

Abstract
Motivation
Kinases play a significant role in diverse disease signaling pathways and understanding kinase inhibitor selectivity, the tendency of drugs to bind to off-targets, remains a top priority for kinase inhibitor design and clinical safety assessment. Traditional approaches for kinase selectivity analysis using biochemical activity and binding assays are useful but can be costly and are often limited by the kinases that are available. On the other hand, current computational kinase selectivity prediction methods are computational intensive and can rarely achieve sufficient accuracy for large-scale kinome wide inhibitor selectivity profiling.
Results
Here, we present a KinomeFEATURE database for kinase binding site similarity search by comparing protein microenvironments characterized using diverse physiochemical descriptors. Initial selectivity prediction of 15 known kinase inhibitors achieved an >90% accuracy and demonstrated improved performance in comparison to commonly used kinase inhibitor selectivity prediction methods. Additional kinase ATP binding site similarity assessment (120 binding sites) identified 55 kinases with significant promiscuity and revealed unexpected inhibitor cross-activities between PKR and FGFR2 kinases. Kinome-wide selectivity profiling of 11 kinase drug candidates predicted novel as well as experimentally validated off-targets and suggested structural mechanisms of kinase cross-activities. Our study demonstrated potential utilities of our approach for large-scale kinase inhibitor selectivity profiling that could contribute to kinase drug development and safety assessment.
Availability and implementation
The KinomeFEATURE database and the associated scripts for performing kinase pocket similarity search can be downloaded from the Stanford SimTK website (https://simtk.org/projects/kdb).
Supplementary information
Supplementary data are available at Bioinformatics online.
1 Introduction
The human kinome consists of ∼500 protein kinases that regulate diverse cellular process including metabolism, apoptosis, immune response, cell growth and proliferation (Braconi Quintaje and Orchard, 2008; Manning et al., 2002). Based on cellular localization and the residue(s) of phosphorylation, human protein kinases can be classified as receptor or non-receptor kinases and tyrosine, serine/threonine, or dual kinases, respectively. Many kinases have been implicated in a wide variety of diseases including cancers, inflammatory diseases and metabolic syndrome and are potential therapeutic targets (Momcilovic et al., 2017; Patterson et al., 2014). Consequently, designing selective kinase inhibitors that target specific cellular pathways, known as ‘targeted therapy,’ has been a major focus area in the pharmaceutical industry. Following the discovery of the Abelson murine leukemia viral oncogene homolog (ABL)-targeting agent imatinib (Gleevec) as a highly successful treatment for chronic myeloid leukemia (CML), numerous other kinase targets have been actively pursued for cancer therapy. Several well-known druggable kinases include stem-cell factor receptor (KIT) for gastrointestinal tract stromal tumors (GIST), proto-oncogene tyrosine-kinase receptor (RET) for medullary thyroid carcinoma (MTC) and fetal liver tyrosine-protein kinase receptor 3 (FLT3) for acute lymphocytic leukemia (ALL) (Akeno-Stuart et al., 2007; Druker et al., 2001; Helguera et al., 2006; Rosnet et al., 1991; Verweij et al., 2004). Since the majority of kinase inhibitors bind within the ATP binding region, unintended inhibitor off-target binding can occur due to high structural homology across kinase ATP binding pockets, resulting in either favorable interactions or adverse events (Hanks et al., 1988). For example, although imatinib was originally developed as a selective ABL inhibitor, the discovery of its unexpected activity against KIT has led to the approval of imatinib for the treatment of GIST (Demetri et al., 2002). Similarly, multi-targeted tyrosine kinase inhibitors like sunitinib, a potent inhibitor of VEGFR1, VEGFR2, FLT3, KIT, PDGFRα and PDGFRβ could counter drug resistance by preventing ‘signal rewiring’ in cancer treatment. However, unintentional off-target drug binding have already proven to contribute to severe side-effects and dose-limiting toxicities from numerous clinical and preclinical studies (Tabernero, 2007). Therefore, an accurate method for comprehensive off-target profiling of kinase drug candidates is essential for kinase inhibitor design and assessment of drug safety and efficacy.
Multiple approaches have been developed to profile the kinase selectivity of small molecules. Experimental methods include biochemical activity and binding assays that measure the effects of kinase inhibitors in large kinase panels (Brandt et al., 2009; Fedorov et al., 2007; Karaman et al., 2008). However, these biochemical methods are often costly, time consuming and limited by the kinases that are available (Sheridan et al., 2009). Furthermore, variations among the assay platforms and the activity metrics assessed can potentially lead to apparent differences in the selectivity measured for the same molecule (Anastassiadis et al., 2011; Fabian et al., 2005; Fedorov et al., 2007). To this end, computational kinase selectivity profiling methods could serve as efficient alternatives for predicting kinase inhibitor selectivity across a larger kinome space. Although kinase sequence similarity searching is a widely used method to predict kinase inhibitor off-target bindings, sequence homology alone cannot fully capture inhibitor selectivity (Fabian et al., 2005; Sheridan et al., 2009). For example, a single residue difference in the binding pocket among the homologous p38 kinase isoforms was enough to define discrete selectivity toward diverse kinase inhibitors (Caffrey et al., 2008). Other computational methods for off-target predictions include chemical similarity inference, inverse docking of inhibitor to multiple kinase structures, or binding site similarity comparison (Caffrey et al., 2008; Kinnings and Jackson, 2009; Kuhn et al., 2006; Sciabola et al., 2008; Subramanian and Sud, 2010; Zahler et al., 2007). Recently, machine learning approaches such as kernel regression have also been developed for kinase activity profiling where a computational model was trained based on descriptors of protein sequence and chemical structures to predict bioactivities of new drugs (Cichonska et al., 2017). While useful, many of these approaches did not fully utilized 3D structure information of the proteins and their prediction outcomes are often dependent on the quality and availability of the structure-activity data or require intensive computation of binding scores and structural superpositions. Furthermore, their performances have not been fully evaluated for kinome-wide prediction (Zahler et al., 2007).
