–1.70)a | |
| Total no. of reflections | 504,988 (24,661)a | 1,076,885 (53,050)a | 2,229,551 (104,039)a | |
| Unique reflections | 132,199 (6,511)a | 151,191 (7,386)a | 356,745 (17,647)a | |
| Completeness (%) | 99.8 (99.9)a | 100.0 (99.9)a | 99.9 (99.9)a | |
| Mean I/σ | 16.6 (2.2)a | 11.4 (2.1)a | 16.4 (2.0)a | |
| Rmergeb | 0.07 (0.64)a | 0.13 (0.93)a | 0.07 (0.91)a | |
| Rmeas | 0.09 (0.74)a | 0.13 (1.00)a | 0.08 (1.00)a | |
| CC1/2 | 0.99 (0.72)a | 0.99 (0.92)a | 0.99 (0.74)a | |
| Multiplicity | 3.8 (3.8)a | 7.1 (7.2)a | 6.2 (5.9)a | |
| Wilson B factor (Å2) | 35 | 30 | 19 | |
| Refinement statistics | ||||
| Rwork/Rfreec | 0.19/0.22 | 0.20/0.23 | 0.17/0.19 | |
| Total non-H-atoms | 19,434 | 21,431 | 23,578 | |
| Protein | 18,366 | 20,100 | 20,228 | |
| Ligand/ions | 16 | 0 | 134 | |
| Water | 1,052 | 1,331 | 3,216 | |
| r.m.s.d. bond lengths (Å) | 0.002 | 0.002 | 0.007 | |
| r.m.s.d. bond angles (°) | 0.58 | 0.64 | 0.89 | |
| Average B factors | 40 | 35 | 24 | |
| Ramachandran plot | ||||
| Favored (%) | 97.95 | 96.70 | 96.61 | |
| Allowed (%) | 2.05 | 3.26 | 3.27 | |
| Outliers (%) | 0.0 | 0.04 | 0.12 | |
| PDB code | 5YZM | 5YZN | 5YZO | |
aValues in parentheses are for the highest-resolution shell.
bRmerge = ΣhklΣi|Ii(hkl) − 〈I(hkl)〉|/ΣhklΣiIi(hkl) where Ii(hkl) and 〈I(hkl)〉 represent the diffraction intensity values of the individual measurement and the corresponding mean value, respectively.
cRfree was calculated using 5% of the data that were excluded from refinement.
Several attempts were made to crystallize the enzyme in alternate conformations. Crystals obtained from another crystallization condition (condition 4: 40 mm potassium phosphate, 16–20% PEG 8000, 20% glycerol) that were soaked in N-Ac-Phe yielded the structure of the active state of the enzyme (PDB code 5YZN; Table 2) at 2.3-Å resolution. In this state, not only was the active site catalytic triad assembled in a conformation conducive to activity, the three loops (residues 42–53, 142–175, and 236–241), which were found to be disordered in the inactive state, were found to be ordered. A higher-resolution (1.7-Å resolution) structure of the active state was obtained using S514A mutant (PDB code 5YZO; Table 2). Although the mutant is inactive due to mutation in the catalytic triad, the crystals of the mutant soaked with Leu-Ala-Ser were found to be in the active state conformation. Both active state structures had residual electron density in the active site; however, the density was not sufficient for confidently modeling the ligands. This active state structure of S514A mutant was solved from crystal grown in condition 4. A crystal of S514A mutant from the same crystallization condition was diffracted without soaking any substrate. Interestingly, the structure solved from this data set was found to be in the inactive state (PDB code 6IGR; Table S1). Crystals of S514A mutant soaked in Met-Ala-Ala also yielded a 2.3-Å resolution structure in the active state conformation (PDB code 6IKG; Table S1). In this structure, the substrate was modeled in the active site with the help of Polder map (28) (Fig. S1).
The space groups of the solved structures were not the same. Some structures were solved in P21 space group, whereas others were solved in P212121 space group. However, all the structures had four monomers in an asymmetric unit. These monomers were observed in the same conformation (i.e. either all active or all inactive) and shared Cα root mean square deviation (r.m.s.d.) of ∼0.2 Å among them. Each time we solved the structure of the enzyme (WT or mutant) without soaking the crystal in substrate or substrate analog, we found the enzyme to be in the inactive state. Because this structure was reproduced from multiple crystallization conditions and from two different space groups, we believe that this inactive state exists in solution and is unlikely to be a crystallographic artifact. Also, crystals of S514A from the same crystallization condition yielded active state structure from crystal soaked in substrate (PDB code 5YZO; Table 2) and inactive state structure from crystal not soaked in substrate (PDB code 6IGR; Table S1). Both were in the same space group, and unit cell parameters were almost identical. This indicates that the observed changes in the loops and active site residues between active state and inactive state conformations are not affected by the crystal environment and could have some functional significance. Moreover, the three structures found in the active state were solved from crystals that had been soaked in substrate/substrate analog and had additional electron density in the active site close to the catalytic triad. We were able to model the substrate in one of the structures (PDB code 6IKG) using Polder map, which excludes the bulk solvent mask for ligand fitting. These data support the hypothesis that the presence of substrate favors the active state. Unless otherwise specified, the structure described in the text is that of the WT active state (PDB code 5YZN).
The S9Cdr sequence is only 27% identical to that of AAP from Pyrococcus horikoshii (PhAAP); however, their tertiary structures are similar to each other in many respects. The monomeric structure of S9Cdr superposes with that of PhAAP (PDB code 4HXE) (26) with a Cα r.m.s.d. of 1.8 Å for 567 aligned atoms (Fig. S2). It adopts a similar two-domain tertiary structure comprising an N-terminal seven-bladed β-propeller (residues 19–389) and C-terminal catalytic α/β-hydrolase domain (residues 7–18 and 394–655) that are connected by a hinge region (residues 390–393) (Fig. 4A). The major differences between the two structures are localized in the propeller domain. In the β-propeller of S9Cdr, a highly charged 12-residue-long loop (gating loop 1; residues 42–53) is present between strands β2 and β3 in the first propeller blade. This loop is absent in PhAAP. The loop (gating loop 2; residues 142–175) connecting β2 and β3 in the third propeller blade is considerably longer in S9Cdr and has 15 charged residues. A unique loop (cavity loop; residues 301–316) (Fig. S3) protruding into the active site cavity is inserted into strand β1 of the sixth blade of S9Cdr (Fig. 4B). The catalytic domain of S9Cdr matches very well with that of PhAAP except that the latter has an extended C terminus (12 residues).
Figure 4.

Structure of S9Cdr monomer. A, cartoon of S9Cdr monomer showing domains (teal and orange) and relevant loops (magenta and purple). The catalytic triad is shown by green sticks. B, a slice through S9Cdr monomer (orientation about as in A) by a vertical plane facing the viewer showing the active site cavity. C, a surface representation of S9Cdr monomer showing the side opening of the active site cavity.
The active site of S9Cdr comprises the typical serine peptidase catalytic triad residues (Ser-514, Asp-597, and His-629), which are present on the α/β catalytic domain. The active site is covered by a dome-shaped propeller domain and buries a large cavity (active site cavity) with solvent-accessible volume of ∼8900 Å3 (29). The cavity loop, a unique feature of S9Cdr protein that is absent in PhAAP, reduces the cavity volume by ∼20% (Fig. 4B). The active site in the cavity can be accessed either from the propeller tunnel (7–11-Å diameter) (Fig. 4B) or from an elliptical side opening (12–17-Å diameter) at the interface of the two domains (Fig. 4C). However, the propeller tunnel is too narrow to allow substrates to freely access the active site. The side opening, which is lined by residues of loops connecting propeller blades 1–3 and by two loops of the catalytic domain, is large enough to allow substrate access. Among these loops, gating loops 1 and 2 emanating from the propeller domain are particularly noticeable as they protrude from the plane of the side opening and modulate the shape of the opening when they become disordered in the inactive state.