Here, we report a new computational approach for large-scale kinase inhibitor selectivity profiling based on structural knowledge of the ligand binding sites (Fig. 1a) (Zhou et al., 2015). To this end, we have constructed a kinase database called ‘KinomeFEATURE,’ which consists of ∼2850 kinase structures from 189 unique human kinases for kinase binding site similarity searching. The kinase binding site similarities were evaluated by comparing protein microenvironments, which were comprised of diverse physiochemical descriptors in a 6-radial shells at binding residues of the co-crystal ligand (Fig. 1a and Section 2) (Liu and Altman, 2011).The degree of pocket similarity between two kinases was determined using a PocketFEATURE score (PFS) based on the presence or absence of shared protein microenvironments (Fig. 1a and Section 2). For a given kinase inhibitor, its binding pocket was compared to kinase pockets in the KinomeFEATURE database and kinase structures with the best (most negative) PFS were identified (Fig. 1a). In contrast to existing approaches, our binding site similarity is evaluated by comparing microenvironments between two pockets independent of their distances. Therefore, our prediction does not necessitate a strong geometric requirement and is highly robust against protein conformational changes.
Fig. 1.
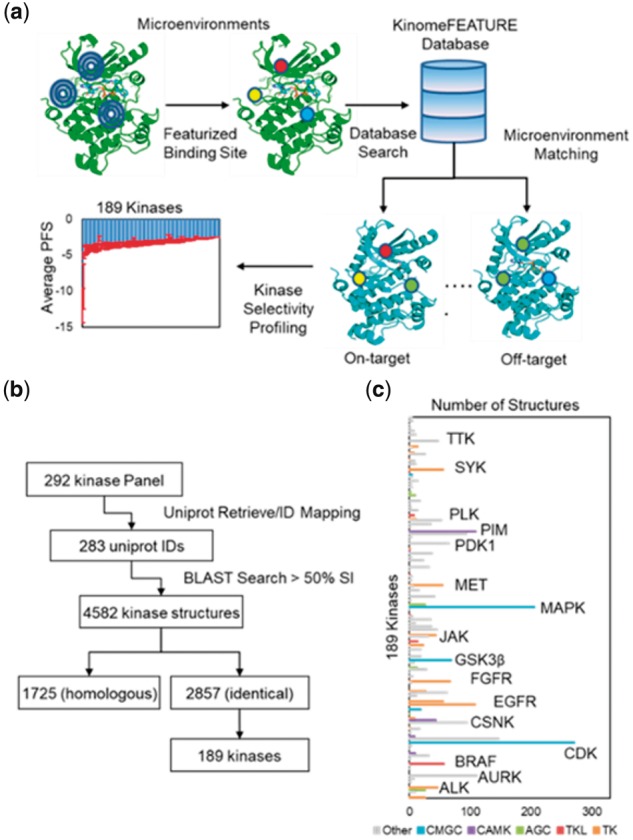
KinomeFEATURE database creation and characterization. (a) Workflow for kinase selectivity profiling using the KinomeFEATURE database. For each kinase pocket in the database, residues within 6 Å of the bound ligand were characterized using several physiochemical descriptors in a 6-concentric radial shell. The degree of binding site microenvironments similarity between two given kinases was evaluated using the PocketFEATURE score (PFS) based on the presence or absence of shared protein microenvironments and the kinase structures with the best (most negative) PFS were identified from multiple kinase structures for a given kinase. (b) Workflow for KinomeFEATURE database creation. 292 kinase genes represented in the SelectScreen® kinase selectivity panel were converted to 283 UniProt ID by UniProt ID mapping. Protein sequence similarity of the PDB identified 4582 protein structures sharing >50% sequence identity. Further UniProt ID matching identified 1725 homologous structures and 2857 identical structures. The 2857 identical structures represented by 189 kinase genes were selected to form the KinomeFEATURE database. (c) Profiling of the KinomeFEATURE database showed that the 2857 kinase drug pockets were distributed among 5 existing kinase categories including tyrosine kinases (TK) (556 structures), tyrosine kinase-like (TKL) kinases (84 structures), protein kinase A, G and C (AGC) (79 structures), calcium/calmodulin-dependent kinases (CAMK) (173 structures), cyclin-dependent kinases (CDK), mitogen-activated protein kinases, glycogen synthase kinases and CDK-like kinases (CMGC) (571 kinases) and others (1394 kinases). Kinases with the most abundant structures are primarily in the TK and CAMK categories including CDK, EGFR and PIM, which are common oncology targets of many existing kinase inhibitors
We tested this approach by profiling the kinase selectivity of 15 known kinase inhibitors and achieved >90% accuracy when comparing our prediction with the biochemical assay data. Additional kinase ATP binding site similarity assessment (120 binding sites) identified 55 kinases with significant promiscuity and revealed unexpected inhibitor cross-activities between PKR and FGFR2 kinases. Kinome-wide selectivity profiling of 11 kinase drug candidates predicted novel as well as experimentally validated off-targets and suggested structural mechanisms of kinase cross-activities. The study demonstrated potential utilities of our approach for accurate and robust kinase inhibitor activity prediction that could contribute to further kinase drug development and safety assessment.
2 Materials and methods
2.1 Compounds
Fifteen kinase known inhibitors (compound 1–15) for validation study were retrieved from the PDB as previously reported. C16 (compound 16), LY2874455 (compound 17) and 11 kinase drug candidates (compounds 18–28) were purchased or synthesized by Genentech, Inc., at greater than 95% purity. For additional compound information, please see Supplementary Tables S3 and S7.
2.2 In vitro kinase activity and binding assays
Kinase inhibitor potency and selectivity was assessed in a panel of 156 recombinant human kinase activity and binding assays, including cytoplasmic and receptor tyrosine kinases, serine/threonine kinases and lipid kinases (SelectScreen® Kinase Profiling Services, Thermo Fisher Scientific, Madison, WI). The kinase activity assays measure peptide phosphorylation (Z’-LYTE®) or ADP production (Adapta®) while the kinase binding assays monitor displacement of ATP site-binding probes (LanthaScreen®). The ATP concentrations used in the activity assays were within 2-fold of the experimentally determined apparent Michaelis constant (Kmapp) value for each kinase while the competitive binding tracer concentrations used in the binding assays were within 3-fold of the experimentally determined dissociation constant (Kd) values. Inhibitors were tested at 0.1 and 1 µM in duplicate against each kinase and the mean % inhibition values are reported. For selected kinases, 10-point inhibitor titrations were carried out using the same kinase assays as used in the single point tests in order to determine the inhibitor concentration providing 50% inhibition (IC50). Details regarding the kinase proteins used and the assay protocols are available online (thermofisher.com).