Quaternary structure
S9Cdr adopts a tetrameric quaternary structure in solution with a molecular mass of ∼265 kDa (30). The four molecules in the asymmetric unit of each crystal structure are equivalent to the native tetramer as analyzed by the PDBePISA server (Fig. 5A) (31). Upon comparing the quaternary structure of S9Cdr with that of PhAAP, we found that their dimeric subunits are similar with a Cα r.m.s.d. of ∼2.4 Å for 1220 aligned atoms. S9Cdr is tetrameric and can be described as a dimer of dimers arranged at an angle of 180°, whereas PhAAP is hexameric and can be described as a trimer of dimers arranged at an angle of 120° (Fig. 5, B and C).
Figure 5.
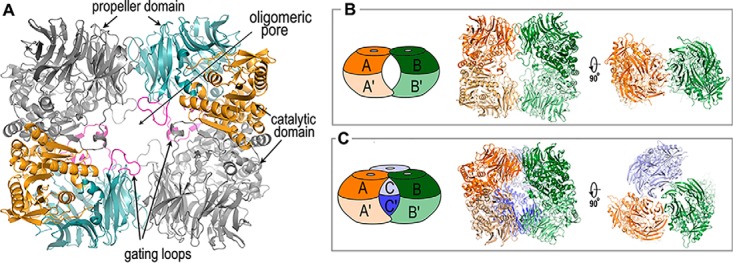
Quaternary structures of S9Cdr and PhAAP. A, cartoon of the S9Cdr tetramer. The dimeric subunits of the tetramer are shown with monomers (gray and colored) interacting with their catalytic domains. The colored monomer follows the color scheme of Fig. 4A. B, oligomerization in S9Cdr. Two dimers (AA′ and BB′) are arranged to form a tetrameric structure. C, oligomerization in PhAAP (PDB code 4HXG). A hexamer is formed by arrangement of three dimers (AA′, BB′, and CC′). In B and C, the left figure shows the schematic of the oligomeric structure and the middle and right figures show the cartoon in two orientations.
In S9Cdr, the dimeric subunit is formed by the extensive interactions of the catalytic domains of the monomers. Additionally, gating loop 2 region (residues 155–159) of the propeller domain forms antiparallel β strand interactions with the terminal β strand (β39) of the catalytic domain from the other monomer. The dimeric interface excludes a large buried surface area of ∼2540 Å2. The two dimeric subunits interact via their propeller domains to form a tetramer. The interactions of propeller domains further exclude buried surface area of ∼640 Å2 at each interface. The tetrameric assembly thus formed creates a pore (oligomeric pore) through the tetramer. This oligomeric pore is bordered by two smaller (gating loop 1) and two larger (gating loop 2) loops on each side. There is a slight change in the organization of the tetramer between the active and the inactive state conformations. The Cα r.m.s.d. differences between active and inactive dimers is 0.88 Å for 1176 aligned atoms, whereas that for the tetramer is 1.77 Å for 2350 aligned atoms.
Active site and substrate binding
In the active state structure, the side chains of Ser-514, Asp-597, and His-629 form the catalytic triad and create a charge relay system for nucleophilic attack by the catalytic residue Ser-514 (Fig. 6, A and C). Ser-514 is located at the sharp turn (nucleophilic elbow) between a strand (β36) and a helix (α6), which is similar to that of POP peptidases. The other catalytic residue, Asp-597, is located on a loop (Asp loop; residues 595–602), and His-629 is located on a conformationally flexible loop (His loop; residues 625–637) (Fig. S3). Site-directed mutagenesis of each residue of the catalytic triad (S514A, H629A, and D597A) led to complete loss of the enzymatic activity, thus confirming their indispensable functional role (Fig. 2D). S514A mutation had no effect on the structure of the active site. The active state structures of S514A mutant and WT enzymes are almost identical (Cα r.m.s.d. of 0.15 Å) except for the absence of electron density for the serine side chain in the mutant structure (Fig. 6B). The main chain nitrogen atom of Gly-434 located on the adjacent loop makes the oxyanion hole.
Figure 6.
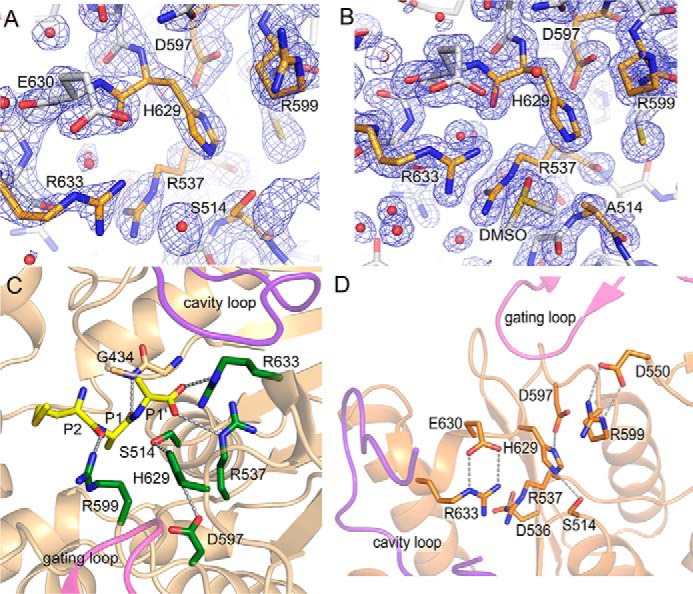
Structure of S9Cdr active sites. A and B, the electron density maps (2mFo − DFc at contour level 1.5σ) near the catalytic triad of the active state of S9Cdr WT (A) and S514A mutant (B). The relevant residues in both cases are shown in orange color. C, the substrate (Met-Ala-Ala; yellow sticks) was modeled in the active site of the active state of S9Cdr S514A mutant using Polder map. This structure was aligned with the active state of WT S9Cdr to make the figure. Substrate-binding arginine residues and catalytic triad residues are colored in green sticks. D, stabilization of side chain conformations of substrate-binding arginine residues by respective aspartate/glutamate residues in the active S9Cdr.
A structure of the S514A mutant of S9Cdr was found to have significant residual electron density in the active site, and the tripeptide substrate Met-Ala-Ala was modeled into the active site of the enzyme (Fig. 6C and Fig. S1). We also modeled a tripeptide into the active site of the S9Cdr enzyme based on superposition with the crystal structure of substrate-bound porcine POP (PDB code 1E8N) (32). The position of the ligand in the model based on the porcine POP structure and that in the solved crystal structure were almost identical. Based on the substrate-bound crystal structure, the substrate-binding subsites can be identified in the active site of S9Cdr (Fig. 6C). The structure revealed the positions of S1 and S1′ subsites with high confidence. The scissile peptide bond is between the last two alanine residues (at P1 and P1′) of the substrate. The carbonyl carbon of P1 alanine is placed at the position of the nucleophilic attack by Oγ of Ser-514, and its carbonyl oxygen is toward the main chain nitrogen of the oxyanion hole residue Gly-434. In this position, the side chain of P1 Ala fits into the S1 pocket, which is lined by hydrophobic residues Trp-481, Ile-539, Phe-545, Ile-551, Phe-555, Trp-556, and Cys-600. The side chain of Ala at P1′ is exposed to the cavity. Three arginine residues (Arg-537, Arg-599, and Arg-633), which bind the substrate peptide, are located near the active site. The side chain of Arg-599 forms a hydrogen bond with the carbonyl oxygen of Met at the P2 position. This arginine is located on the Asp loop and is conserved in POP, OPB, and AAP peptidases (24). The free α-carboxylate of Ala at the P1′ position interacts with side chains of Arg-537 and Arg-633 (Fig. 6C). These two arginine residues are unique features of the S9Cdr enzyme and have not been conserved in other structurally characterized POP peptidases. Arg-537 is located on a loop between strand β37 and helix α7 adjacent to the nucleophilic elbow, and Arg-633 is located on the His loop (Fig. S3). Interestingly, the side chains of the substrate-binding arginine residues (Arg-537, Arg-599, and Arg-633) are stabilized by the electrostatic interactions with those of Asp-536, Asp-550, and Glu-630, respectively (Fig. 6D). The individual point mutagenesis of the substrate-binding arginine residues (R537A, R599A, and R633A) leads to acute loss of the enzymatic activity in S9Cdr (Fig. 2D), thus confirming their importance for activity of the enzyme. The importance of the interacting acidic residues was also confirmed by making individual point mutants (D536A, D550A, and E630A) that were found to have reduced enzymatic activities.
Based on the substrate-bound structure, some aspects of substrate specificity can be understood. The P1 pocket is very hydrophobic, and the presence of a hydrophilic residue at the P1 position could be unfavorable (Fig. 6C). Indeed, the enzyme had lower activity against peptides with the hydrophilic residue Ser at the P1 position, whereas residues Ala and Leu were preferred at this position (Fig. 2B). The P2 position is stabilized by hydrogen bonding of the carbonyl oxygen with Arg-599. The N-terminal residue of the tripeptide substrate is oriented in a way that the side chain is in close proximity with Arg-599 (Fig. 6C). Hence, positively charged residues at the N terminus of tripeptide substrates should be unfavorable. This was verified by the low activity of the enzyme toward Lys-Ala-Ser substrate, whereas the activity against Leu-Ala-Ser substrate was quite high (Fig. 2B). Two arginine residues (Arg-537 and Arg-633) bind to the free α-carboxylate of the peptide substrate, thus facilitating substrate binding (Fig. 6C). Furthermore, the cavity loop occupies the space in the active site cavity at positions P2′ and onward (Fig. 4B), which orients substrates for carboxypeptidase activity.
Comparison of active and inactive state structures
The Cα r.m.s.d. differences between structures solved in active and inactive states are 0.59 Å for 591 aligned atoms of the monomer and 1.77 Å for 2350 aligned atoms of the tetramer. Although the Cα r.m.s.d. values are low, there are significant local differences between the active and inactive state structures of the WT enzyme (Fig. S4). In the active state structure, the gating loops are ordered, and the active site is assembled. In contrast, the gating loops of each monomer of the inactive state structure are disordered, and the active site is completely disassembled. The disassembly of active site is achieved via two modes: (i) disruption of the Ser-His-Asp catalytic triad and (ii) dismantling of the substrate-binding site. This process is enabled by the following conformational changes (Fig. 7A). The His loop with loop-helix-loop structure changes its conformation and is displaced by ∼8 Å from its position in the active structure. This change displaces Cα atoms of the catalytic His-629 and substrate-binding Arg-633 residues by ∼6.2 and 10.5 Å, respectively. The Asp loop moves slightly outward, thus displacing the catalytic Asp-597 by ∼1.2 Å from its position in the active structure. A critical arginine (Arg-537; catalytic triad–disrupting arginine) located on the adjacent loop of the nucleophilic elbow alters its side chain conformation (from ptt-85 where χ1 = 62°, χ2 = 180°, χ3 = 180°, and χ4 = −85° to ttp180 where χ1 = −177°, χ2 = 180°, χ3 = 65°, and χ4 = −175°) and moves (by 6.7 Å) to a location where it is positioned between the residues of the catalytic triad in the inactive structure. It forms a salt bridge with the catalytic residue Asp-597. The nucleophile Ser-514 changes its side chain rotamer in a way that disfavors catalytic triad formation. Thus, in the inactive state structure of S9Cdr, the catalytic triad is completely disrupted and is not competent to perform catalysis (Fig. 7B). The substrate-binding residues (Arg-537, Arg-599, and Arg-633) also adopt alternate conformations in the inactive structure that render them incompetent for substrate binding. Moreover, the acidic residues (Asp-536, Asp-550, and Glu-630) that stabilize the side chain conformations of these arginine residues in the active structure cease to interact with them in the inactive structure. Thus, the substrate-binding site is dismantled in the inactive structure. The side chain of Arg-537 is particularly interesting as it switches its conformation from the substrate-binding position in the active structure to the catalytic triad-disruptive position in the inactive structure, thus playing critical roles in each conformation.
Figure 7.
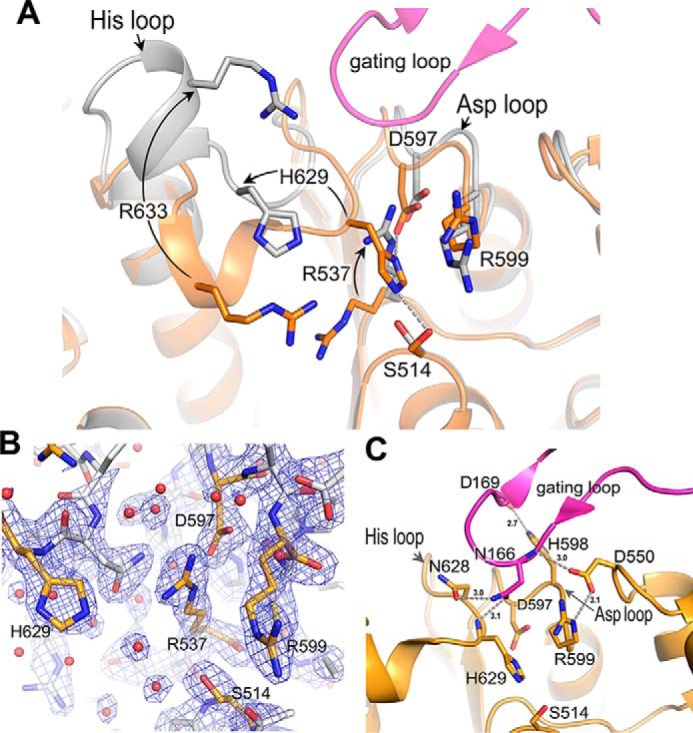
Active and inactive conformations of S9Cdr active site. A, superposition of the active sites of the active (orange) and inactive (gray) enzymes. The active and inactive structures show the assembly and disassembly of the active site, respectively. The critical residues are shown as sticks. The arrows mark the conformational changes from active to inactive structures. B, the electron density maps (2mFo − DFc at contour level 1.5σ) near the catalytic triad of the inactive state of S9Cdr WT. The relevant residues are shown in orange color. C, important interactions of the gating loop with the residues of the His and Asp loops in the active enzyme.