2.3 Statistical analysis
IC50 values were determined by plotting the % of control activity or binding data (mean of duplicate measurements) against the log of the inhibitor concentration and fitting the data by non-linear regression to the variable slope 4-parameter sigmoidal inhibition model using GraphPad Prism version 5.0f for Mac OS X (GraphPad Software, San Diego, CA USA, www.graphpad.com). Receiver operating characteristic (ROC) curves and the related area under the curve (AUC) for the kinase-panel selectivity analysis were obtained using the ROC curve node in the KNIME program (version 3.3.1) (Beisken et al., 2013). AUC was used to estimate the accuracy in binding class prediction for both sequence similarity and PFS. The AUC values were calculated for the entire set as well as for all the individual 17 targets with at least one active kinase inhibitor using the ‘ROC curve’ node in KNIME. To estimate the error rates for the AUC values of the two virtual screening methods, an independent group t-test was performed by evaluating AUC values for each 17 targets to determine a significant P-value. The statistical analysis was performed using the ‘independent group t-test’ node in KNIME.
2.4 Software
The heatmap analysis was performed using R statistical package (version 3.2.3). The network analysis was performed using Cytoscape software (version 3.4.0). The structural alignment was performed using the FeatureViz web-based visualization program (https://simtk.org/projects/feature-viz/).
3 Results
3.1 Creation and characterization of KinomeFEATURE database
To create the KinomeFEATURE database for kinase binding pocket similarity comparison, we performed a BLAST sequence similarity search of the commercial available kinases in the SelectScreen® kinase selectivity panel (ThermoFisher) against the protein data bank (PDB) and identified 2857 kinase structures represented by 189 unique human kinases (Fig. 1b and Supplementary Text S1) (Berman et al., 2003; Braconi Quintaje and Orchard, 2008). Inspection of the KinomeFEATURE database indicated that the 2857 binding pockets were distributed among 5 existing protein kinase categories including tyrosine kinases (TK) (556 structures), tyrosine kinase-like (TKL) kinases (84 structures), protein kinase A, G and C (AGC) (79 structures), calcium/calmodulin-dependent kinases (CAMK) (173 structures), cyclin-dependent kinases, mitogen-activated protein kinases, glycogen synthase kinases, CDK-like kinases (CMGC) (571 structures) and others (1394 structures) (Fig. 1c and Supplementary Table S1). As expected, kinases with the most abundant structures are primarily in the CMGC, TK and CAMK groups including CDK, MAPK, EGFR and PIM, which are common therapeutic targets of many drugs in preclinical and clinical development for cancer treatments (Arora and Scholar, 2005; Cicenas and Valius, 2011; Swords et al., 2011).
For each kinase pocket in the database, residues within 6 Å of the bound ligand were identified and subsequently characterized using diverse physiochemical descriptors in 6-concentric radial shells using the PocketFEATURE program (Fig. 1a and Section 2) (Liu and Altman, 2011). To compare two kinase binding sites, residues within 6 Å of the bound ligand in the kinase structure were identified to define a drug binding pocket (Liu and Altman, 2011). For each residue in the binding site, the geometric center of the residues was first determined based on the location of the heavy atom. The FEATURE program was used to compute the residue microenvironments in 6-concentric radial shells where each shell contains 180 physiochemical descriptors (Zhou et al., 2015). To evaluate the similarity between two microenvironments, a Tanimoto-like score was computed to identify the shared bits between two feature vectors of 1280 length. To evaluate the pocket similarity between two sites, the microenvironments were sequentially compared in a combinatorial fashion to maximize the total matching scores. A PocketFEATURE score (PFS) was calculated by normalizing the summed Tanimoto scores against a random background of all permissible microenvironment pairs.
We validated our approach by predicting kinase off-target bindings of 15 known kinase inhibitors, which encompass diverse kinase drug classes. These molecules have been previously tested against 280 unique kinases for their inhibition of kinase activity or binding affinity, and their co-crystal structures are available in the PDB (Karaman et al., 2008).The selected compounds include 2 ABL inhibitors: dasatinib (compound 1) and imatinib (compound 2); 2 multi-target inhibitors: sorafenib (compound 3) and staurosporine (compound 4); 3 P38α inhibitors: VX-745 (compound 5), SB203580 (compound 6) and BIRB-796 (compound 7); 3 EGFR inhibitors: lapatinib (compound 8), gefitinib (compound 9) and erlotinib (compound 10); 2 CDK inhibitors: flavopiridol (compound 11) and roscovitine (compound 12); 1 PRKß inhibitor: LY-333531 (compound 13); 1 AurA inhibitor: VX-680 (compound 14) and 1 KIT inhibitor: sunitinib (compound 15). To identify the optimal score cutoff, we analyzed the PFS of 8 selected kinase inhibitors: dasatinib, imatinib, VX-745, SB203580, lapatinib, gefitinib, flavopiridol and sunitinib at three binding affinity (Kd) thresholds: 100 nM, 1 µM and 10 µM, as measured in the biochemical assays (Fig. 2a) (Karaman et al., 2008). The PFS distribution showed that the KinomeFEATURE database search achieved the highest sensitivity for differentiating primary targets from off-targets at 100 nM potency for 8 selected molecules using the optimal PFS score cutoff (PFS < −5) (Fig. 2a). The observation was supported by evaluating the receiver operating characteristics (ROC) curves where the area-under-curve (AUC) at three concentration thresholds were all greater than 0.7 (AUC100nM = 0.78, AUC1µM = 0.72 and AUC10µM = 0.7) (Fig. 2b). Retrospective performance comparison for the 15 kinase inhibitors showed that our method improved kinase selectivity prediction over the binding site signature (BSS) method and yielded an average accuracy and specificity of 91 and 96% using the optimal PFS score cutoff (Fig. 2c, e and Supplementary Text S3) (Subramanian and Sud, 2010). Notably, imatinib achieved the highest prediction accuracy and specificity of ∼97% potentially due to the robustness gained from evaluating multiple kinase-inhibitor co-crystal complexes. Linear regression analysis of imatinib kinase off-target profile established a significant correlation between the PFS and binding affinity (logKd) (R2 = 0.81) and the predicted binding affinity were consistent with the observed binding data as visualized by the kinase phylogenetic tree (Fig. 2d and e). With the exception of staurosporine, each kinase inhibitor has an R2 value > 0.5 with an average R2 value of 0.63 (Fig. 2c and Supplementary Fig. S2).