In the active state structure, gating loop 2 adopts a conformation in which one region of the loop (residues 155–159) forms antiparallel β strand interactions with a solvent-exposed β strand (β39; β edge) of the catalytic domain, which helps in the formation of a dimeric subunit of a tetramer. This interaction is conserved in the structure of PhAAP (PDB code 4HXG) (26). Residues 164–169 of the gating loop form hydrogen-bonding interactions with those from the His loop (625–637), Asp loop (595–602), and another loop (548–554) of the catalytic domain; these are Asn-166 (Nδ2) with Asn-628 (Oδ1), Asn-166 (Oδ1) with His-629 (N), Asp-169 (Oδ1) with His-598 (Nϵ2), Arg-164 (Nϵ2) with Thr-548 (O), and Asn-166 (N) with Asp-550 (Oδ2) (Fig. 7C). In the inactive state structure, the disordering of gating loop 2 leads to loss of these interactions. This is concomitant with the conformational changes in the His and Asp loops that destabilize the catalytic triad and substrate-binding site. We observed significant contribution of gating loops 1 and 2 in regulating the size of the oligomeric pore. The active state tetramer with structured gating loops has a pore size of 9–12 Å (Fig. 5A). In the inactive state tetramer, these loops are disordered; thus, the pore size is increased considerably. The importance of the gating loop is supported by our mutagenesis result where N166A mutation caused a 90% loss of enzymatic activity (Fig. 2D). The backbone and sidechain of Asn-166 make multiple polar interactions in the closed conformation, and these interactions seem to be critical for closing the loop and activity of the enzyme. Asp-169 of the gating loop also makes a polar interaction in the closed conformation, but it is not as centrally located as Asn-166. Mutagenesis data show that Asp-169 is not as important for the gating because the D169A mutation is almost as active as the native enzyme (Fig. 2D). Asp-550 interacts with the gating loop and with the substrate-binding residue Arg-599. D550A mutation leads to acute loss of enzymatic activity. This could be due to its role in gating or its role in stabilizing Arg-599. However, Asp-536 and Glu-630 are also involved in stabilization of other substrate-binding arginine residues, and mutations at these acidic residues (D536A and E630A) have a modest effect on the activity of the enzyme. This suggests that the acute loss of activity in the case of the D550A mutation is due to its interaction with the gating loop. Acute loss of activity upon mutation of residues tethering the gating loop to the active site highlights the importance of closing the gating loop.
Verification of activity in S9Cdr homolog
Based on analysis of S9Cdr structure, the main unique features of the enzyme that allow its carboxypeptidase activity are the cavity loop, conserved active site arginine residues, and aspartate/glutamate residues stabilizing the important arginine residues. Homologs of S9Cdr were searched using BLAST. Forty-two homologs were found to possess the important features mentioned above, including the S9C enzyme from Geobacillus stearothermophilus (S9Cgs; UniParc accession number UPI0006AC02FD, GenBankTM accession number WP_053532245). This enzyme was cloned, expressed, and purified. Although the enzyme was found to be active toward MLSAA peptide, it was unable to cleave MLSAA-NH2. The peptide cleavage profile of the enzyme confirmed that it is a carboxypeptidase (Fig. 8).
Figure 8.
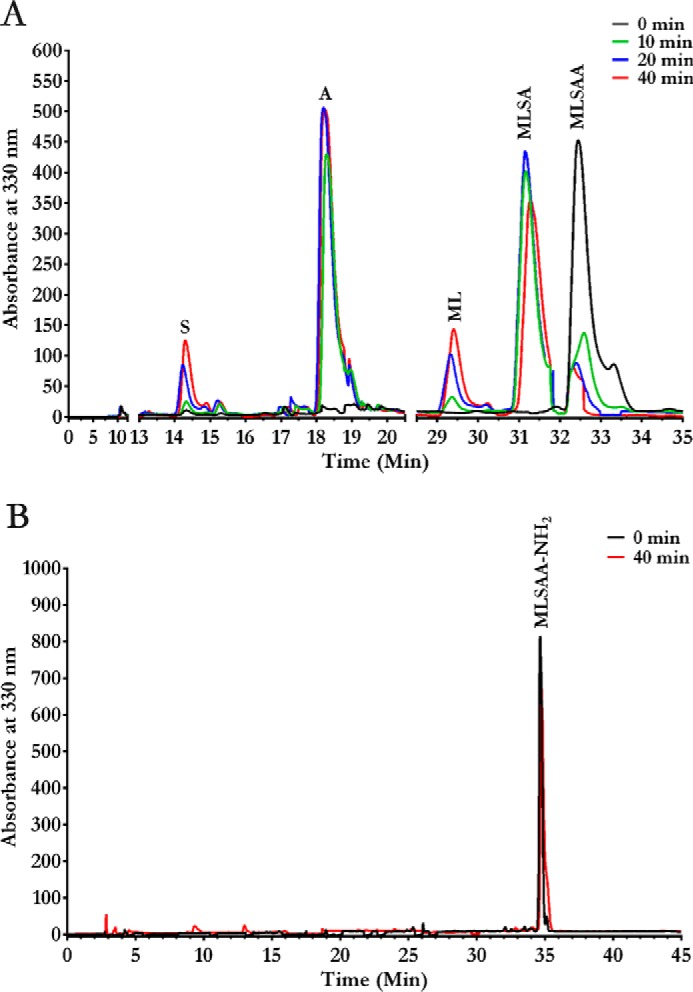
HPLC elution profiles showing carboxypeptidase activity of homologous protein S9Cgs. The enzyme was incubated with individual peptides, and the reaction products were monitored at different time intervals by subjecting them to a reverse-phase C18 column chromatography after derivatizing with o-phthalaldehyde reagent. The elution profiles of the hydrolyzed products for MLSAA (A) and MLSAA-NH2 (B) peptides are shown.
Discussion
Enzymes of the POP family are grouped into different subfamilies based on their sequence similarities and substrate specificities (Fig. 1) (9). Endopeptidase, dipeptidyl peptidase, tripeptidyl peptidase, and acylaminoacyl peptidase activities have been reported previously from the POP family; however, carboxypeptidase activity has never been assigned earlier to any peptidase of this family. Based on sequence similarity, the S9Cdr enzyme is annotated as AAP in the NCBI and UniProt databases. However, our enzymatic activity data clearly show that both acylaminoacyl peptidase and endopeptidase activities attributed to AAP are not observed for this protein. Moreover, our enzymatic assays and product elution profiles conclusively demonstrate that S9Cdr is a carboxypeptidase. The enzyme catalyzes sequential removal of C-terminal residues with free α-carboxylate (Fig. 3). The S9Cdr enzyme, however, is still a member of POP family as confirmed by sequence similarity and structural similarity. Furthermore, this enzyme lacks the ability to remove the N-terminal amino acid residue from peptides with a free α-amino group, which is a characteristic feature of the peptidases from the POP family (9). Thus, this is the first report of a carboxypeptidase from the POP or S9 family.
The S9Cdr enzyme achieves carboxypeptidase activity via structural adaptations illuminated by the crystal structures reported here. A cavity loop protrudes from the propeller domain and is placed near the binding site for the C terminus of the peptide substrate. The loop fills the void in the cavity beyond the P1′ position (Fig. 4B), thus crowding positions P2′ and onward, which are present in substrates of other POP enzymes but absent in carboxypeptidase substrates. This could help to channelize the substrate to the active site and reduce the nonspecific interactions in the cavity. The substrate-bound structure has uncovered two arginine residues (Arg-537 and Arg-633) that bind to free α-carboxylate of the peptide substrate and facilitate substrate binding (Fig. 6C). Mutagenesis studies confirm that these arginine residues are essential for the enzymatic activity (Fig. 2D). Thus, insertion of the cavity loop and strategic placement of arginine residues in the substrate-binding cavity enable appropriate positioning of the substrate for carboxypeptidase activity of S9Cdr.
The peptidases of the POP family have adopted sophisticated mechanisms to regulate access to the buried active site and permit smaller peptides into the active site cavity while excluding the longer or structured peptides of the cell to prevent their nontargeted digestion. In mammalian POPs, a flexible loop structure at the domain interface has been proposed to regulate substrate entry to the active site (20, 21). In prokaryotic POPs and OPBs, the structural and biochemical evidence clearly shows that smaller size peptides can enter through a transient opening formed by hinge-like movement of the two domains (22–24). In AAP enzymes, two different mechanisms for substrate screening have been proposed. Dimeric AAP (ApAAP) from Aeropyrum pernix shows dynamic opening and closing of the enzyme that are rather similar to observations in prokaryotic POPs and OPBs (25). In contrast, hexameric PhAAP shows the double-gated entry mechanism. In this mechanism, substrates can access the active site by double check-in: first by passing through the major opening of the oligomer and then through a smaller side opening of the monomer (26). The physiologically relevant mammalian AAP is a tetrameric enzyme and is expected to use a different mechanism for substrate screening. The structure of the mammalian AAP is not known despite two reports on its crystallization several years ago (33, 34). Until now there was no suitable tetrameric structure in the POP family that could act as a reference model for this enzyme. Because S9Cdr adopts a tetrameric structure and its monomer is structurally close to that of PhAAP, it could act as a structural template for the physiologically relevant tetrameric mammalian AAP.
Based on our structural and mutagenic data, we propose the following mechanism for substrate screening in S9Cdr (Fig. 9). The active site cavity can be accessed in two steps: first via the oligomeric pore of the tetramer and later through the side opening of the monomer. The oligomeric pore and the side opening of the monomer seem to be regulated by the flexible gating loops that consist of a large number of charged residues. Substrate binding may follow either an induced fit or a conformational selection model (35). In the case of the induced fit model, the substrate could diffuse into the inactive structure through the widened oligomeric pore facilitated by the disordering of the gating loops. The shape of the side opening is also modulated, which may further assist entry of substrate. In the induced fit model, binding of the substrate into the active site cavity would be coupled with large scale conformational changes, including ordering of the gating loops and assembly of the active site (Fig. 9B). It is likely that these two processes that render the binding of substrate into a productive encounter are linked because the loops responsible for assembly of the active site interact with the gating loops (Fig. 7C). The disordered gating loops in the inactive structure may also allow the C terminus of larger and structured peptides to access the active site cavity. However, in such a scenario, steric hindrance posed by large size of the polypeptide would disallow ordering of gating loops. Consequently, the active site will remain disassembled, thus rendering such unintended binding unproductive (Fig. 9C). Another mode of substrate binding would be conformational selection. In this possibility, the enzyme would exist alternately in active and inactive conformations. Small substrates could diffuse into the active structure via the side opening. However, direct access to the assembled active site would be disallowed for larger and structured peptides due to a smaller oligomeric pore and shielded side opening created by ordered gating loops (Fig. 9B). Hence, the nontargeted proteins of the cytoplasm would be prevented from digestion by restricting access to the active site cavity in the active state and by disassembly of the catalytic triad in the inactive state where access to the active site cavity is not restricted. This mechanism of substrate screening involving gating coupled with active site assembly/disassembly is entirely different from those proposed for other POP family peptidases.
Figure 9.

Substrate-screening mechanism in S9Cdr carboxypeptidase. A, surface representation of the tetramer of the active enzyme showing the gate of the enzyme formed by the oligomerization of the monomers. The gating loops are colored in magenta and light pink, and rest of the color scheme of subunits follows Fig. 5A. B, schematic of the active state of the enzyme wherein the gating loops are ordered and the active site is assembled. It is conducive for cleavage of smaller peptides but prevents the entry of larger peptides or proteins into the active site cavity. C, schematic of the inactive state of the enzyme in which the gating loops are disordered and the active site is disassembled. The structured peptides or proteins may enter through the oligomeric pore and their C′-end may have access to the active site cavity via the side opening of the monomer, but they cannot be hydrolyzed due to the disassembled active site.
The mechanism proposed here for S9Cdr is different from that of prokaryotic POP, OPB, and ApAAP as it does not involve any movement of domains of a monomer (23–25). The substrate screening in hexameric PhAAP enzyme is somewhat similar to that proposed here for S9Cdr with respect to the double-gated entry mechanism (26); however, PhAAP lacks the association of the gating mechanism with assembly/disassembly of the active site, and there is no evidence of an inactive state equivalent to that of S9Cdr in PhAAP. The loop equivalent to gating loop 1 and the residues equivalent to Arg-537 (catalytic triad–disrupting arginine) and Arg-633 are absent in PhAAP. The loop equivalent to gating loop 2 is found in POP and PhAAP enzymes (Fig. S5). This loop in mammalian POP has already been proposed to play an important role for access of substrate to the active site (20, 21). However, comparison of S9Cdr with PhAAP and other members of the POP family suggests that gating loop 1 is a unique feature of S9Cdr.
Our study provides significant structural details of this novel carboxypeptidase from the POP family. Based on the structural and mutagenesis analyses, we found that two arginine residues (Arg-537 and Arg-633) are important for carboxypeptidase activity. Furthermore, the arginine side chains are stabilized by nearby acidic residues. These conserved features were used to locate prospective serine carboxypeptidases among POP family peptidases. We found 42 prokaryotic homologs in the NCBI database that have greater than 30% sequence identities and possess the conserved arginine residues of the active site cavity (Fig. S3). We expect all 42 homologs to exhibit carboxypeptidase activity. To test our hypothesis, we cloned and purified one of the homologs from G. stearothermophilus (S9Cgs; 37% sequence identity with S9Cdr) and performed the enzymatic assays. This protein is annotated as a dipeptidyl aminopeptidase or acylaminoacyl peptidase in the NCBI database. In contrast to this annotation, our results confirmed that S9Cgs is a carboxypeptidase (Fig. 8). Thus, based on the structural features we have highlighted, new and existing putative peptidases of the POP family can be annotated more accurately. Our studies thus provide a new dimension to the enzymatic activities that could be considered while interpreting the biological role of the POP peptidases. Moreover, the substrate-screening mechanism proposed in this study has implications for peptidases in general. Proteins and peptides are so prevalent in the cellular environment that it is essential for peptidases to utilize stringent substrate specificity mechanisms to prevent nonspecific hydrolysis of closely related molecules. The way in which the gating of substrates is linked with assembly/disassembly of the active site in S9Cdr is, to the best of our knowledge, unprecedented in the literature for peptidases in general. However, because the dynamic aspects of gating and the structural details of alternate conformations of enzymes can be elusive, it is likely that future investigations may reveal such mechanisms in other proteases and peptidases as well.
Experimental procedures
Materials
Oligonucleotide primers were purchased from Eurofins Genomics (India). The QuikChange site-directed mutagenesis kit was purchased from Stratagene. Various custom-synthesized peptides were purchased from Sigma-Aldrich or Genic Bio (China).