Fig. 2.
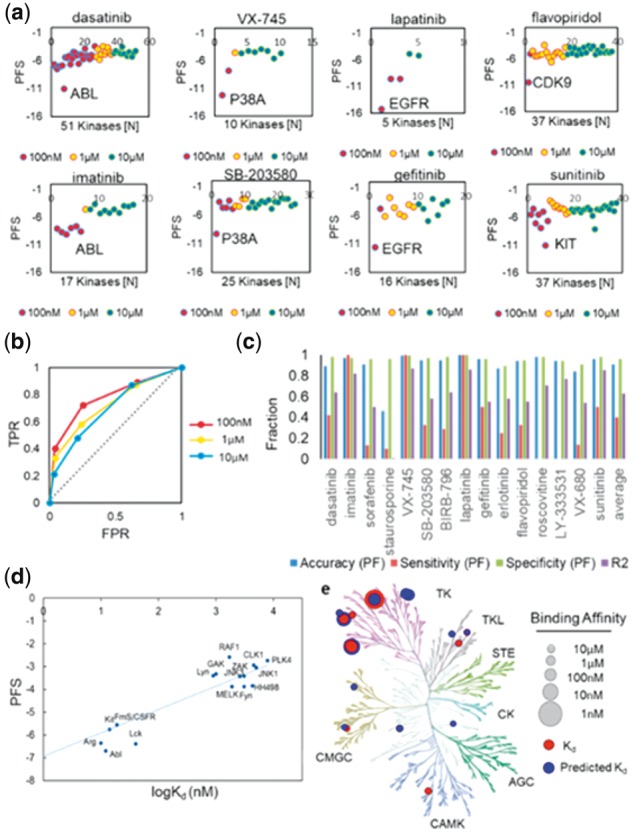
Computational off-target profiling of known kinase inhibitors. (a) The PocketFEATURE score distribution of 6 selected kinase inhibitors: lapatinib, sunitinib, flavopiridol, VX-745, imatinib and gefitinib showed that the KinomeFEATURE database can identify primary targets and other off-targets with high selectivity, particularly for kinase-ligand interactions with Kd < 100 nM. Note that the x-axis indicates the rank order of the kinases based on the PFS. Kianses that mapped back to the ligand with negative PFS are shown. (b) Performance assessment for selectivity profiling of 15 known kinase inhibitors using the PocketFEATURE algorithm. The area-under-curve (AUC) values from the receiver operating characteristics (ROC) curves evaluated at three concentration thresholds were greater than 0.7 (AUC100nM = 0.78, AUC1µM = 0.72, AUC10µM = 0.7). (c) The prediction accuracy, sensitivity and specificity for 15 known kinase dugs were assessed using the optimal scoring cutoff (PFS = −5) (see Supplementary Text S3). The KinomeFEATURE database search achieved an average of 90% accuracy, 96% specificity and 40% sensitivity at the 100-nM potency threshold. (d) Validation of predicted off-targets of imatinib. Linear regression analysis of the predicted off-targets of imatinib showed a high correlation (R2 > 0.81) between the predicted PFS, and binding affinity (logKd) and was used to construct a linear model to predicted kinase binding affinities. (e) Comparison of predicted (color: blue) and experimental validated (color: red) off-target binding affinities visualized by the kinase phylogenetic tree. Note that the size of the node is proportional to the binding affinity (and inversely proportional to the Kd)
3.2 Computational estimation of kinase promiscuity
Since most kinase inhibitors target the ATP binding site through competitive inhibition, site similarity analyses of the kinase ATP binding pocket will offer critical information on the kinase promiscuity in the absence of bound ligands. To estimate the ligand selectivity of druggable kinases, we profiled the off-target binding of 120 kinase ATP binding sites retrieved from the KinomeFEATURE database and compared their pocket similarity based on the PFS. Network clustering of kinase ATP binding sites with a PFS threshold < −5 identified 55 highly connected nodes with potential kinase cross-activities. The identified kinases included common drug targets for cancer therapy like EGFR, RET, KIT and others (Fig. 3a). Interestingly, promiscuity estimation based the node degree e.g. number of neighbors connected to each node, showed that kinases such as GS3K3β, FGFR2 and TYK2 were highly promiscuous and their binding site features were similar to 40–60 other kinase ATP pockets (Fig. 3b). In comparison, KIT, PLK1, AurA and PIM1 kinase pockets appear to be more selective toward ligand binding based on our assessment (Fig. 3b). To further substantiate these predictions, we analyzed the predicted PFS values of 55 selected kinases using a pairwise similarity map with an optimal score cutoff (PFS < −5) and compared to that by protein sequence similarity search using a sequence identity (SI) threshold commonly used for predicting protein structure homology (SI > 35%)(Fig. 3c, Supplementary Fig. S3 and Supplementary Tables S4 and S5) (Forrest et al., 2006). To validate the predicted off-targets using either approaches, we identified 17 kinases that have at least one specific inhibitor tested in a previous kinase assay panel and showed that both similarity score cutoffs achieved the optimal accuracy, specificity and sensitivity evaluated by multiple threshold values (Supplementary Fig. S4 and Supplementary Table S6) (Davis et al., 2011). The ROC curves indicated that PFS achieved a higher AUC values than SI by either including primary targets (AUCPFS = 0.78 versus AUCSI = 0.72) or excluding primary targets (AUCPFS = 0.74 versus AUCSI = 0.66) (Fig. 3c, Supplementary Fig. S5a and b). The average AUC values for each 17 targets is 0.81 (STD = 0.11) for PFS and 0.72 (STD = 0.13) for SI respectively. A Statistical group t-test showed that the difference between the two methods is statistical significant (P-value = 0.04) (Section 2). Notably, the removal of primary targets equally affected the performance using either approach (ΔAUC < 0.05), suggesting that the improvement in prediction by PFS was not attributed to the on-target predictions.
Fig. 3.