Cloning and mutagenesis
S9Cdr gene was cloned in pST50Tr expression vector (36) using BamHI and BsrGI restriction enzyme sites and was reported in Are et al. (30). Point mutations in this recombinant plasmid were introduced using a PCR-based QuikChange site-directed mutagenesis kit (Stratagene) according to the manufacturer's protocol using plasmid DNA as a template and appropriate pairs of mutagenic primers. The S9Cgs gene was cloned into a modified pET21a vector using KpnI and HindIII restriction enzyme sites. All constructs were cloned in fusion with N-terminal His6 tag. All mutants were verified by plasmid DNA sequencing.
Protein preparation
Recombinant S9Cdr protein and its mutants were expressed in Escherichia coli BL21(DE3) pLysS cells. Recombinant S9Cgs protein was produced in E. coli Rosetta (DE3) pLysS cells. The proteins were purified according to protocols published previously (30). The proteins were purified using immobilized nickel-nitrilotriacetic acid–affinity and size-exclusion column chromatography. Although the His6 tags of S9Cdr and its mutants were cleaved using tobacco etch virus protease after purification, the tag for S9Cgs was not cleaved.
Enzyme activity
Activity of the enzyme was determined using various custom-synthesized peptides at pH 8.0 and 37 °C. To determine the substrate specificity toward various peptides, purified enzyme (15–80 ng) was incubated with substrates (2 mm) in an 80-μl reaction composed of 50 mm Tris-Cl, pH 8.0, at 37 °C for 10 min. The assay was designed so that it gives initial steady-state rate without substrate depletion. The activity of the enzyme was determined in terms of release of free amino acids by the enzyme, which was estimated using a modified ninhydrin assay that involves the use of Cd-ninhydrin reagent to detect the free α-amino group of liberated amino acids from peptides (37). The Cd-ninhydrin reagent (320 μl) was added to the reaction mixture at the end of the reaction followed by boiling in a 100 °C water bath for 4 min to develop the color. After cooling, the color intensity produced was measured at 504 nm. To determine specificities toward N-blocked para-nitroanilide (pNA) substrates, the enzyme was incubated with substrate at 37 °C for 10 min. Color intensity developed due to release of pNA was determined by measuring the absorbance at 410 nm. The specific activity was expressed as μmol of Gly eq. or pNA mg−1 min−1. Enzyme assays were performed in triplicates. The enzymatic parameters were determined for tripeptides. Kinetic constants (Km and kcat) were determined by linear regression (Michaelis–Menten) curve fitting using Graph Pad Prism (version 6.0).
Detection of cleavage sites in peptides
Cleavage patterns of substrates were monitored by reverse-phase HPLC chromatography using a C18 column after derivatizing the products with o-phthalaldehyde reagent. Initially, peptides with or without a free C terminus were incubated with S9Cdr (or S9Cgs) enzyme for proteolytic cleavage. Typically, 5 mm substrates were incubated with 5 μg of S9Cdr enzyme at 37 °C, and an aliquot of 40 μl from the reaction mixture was taken out at different time intervals. The reaction product was first treated with 60 μl of 1:2 diluted o-phthalaldehyde reagent (0.5% (w/v) o-phthalaldehyde prepared in a solution comprising 200 mm borate, pH 10.4, 80% methanol, 25 μm 3-mercaptopropionic acid) for 3 min to derivatize the products and thereafter loaded on a reverse-phase C18 column (ODS Hypersil, Thermo Scientific) equilibrated with 12.5 mm phosphate buffer, pH 7.2 (Eluent A). A Gradient of 50% acetonitrile in the same buffer (Eluent B) was used for elution of hydrolyzed products at a flow rate of 1 ml/min for 30 min. The elution profiles were monitored at 330 nm and compared with the elution profiles obtained for different standard amino acids and peptides.
Crystallization of S9Cdr WT and S514A mutant protein
Both S9Cdr WT and S514A mutant proteins were crystallized in 96-well U-bottom plates at 14 mg/ml and 21 °C using a microbatch under oil method (38). The initial crystallization condition for the inactive state of WT S9Cdr protein was reported previously (30). The crystals of WT S9Cdr protein in inactive state were obtained in 50 mm sodium acetate, pH 4.5, 200 mm NaCl, 10 mm MgCl2, 12% (w/v) PEG 3350. The crystals of the active state of WT S9Cdr were obtained from the protein incubated overnight with 2 mm phenylmethylsulfonyl fluoride. Initial crystal hits were obtained as fine needles, which were introduced as microseeds into drops pre-equilibrated with protein (39). The best diffraction quality crystals for active WT protein were obtained in 40 mm KH2PO4, 20% (v/v) glycerol, 16% (w/v) PEG 8000 and later soaked in 50 mm N-Ac-Phe for 1 h. The crystals of the active state conformation of S514A mutant protein were grown in 40 mm KH2PO4, 20% (v/v) glycerol, 20% (w/v) PEG 8000 by microseeding and further soaked in 10 mm Leu-Ala-Ser substrate for 1 h. The crystallization conditions for the other reported structures are summarized in Table S2. All the crystals were cryoprotected with Parabar 10312 oil and stored in liquid nitrogen prior to their diffraction.
Data collection, processing, and structure determination
The diffraction data sets were collected at the protein crystallography beamline (PX-BL21), Indus-2 synchrotron, Raja Ramanna Centre for Advanced Technology (RRCAT), India (40) from cryocooled crystals (100 K). Data were indexed and integrated using XDS (41) and subsequently scaled using AIMLESS (42). The phase problem was solved by molecular replacement using the Phaser suite (43) on CCP4. The molecular replacement search model was prepared from a single chain of PhAAP (PDB code 4HXE; sequence identity, 27%). Automated model building was performed using PHENIX Autobuild (44). The model was improved by multiple rounds of manual refitting using Coot (45) and refinement using both REFMAC5 (46) and phenix.refine (44). Substrate and solvent molecules were selected by examination of 2Fo − Fc and Fo − Fc maps contoured at 1σ and 3.5σ, respectively. The quality of the model was analyzed with MolProbity (47). The structural analysis and illustrations were performed using PyMOL (48) and the UCSF Chimera package (49). The statistics of data processing and refinement are summarized in Table 2 and Table S1.
Author contributions
P. Y., V. D. G., A. K., S. M. G., S. N. J., and R. D. M. data curation; P. Y., N. K. G., A. K., and S. N. J. investigation; P. Y., V. D. G., and R. D. M. writing-original draft; P. Y., V. D. G., A. K., S. M. G., S. N. J., and R. D. M. writing-review and editing; S. N. J. and R. D. M. conceptualization; S. N. J. and R. D. M. supervision.
Supplementary Material
Acknowledgments
We are grateful to Dr. S. M. Sharma for support during this project. We thank the staff at Indus-2 (Raja Ramanna Centre for Advanced Technology (RRCAT), India) for providing synchrotron beam time. We acknowledge the use of the PX-BL21 beamline and its associated biochemical facilities. We thank Shravan D. Kumbhare (project assistant) for initial work on S9Cgs protein.
The authors declare that they have no conflicts of interest with the contents of this article.
This article contains Figs. S1–S5 and Tables S1 and S2.
- POP
- prolyl oligopeptidase
- OPB
- oligopeptidase B
- AAP
- acylaminoacyl peptidase
- S9Cdr
- serine carboxypeptidase from D. radiodurans R1
- PhAAP
- acylaminoacyl peptidase from P. horikoshii
- ApAAP
- acylaminoacyl peptidase from A. pernix
- S9Cgs
- serine carboxypeptidase from G. stearothermophilus
- PDB
- Protein Data Bank
- pNA
- para-nitroanilide
- r.m.s.d.
- root mean square deviation.