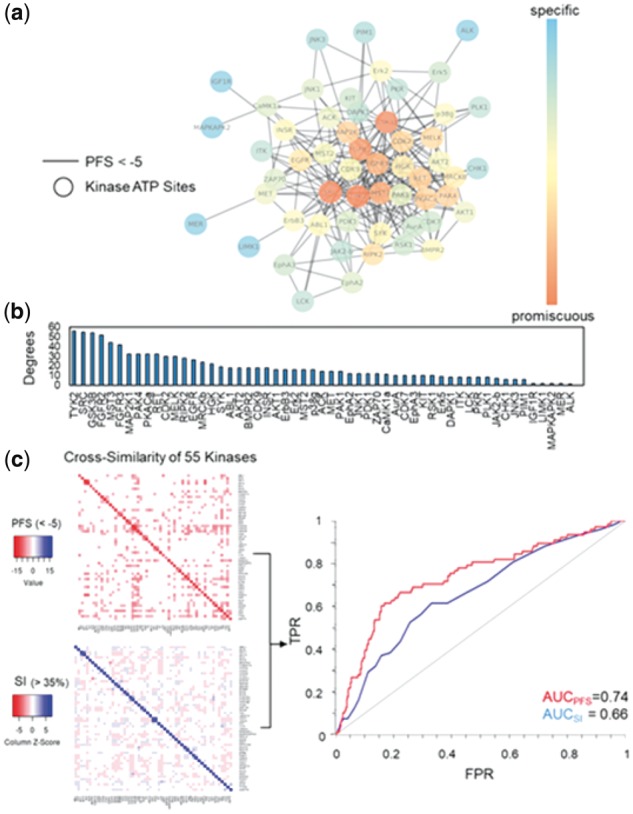
Computational estimation of intrinsic kinase promiscuity. (a) Network similarity clustering of 120 kinase ATP binding pockets from the KinomeFEATURE database based on a optimal PFS threshold (<-5) identified 55 kinases of potential inhibitor cross-activities. (b) To estimate kinase promiscuity, the number of neighbors connected to each node (degree) was used to determine the number of direct interacting partners. The node color in the network correlates with the degree of connectivity. (c) The cross-similarity of 55 kinases were evaluated based on the PFS or SI visualized using heatmaps and the performance were validated by comparing predictions with 17 kinases that have at least one specific inhibitor tested in a previous kinase assay panel (see Fig. 4a and Supplementary Table S6). Quantitative performance assessment based on the ROC curves shows that PFS achieved a higher PFS than SI by either including primary targets (AUCPFS = 0.79 versus AUCSI = 0.72) or excluding primary targets (AUCPFS = 0.74 versus AUCSI = 0.66) (Supplementary Fig. S5a and b)
Overall, our assessment showed that among the 935 total off-target pairs, 109 off-target pairs were predicted by pocket microenvironments similarity (PFS < −5), 110 off-target pairs were predicted by sequence homology (SI > 35%), 75 off-target pairs were predicted by both approaches, while ∼600 off-target pairs were not predicted to have substantial structural or sequence similarities. Furthermore, 123 off-target pairs predicted by the PFS were confirmed by the kinase biochemical assay data, in comparison to 94 off-target pairs by sequence homology search, and 75 off-target pairs by both approaches. To test if our prediction can reveal novel kinase inhibitor off-target interactions, we experimentally validated the predicted compound cross-activity pair between FGFR2 and PKR kinases (PFS = −5.8) that have not been confirmed by the experiment (Fig. 4a and Supplementary Table S4). As an initial assessment, we evaluated the effects of the PKR selective inhibitor, C16 (compound 16), and the FGFR2 selective inhibitor, LY2874455 (compound 17), on PKR and FGFR2 kinases at 1 µM (Fig. 4b and Section 2) (Jammi et al., 2003; Zhao et al., 2011). C16 achieved 95% inhibition of FGFR2, which compares with 99% inhibition of this kinase by LY2874455 (Fig. 4b). On the other hand, LY2874455 exhibited 46% inhibition of PKR, whereas C16 inhibited PKR by 85% (Fig. 4b). To further quantify these initial observations, we carried out full inhibitor titrations and found that the PKR inhibitor C16 inhibited its primary target with an IC50 of 141 nM, but it also inhibited the FGFR2 kinase with an IC50 of 31.8 nM (Fig. 4c and Supplementary Fig. S6). The FGFR2 kinase inhibitor LY2874455 was more selective for its primary target, and showed an IC50 of 1.27 nM against FGFR2 and an IC50 of 843 nM against PKR (Fig. 4c and Supplementary Fig. S6). Notably, the sequence similarity between FGFR2 and PKR kinase is only 23% and no significant chemical similarity observed between C16 and LY2874455 ligands (Lo et al., 2015, 2016, 2017). Thus, the compound cross-activity between PKR and FGFR2 could not have been predicted by conventional kinase selectivity computational prediction methods. To further test the compound cross-activities of three kinase triplicate pairs: FGFR2/FGFR3/PKR, FGFR2/RET/PKR and FGFR2/Aurora A/PKR, we experimentally evaluated IC50 values of PKR inhibitor C16 and FGFR2 inhibitor LY2874455 on FGFR3, RET and Aurora A (Fig. 4c and Supplementary Fig. S6). Using a PFS cutoff of -5 and an activity cutoff of 100 nM, we showed that the PKR inhibitor C16 is active against FGFR2 (PFS = −5.46, IC50 = 31.8 nM) but not FGFR3 (PFS = −4.68, IC50 = 478 nM) as predicted by their pocket similarities with the PKR kinase. Similarly, the FGFR2 inhibitor LY2874455 was capable of inhibiting the Aurora A kinase due to a high pocket similarity with FGFR2 (PFS = −5.41, IC50 = 11.1 nM) while the same compound did not inhibit the PKR activity significantly (PFS = −2.73, IC50 = 114 nM). Notably, the cross-activity between FGFR2 and Aurora A would not have been predicted by the sequence-based approach (SI = 26%). Although the PKR inhibitor C16 has a moderate inhibition on RET (PFS = −3.96, IC50 = 33.8 nM), the compound activity is >10 fold lower than LY2874455 against RET, which shared a higher pocket similarity to FGFR2 (PFS = − 7.8, IC50 = 2.56 nM). Overall, the experimental data is consistent with our computational prediction of kinase selectivity.
Fig. 4.
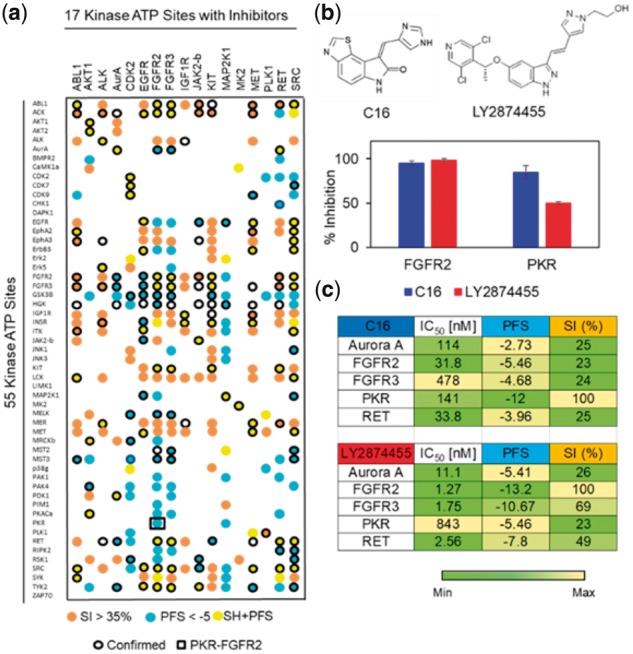
Computational identification of known and novel kinase off-target. (a) Pair-wise cross similarity map of 55 kinase ATP pockets predicted by the PocketFEATURE score (<−5) (color: orange), sequence similarity measure (>35%) (color: blue), both approaches (color: yellow) or neither approaches (color: white). 17 kinases (column) have a least one specific inhibitor tested in a previous SelectScreen® kinase selectivity panel and the confirmed off-target pairs (border: black) were highlighted in the heatmap. The PocketFEATURE algorithm predicted a high binding site similarity (PFS = −5.8) between FGFR2 and PKR. (b) Experimental validation of compound cross-activity between FGFR2 and PKR kinases. The PKR selective inhibitor, C16 (compound 16), and the FGFR2 selective inhibitor, LY2874455 (compound 17), were tested at 1 µM for their interactions with FGFR2 and PKR kinases, respectively. C16 (compound 16) showed 93% FGFR2 inhibition as compared with 97% inhibition by LY2874455 (compound 17). On the other hand, LY2874455 displayed 46% inhibition of PKR in comparison to 88% inhibition of PKR by C16. (c) IC50 evaluation [nM], PFS and SI (%) of C16 (compound 16) and LY2874455 (compound 17) against Aurora A, FGFR2, FGFR3, PKR and RET kinases
3.3 Off-target identification of kinase drug candidates
Since kinase inhibitor off-target interactions have been a major cause of severe adverse events in patients, determining compound selectivity is essential for kinase inhibitor design prior to clinical studies (Force et al., 2007). Here, we applied the KinomeFEATURE database to profile the off-targets of 11 kinase drug candidates in preclinical and clinical development that were designed to bind selectively to 5 kinase targets: PIM1/2, JAK, cMET, BRAF and PI3K (Supplementary Table S7). Using co-crystal structures of the kinase drug candidates, we evaluated the binding site microenvironments similarity of these compounds against the database to identify potential kinase off-target interactions. As an initial validation, the primary targets of the 11 kinase drug candidates were all correctly predicted by the best PFS (Supplementary Table S7, Supplementary Figs S7 and S8). Overall, KinomeFEATURE database profiling of the compounds identified more than 220 primary targets and novel off-targets above the significant PFS threshold of <−5 (Supplementary Table S8).
To further validate kinase off-target prediction, we evaluated the inhibitor potency (logIC50) of 11 kinase drug candidates against 111 kinases with protein crystal structures in PDB and compared the predicted and observed binding profile using heatmaps (Fig. 5a, Supplementary Fig. S9, 10, 11 and Supplementary Text S4). Consistent with the experimental observations, the PFS revealed promiscuous kinase off-target bindings for PIM inhibitors (compounds 18, 19 and 20), JAK inhibitors (compounds 21, 22 and 23) and cMET inhibitors (compounds 24 and 25) whereas the binding interactions for BRAF inhibitors (compounds 26 and 27), and the PI3K inhibitor (compound 28) are relatively specific (Fig. 5b, Supplementary Figs S10 and S11). Linear regression analyses showed that multiple predicted kinase off-targets with significant PFS (PFS < −5) were confirmed by the in vitro biochemical assay data, including CHK1, DAPK1 and GSK3β for PIM inhibitors (compounds 18, 19 and 20), TYK2 and CSF1R for JAK inhibitors (compounds 21, 22 and 23), TRK, LCK and FLT3 for cMET inhibitors (compounds 24 and 25), and LCK, ZAK and RIPK2 for BRAF inhibitors (compounds 26 and 27) (Fig. 5b, Supplementary Figs S9 and S10). On the other hand, high target selectivity was confirmed for the PI3K inhibitors (compound 28) (Supplementary Fig. S11).
Fig. 5.