References
- 1. Polgár L. (2002) The prolyl oligopeptidase family. Cell. Mol. Life Sci. 59, 349–362 10.1007/s00018-002-8427-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Szeltner Z., and Polgár L. (2008) Structure, function and biological relevance of prolyl oligopeptidase. Curr. Protein Pept. Sci. 9, 96–107 10.2174/138920308783565723 [DOI] [PubMed] [Google Scholar]
- 3. Hughes T. E., Mone M. D., Russell M. E., Weldon S. C., and Villhauer E. B. (1999) NVP-DPP728 (1-[[[2-[(5-cyanopyridin-2-yl)amino]ethyl]amino]acetyl]-2-cyano-(S)-pyrrolidine), a slow-binding inhibitor of dipeptidyl peptidase IV. Biochemistry 38, 11597–11603 10.1021/bi990852f [DOI] [PubMed] [Google Scholar]
- 4. Morty R. E., Troeberg L., Powers J. C., Ono S., Lonsdale-Eccles J. D., and Coetzer T. H. (2000) Characterisation of the antitrypanosomal activity of peptidyl alpha-aminoalkyl phosphonatediphenyl esters. Biochem. Pharmacol. 60, 1497–1504 10.1016/S0006-2952(00)00459-7 [DOI] [PubMed] [Google Scholar]
- 5. Yamin R., Zhao C., O'Connor P. B., McKee A. C., and Abraham C. R. (2009) Acyl peptide hydrolase degrades monomeric and oligomeric amyloid-β peptide. Mol. Neurodegener. 4, 33 10.1186/1750-1326-4-33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Santhoshkumar P., Xie L., Raju M., Reneker L., and Sharma K. K. (2014) Lens crystallin modifications and cataract in transgenic mice overexpressing acylpeptide hydrolase. J. Biol. Chem. 289, 9039–9052 10.1074/jbc.M113.510677 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Scaloni A., Jones W., Pospischil M., Sassa S., Schneewind O., Popowicz A. M., Bossa F., Graziano S. L., and Manning J. M. (1992) Deficiency of acylpeptide hydrolase in small-cell lung carcinoma cell lines. J. Lab. Clin. Med. 120, 546–552 [PubMed] [Google Scholar]
- 8. Zeng Z., Rulten S. L., Breslin C., Zlatanou A., Coulthard V., and Caldecott K. W. (2017) Acylpeptide hydrolase is a component of the cellular response to DNA Damage. DNA Repair. 58, 52–61 10.1016/j.dnarep.2017.08.008 [DOI] [PubMed] [Google Scholar]
- 9. Rawlings N. D., Waller M., Barrett A. J., and Bateman A. (2014) MEROPS: the database of proteolytic enzymes, their substrates and inhibitors. Nucleic Acids Res. 42, D503–D509 10.1093/nar/gkt953 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Wilk S. (1983) Prolyl endopeptidase. Life Sci. 33, 2149–2157 10.1016/0024-3205(83)90285-0 [DOI] [PubMed] [Google Scholar]
- 11. Polgár L. (1997) A potential processing enzyme in prokaryotes: oligopeptidase B, a new type of serine peptidase. Proteins. 28, 375–379 [DOI] [PubMed] [Google Scholar]
- 12. Kenny A. J., Booth A. G., George S. G., Ingram J., Kershaw D., Wood E. J., and Young A. R. (1976) Dipeptidyl peptidase IV, a kidney brush-border serine peptidase. Biochem. J. 157, 169–182 10.1042/bj1570169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Banbula A., Mak P., Bugno M., Silberring J., Dubin A., Nelson D., Travis J., and Potempa J. (1999) Prolyl tripeptidyl peptidase from Porphyromonas gingivalis: a novel enzyme with possible pathological implications for the development of periodontitis. J. Biol. Chem. 274, 9246–9252 10.1074/jbc.274.14.9246 [DOI] [PubMed] [Google Scholar]
- 14. Jones W. M., Manning L. R., and Manning J. M. (1986) Enzymic cleavage of the blocked amino terminal residues of peptides. Biochem. Biophys. Res. Commun. 139, 244–250 10.1016/S0006-291X(86)80105-X [DOI] [PubMed] [Google Scholar]
- 15. Kobayashi K., and Smith J. A. (1987) Acyl-peptide hydrolase from rat liver: characterization of enzyme reaction. J. Biol. Chem. 262, 11435–11445 [PubMed] [Google Scholar]
- 16. Fülöp V., Böcskei Z., and Polgár L. (1998) Prolyl oligopeptidase: an unusual beta-propeller domain regulates proteolysis. Cell 94, 161–170 10.1016/S0092-8674(00)81416-6 [DOI] [PubMed] [Google Scholar]
- 17. Rasmussen H. B., Branner S., Wiberg F. C., and Wagtmann N. (2003) Crystal structure of human dipeptidyl peptidase IV/CD26 in complex with a substrate analog. Nat. Struct. Biol. 10, 19–25 10.1038/nsb882 [DOI] [PubMed] [Google Scholar]
- 18. Bartlam M., Wang G., Yang H., Gao R., Zhao X., Xie G., Cao S., Feng Y., and Rao Z. (2004) Crystal structure of an acylpeptide hydrolase/esterase from Aeropyrum pernix K1. Structure 12, 1481–1488 10.1016/j.str.2004.05.019 [DOI] [PubMed] [Google Scholar]
- 19. McLuskey K., Paterson N. G., Bland N. D., Isaacs N. W., and Mottram J. C. (2010) Crystal structure of Leishmania major oligopeptidase B gives insight into the enzymatic properties of a trypanosomatid virulence factor. J. Biol. Chem. 285, 39249–39259 10.1074/jbc.M110.156679 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kaszuba K., Róg T., Danne R., Canning P., Fülöp V., Juhász T., Szeltner Z., St Pierre J. F., García-Horsman A., Männistö P. T., Karttunen M., Hokkanen J., and Bunker A. (2012) Molecular dynamics, crystallography and mutagenesis studies on the substrate gating mechanism of prolyl oligopeptidase. Biochimie 94, 1398–1411 10.1016/j.biochi.2012.03.012 [DOI] [PubMed] [Google Scholar]
- 21. Szeltner Z., Juhász T., Szamosi I., Rea D., Fülöp V., Módos K., Juliano L., and Polgár L. (2013) The loops facing the active site of prolyl oligopeptidase are crucial components in substrate gating and specificity. Biochim. Biophys. Acta 1834, 98–111 10.1016/j.bbapap.2012.08.012 [DOI] [PubMed] [Google Scholar]
- 22. Shan L., Mathews I. I., and Khosla C. (2005) Structural and mechanistic analysis of two prolyl endopeptidases: role of interdomain dynamics in catalysis and specificity. Proc. Natl. Acad. Sci. U.S.A. 102, 3599–3604 10.1073/pnas.0408286102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Li M., Chen C., Davies D. R., and Chiu T. K. (2010) Induced-fit mechanism for prolyl endopeptidase. J. Biol. Chem. 285, 21487–21495 10.1074/jbc.M109.092692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Canning P., Rea D., Morty R. E., and Fülöp V. (2013) Crystal structures of Trypanosoma brucei oligopeptidase B broaden the paradigm of catalytic regulation in prolyl oligopeptidase family enzymes. PLoS One 8, e79349 10.1371/journal.pone.0079349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Harmat V., Domokos K., Menyhárd D. K., Palló A., Szeltner Z., Szamosi I., Beke-Somfai T., Náray-Szabó G., and Polgár L. (2011) Structure and catalysis of acylaminoacyl peptidase: closed and open subunits of a dimer oligopeptidase. J. Biol. Chem. 286, 1987–1998 10.1074/jbc.M110.169862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Menyhárd D. K., Kiss-Szemán A., Tichy-Rács É, Hornung B., Rádi K., Szeltner Z., Domokos K., Szamosi I., Náray-Szabó G., Polgár L., and Harmat V. (2013) A self-compartmentalizing hexamer serine protease from Pyrococcus horikoshii: substrate selection achieved through multimerization. J. Biol. Chem. 288, 17884–17894 10.1074/jbc.M113.451534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Bar-Even A., Noor E., Savir Y., Liebermeister W., Davidi D., Tawfik D. S., and Milo R. (2011) The moderately efficient enzyme: evolutionary and physicochemical trends shaping enzyme parameters. Biochemistry 50, 4402–4410 10.1021/bi2002289 [DOI] [PubMed] [Google Scholar]