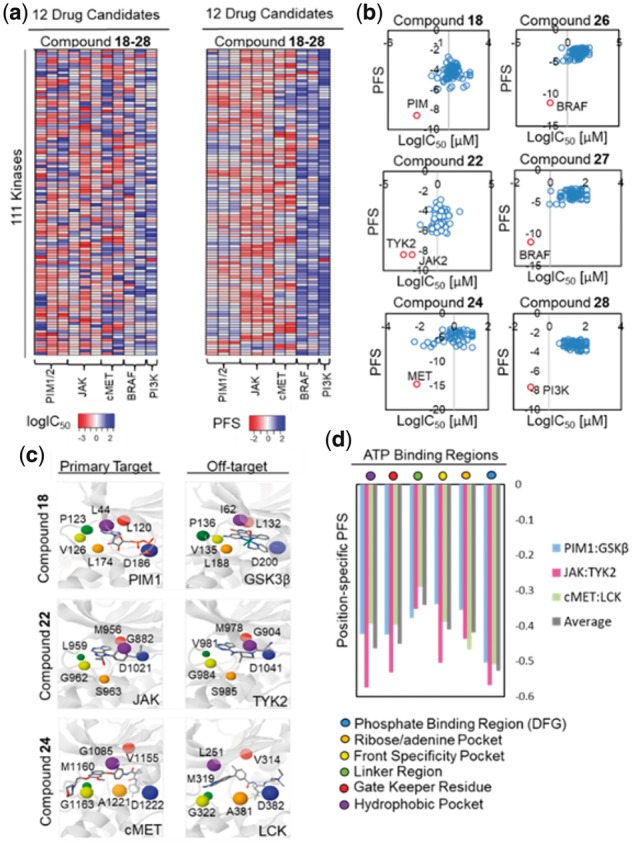
Off-target identification of kinase drug candidates. (a) Heatmaps of kinase off-target binding profile generated based on PFS values or experimentally observed IC50 values (logIC50) for the 11 kinase drug candidates (compounds 18–28). Consistent with the experimental data, predicted PFS revealed promiscuous kinase off-target binding for PIM inhibitors (compounds 18, 19 and 20), JAK inhibitors (compounds 21, 22 and 23), and cMET inhibitors (compounds 24 and 25) while binding for BRAF inhibitors (compounds 26 and 27), and a PI3K inhibitor (compound 28) were highly specific. (b) Scatterplots of predicted PFS values and experimental IC50 values (logIC50) for PIM inhibitor (compound 18), JAK inhibitor (compound 22), cMET inhibitor (compound 24), BRAF inhibitor (compound 26 and 27), and PI3K inhibitor (compound 28). Note that the primary targets with the highest binding affinity (lowest logIC50 values) were predicted by the optimal (most negative) PFS. (c) Structural alignments between primary targets: PIM1 (PDB: 1XR1), JAK (PDB: 4IVD), and cMET (PDB: 3LQ8) and off-targets: GSK3β (PDB: 3PUP), TYK2 (PDB: 3LXN), and LCK (PDB: 2OG8) with CHK1 kinase (PDB: 1NVR) identified critical residues pair involved in kinase off-target binding from the multiple-sequence alignment (see Supplementary Fig. S12). (d) The identified critical binding regions include the phosphate binding region/DFG, the ribose/adenine pocket, the front specificity pocket, the linker region, the gate keeper residue, and the hydrophobic pocket region. Site similarity comparison based on the PocketFEATURE algorithm showed that phosphate binding region/DFG, hydrophobic pocket, and gate keeper regions have the highest average PS-PFS, suggesting their important functional roles in modulating compound cross-activities (Fig. 5d and Supplementary Fig. S12)
Importantly, the identified off-targets revealed additional pharmacological mechanisms for the analyzed kinase drug candidates. For example, off-target ZAK inhibition by BRAF inhibitors has been implicated in the suppression of JNK pathway activation and apoptosis during the treatment of melanoma, which is thought to result in the secondary cutaneous squamous cell carcinoma (cSCC) (Vin et al., 2013). On the other hand, inhibition of GSK3β, a known off-target of multiple PIM inhibitors, was recently shown to have synergy with PIM inhibition in the treatment of prostate cancers (Santio et al., 2016). To determine the structural basis of off-target interactions for PIM (compound 18), JAK (compound 22) and cMET (compound 24) kinase drug candidates, we performed multiple sequence alignments followed by structural superposition to identify critical residues involved in 6 regions of the kinase inhibitor ATP binding site including phosphate binding region (DFG), ribose/adenine pocket, front specificity pocket, linker region, gate keeper residue and hydrophobic pocket (Supplementary Fig. S12 and Supplementary Text S5). Further structural alignments between predicted intended targets: PIM1 (PDB: 1XR1), JAK (PDB: 4IVD) and cMET (PDB: 3LQ8) and predicted off-targets: GSK3β (PDB: 3PUP), TYK2 (PDB: 3LXN) and LCK (PDB: 2OG8) from the KinomeFEATURE database search was used to determine the position specific-PFS (PS-PFS) value between identified residue pairs. PocketFEATURE analysis showed that the DFG (PS-PFSAverage = −0.53), the hydrophobic pocket (PS-PFSAverage = −0.46), and the gate keeper regions (PS-
PFSAverage = −0.45) have the highest average PS-PFS than the ribose/adenine pocket (PS-PFSAverage = −0.42), the front specificity pocket (PS-PFSAverage = −0.41) or the linker region (PS-FPSAverage = −0.34), suggesting their important functional roles in modulating compound cross-activities (Supplementary Text S5 and Fig. 5d) (Patel and Doerksen, 2010; Sohl et al., 2015). Interestingly, each kinase has slight variations in the PS-PFS in different regions of the ATP binding site, which likely reflects the unique binding chemistry of distinct kinases. Thus, the mechanistic knowledge derived from our analysis can potentially be leveraged to design inhibitors target selective kinases to maximize therapeutic effects or reduce side effects.
4 Discussion
Kinases play a significant role in diverse biological pathways and aberrant kinase activity has been implicated in a multitude of diseases. Therefore, methods to inform and ultimately improve the design of selective kinase inhibitors are of tremendous value to develop more potent and safer drugs. While kinase inhibitor selectivity can be experimentally measured using biochemical and kinase binding assay panels, high costs and variability between assay platforms can hinder large-scale kinase selectivity studies or profile comparisons. To this end, we report a new kinase off-target prediction method based on the comparison of binding site microenvironments similarity characterized by diverse physiochemical descriptors. To enable large-scale kinome analysis, we have constructed the KinomeFEATURE database of ∼2850 kinase pocket structures from 189 unique human kinases from the Invitrogen panel and can directly complement experimental validation. In a retrospective analysis, we have showed that our KinomeFEATURE database profiling improved kinase off-target binding prediction accuracy and specificity over the commonly used binding site signature or sequence homology search methods. To further demonstrate the utility of this approach, we applied the algorithm to predict the compound cross-activity between the FGFR2 and PKR kinase inhibitors, LY2874455 and C16, and identified FGFR2 inhibitor LY2874455 as a potent inhibitor of PKR. To date, only C16 had been reported as a selective inhibitor of PKR; thus, our finding suggested that LY2874455 could serve as an initial lead for further PKR inhibitor development. To evaluate the feasibility of our approach for kinome-wide kinase drug selectivity prediction, we computationally profiled the kinase off-target binding activities of 11 kinase drug candidates and identified novel and experimentally validated kinase off-targets. Further structural analysis identified residue pairs critical for modulating kinase cross-activities, which led to further insight into kinase inhibitor promiscuity and pointed to potential strategy for kinase inhibitor design at the kinome scale.