- 28. Liebschner D., Afonine P. V., Moriarty N. W., Poon B. K., Sobolev O. V., Terwilliger T. C., and Adams P. D. (2017) Polder maps: improving OMIT maps by excluding bulk solvent. Acta Crystallogr. D Struct. Biol. 73, 148–157 10.1107/S2059798316018210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Voss N. R., and Gerstein M. (2010) 3V: cavity, channel and cleft volume calculator and extractor. Nucleic Acids Res. 38, W555–W562 10.1093/nar/gkq395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Are V. N., Ghosh B., Kumar A., Yadav P., Bhatnagar D., Jamdar S. N., and Makde R. D. (2014) Expression, purification, crystallization and preliminary X-ray diffraction analysis of acylpeptide hydrolase from Deinococcus radiodurans. Acta Crystallogr. F Struct. Biol. Commun. 70, 1292–1295 10.1107/S2053230X14017944 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Krissinel E., and Henrick K. (2007) Inference of macromolecular assemblies from crystalline state. J. Mol. Biol. 372, 774–797 10.1016/j.jmb.2007.05.022 [DOI] [PubMed] [Google Scholar]
- 32. Szeltner Z., Rea D., Renner V., Juliano L., Fülop V., and Polgár L. (2003) Electrostatic environment at the active site of prolyl oligopeptidase is highly influential during substrate binding. J. Biol. Chem. 278, 48786–48793 10.1074/jbc.M309555200 [DOI] [PubMed] [Google Scholar]
- 33. Feese M., Scaloni A., Jones W. M., Manning J. M., and Remington S. J. (1993) Crystallization and preliminary X-ray studies of human erythrocyte acylpeptide hydrolase. J. Mol. Biol. 233, 546–549 10.1006/jmbi.1993.1531 [DOI] [PubMed] [Google Scholar]
- 34. Wright H., Kiss A. L., Szeltner Z., Polgár L., and Fülöp V. (2005) Crystallization and preliminary crystallographic analysis of porcine acylaminoacyl peptidase. Acta Crystallogr. Sect F. Struct. Biol. Cryst. Commun. 61, 942–944 10.1107/S1744309105029222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Vogt A. D., and Di Cera E. (2012) Conformational selection or induced-fit? A critical appraisal of the kinetic mechanism. Biochemistry 51, 5894–5902 10.1021/bi3006913 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Tan S., Kern R. C., and Selleck W. (2005) The pST44 polycistronic expression system for producing protein complexes in Escherichia coli. Protein Expr. Purif. 40, 385–395 10.1016/j.pep.2004.12.002 [DOI] [PubMed] [Google Scholar]
- 37. Doi E., Shibata D., and Matoba T. (1981) Modified colorimetric ninhydrin methods for peptidase assay. Anal. Biochem. 118, 173–184 10.1016/0003-2697(81)90175-5 [DOI] [PubMed] [Google Scholar]
- 38. Chayen N. E., Stewart P. D. S., and Blow D. M. (1992) Microbatch crystallization under oil a new technique allowing many small-volume crystallization trials. J. Cryst. Growth 122, 176–180 10.1016/0022-0248(92)90241-A [DOI] [Google Scholar]
- 39. D'Arcy A., Villard F., and Marsh M. (2007) An automated microseed matrix-screening method for protein crystallization. Acta Crystallogr. D Biol. Crystallogr. 63, 550–554 10.1107/S0907444907007652 [DOI] [PubMed] [Google Scholar]
- 40. Kumar A., Ghosh B., Poswal H. K., Pandey K. K., Jagannath, Hosur M. V., Dwivedi A., Makde R. D., and Sharma S. M. (2016) Protein crystallography beamline (PX-BL21) at Indus-2 synchrotron. J. Synchrotron Radiat. 23, 629–634 10.1107/S160057751600076X [DOI] [PubMed] [Google Scholar]
- 41. Kabsch W. (2010) XDS. Acta Crystallogr. D Biol. Crystallogr. 66, 125–132 10.1107/S0907444909047337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Winn M. D., Ballard C. C., Cowtan K. D., Dodson E. J., Emsley P., Evans P. R., Keegan R. M., Krissinel E. B., Leslie A. G., McCoy A., McNicholas S. J., Murshudov G. N., Pannu N. S., Potterton E. A., Powell H. R., et al. (2011) Overview of the CCP4 suite and current developments. Acta Crystallogr. D Biol. Crystallogr. 67, 235–242 10.1107/S0907444910045749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. McCoy A. J., Grosse-Kunstleve R. W., Adams P. D., Winn M. D., Storoni L. C., and Read R. J. (2007) Phaser crystallographic software. J. Appl. Crystallogr. 40, 658–674 10.1107/S0021889807021206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Adams P. D., Afonine P. V., Bunkóczi G., Chen V. B., Davis I. W., Echols N., Headd J. J., Hung L. W., Kapral G. J., Grosse-Kunstleve R. W., McCoy A. J., Moriarty N. W., Oeffner R., Read R. J., Richardson D. C., et al. (2010) PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213–221 10.1107/S0907444909052925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Emsley P., and Cowtan K. (2004) Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60, 2126–2132 10.1107/S0907444904019158 [DOI] [PubMed] [Google Scholar]
- 46. Murshudov G. N., Skubák P., Lebedev A. A., Pannu N. S., Steiner R. A., Nicholls R. A., Winn M. D., Long F., and Vagin A. A. (2011) REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr. D Biol. Crystallogr. 67, 355–367 10.1107/S0907444911001314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Chen V. B., Arendall W. B. 3rd, Headd J. J., Keedy D. A., Immormino R. M., Kapral G. J., Murray L. W., Richardson J. S., and Richardson D. C. (2010) MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr. D Biol. Crystallogr. 66, 12–21 10.1107/S0907444909042073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. DeLano W. L. (2012) The PyMOL Molecular Graphics System, version 1.5.0.3, Schroedinger, LLC, New York [Google Scholar]
- 49. Pettersen E. F., Goddard T. D., Huang C. C., Couch G. S., Greenblatt D. M., Meng E. C., and Ferrin T. E. (2004) UCSF Chimera—a visualization system for exploratory research and analysis. J. Comput. Chem. 25, 1605–1612 10.1002/jcc.20084 [DOI] [PubMed] [Google Scholar]
- 50. Schechter I., and Berger A. (1967) On the size of the active site in proteases. I. Papain. Biochem. Biophys. Res. Commun. 27, 157–162 10.1016/S0006-291X(67)80055-X [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.