To define the limitations of the KinomeFEATURE database for kinase selectivity profiling, we investigated one predicted kinase off-target, MER, of one cMET kinase drug candidate (compound 24) whose PFS values deviate from the linear regression model (Supplementary Fig. S11). Further structural alignments between cMET and MER kinase structures suggested that conformational changes upon ligand binding involving α4 could contribute to a reduced microenvironment match between two binding sites (Supplementary Fig. S13). Therefore, developing a probabilistic approach that incorporates protein flexibility may improve the prediction. Like other binding site similarity comparison algorithms, one potential challenge of kinase selectivity profiling using the KinomeFEATURE database remains the availability of protein structure data. However, this limitation can be alleviated by recent advances in homology modeling, de novo protein design techniques as well as the rapidly growth of high throughput structural genomic initiatives (Sali and Blundell, 1993). Although we have shown that most kinase inhibitors activity can be predicted from their binding site, several reports described the compound ‘activity cliff’ where a small structural modification results in drastic changes in their binding profile despite of similarities of their native binding microenvironments (Hu et al., 2013; Stumpfe et al., 2014). This problem can be potentially addressed by combining chemical similarity metrics with PFS to generate a stronger signal for similarity detection (Guha, 2012; Liu and Altman, 2011). Still, our KinomeFEATURE database has attempted to cover existing kinase structures to the fullest extent possible and is directly applicable for kinome-wide kinase inhibitor selectivity profiling. In addition, our strategy can be further expanded to analyze inhibitors targeting kinase mutations as well as non-kinase off targets. Most importantly, we anticipate that our method will complement experimental assays and aid in the design of selective kinase inhibitors to mitigate liabilities associated with kinase off-target binding.
Supplementary Material
Acknowledgements
We thank Kenta Yoshida, Charlie Eigenbrot, Andrew Erdman, Donna Dambach, Dan Ortwine, James Kiefer, James Crawford and Dolo Diaz at Genentech and all members of the Helix group at Stanford University for their helpful feedback and suggestions. We are grateful to Gina Wang, Mark Zak, Daniel Sutherlin, Joachim Rudolph, Tim Heffron, Tamara Kale, James Nesbitt, and Chi Sullivan at Genentech for the help during the manuscript preparation.
Funding
The project was supported by Genentech and the following funding sources: NIH GM102365, LM05652 and HL117798.
Conflict of Interest: none declared.
References
- Akeno-Stuart N., et al. (2007) The RET kinase inhibitor NVP-AST487 blocks growth and calcitonin gene expression through distinct mechanisms in medullary thyroid cancer cells. Cancer Res. ,67, 6956–6964. [DOI] [PubMed] [Google Scholar]
- Anastassiadis T., et al. (2011) Comprehensive assay of kinase catalytic activity reveals features of kinase inhibitor selectivity. Nat. Biotechnol. ,29, 1039–1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arora A., Scholar E.M. (2005) Role of tyrosine kinase inhibitors in cancer therapy. J. Pharmacol. Exp. Ther. ,315, 971–979. [DOI] [PubMed] [Google Scholar]
- Beisken S., et al. (2013) KNIME-CDK: workflow-driven cheminformatics. BMC Bioinformatics ,14, 257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berman H., et al. (2003) Announcing the worldwide Protein Data Bank. Nat. Struct. Biol. ,10, 980. [DOI] [PubMed] [Google Scholar]
- Braconi Quintaje S., Orchard S. (2008) The annotation of both human and mouse kinomes in UniProtKB/Swiss-Prot: one small step in manual annotation, one giant leap for full comprehension of genomes. Mol. Cell Proteomics ,7, 1409–1419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brandt P., et al. (2009) Small kinase assay panels can provide a measure of selectivity. Bioorg. Med. Chem. Lett. ,19, 5861–5863. [DOI] [PubMed] [Google Scholar]
- Caffrey D.R., et al. (2008) Prediction of specificity-determining residues for small-molecule kinase inhibitors. BMC Bioinformatics ,9, 491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cicenas J., Valius M. (2011) The CDK inhibitors in cancer research and therapy. J. Cancer Res. Clin. Oncol. ,137, 1409–1418. [DOI] [PubMed] [Google Scholar]
- Cichonska A., et al. (2017) Computational-experimental approach to drug-target interaction mapping: a case study on kinase inhibitors. PLoS Comput. Biol. ,13, e1005678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis M.I., et al. (2011) Comprehensive analysis of kinase inhibitor selectivity. Nat. Biotechnol. ,29, 1046–1051. [DOI] [PubMed] [Google Scholar]
- Demetri G.D., et al. (2002) Efficacy and safety of imatinib mesylate in advanced gastrointestinal stromal tumors. N. Engl. J. Med. ,347, 472–480. [DOI] [PubMed] [Google Scholar]
- Druker B.J., et al. (2001) Efficacy and safety of a specific inhibitor of the BCR-ABL tyrosine kinase in chronic myeloid leukemia. N. Engl. J. Med. ,344, 1031–1037. [DOI] [PubMed] [Google Scholar]
- Fabian M.A., et al. (2005) A small molecule-kinase interaction map for clinical kinase inhibitors. Nat. Biotechnol. ,23, 329–336. [DOI] [PubMed] [Google Scholar]
- Fedorov O., et al. (2007) A systematic interaction map of validated kinase inhibitors with Ser/Thr kinases. Proc. Natl. Acad. Sci. USA ,104, 20523–20528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Force T., et al. (2007) Molecular mechanisms of cardiotoxicity of tyrosine kinase inhibition. Nat. Rev. Cancer ,7, 332–344. [DOI] [PubMed] [Google Scholar]
- Forrest L.R., et al. (2006) On the accuracy of homology modeling and sequence alignment methods applied to membrane proteins. Biophys. J. ,91, 508–517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guha R. (2012) Exploring uncharted territories: predicting activity cliffs in structure-activity landscapes. J. Chem. Inf. Model. ,52, 2181–2191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanks S.K., et al. (1988) The protein kinase family: conserved features and deduced phylogeny of the catalytic domains. Science ,241, 42–52. [DOI] [PubMed] [Google Scholar]
- Helguera G., et al. (2006) Cytokines fused to antibodies and their combinations as therapeutic agents against different peritoneal HER2/neu expressing tumors. Mol. Cancer Ther. ,5, 1029–1040. [DOI] [PubMed] [Google Scholar]
- Hu Y., et al. (2013) Advancing the activity cliff concept. F1000Res ,2, 199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jammi N.V., et al. (2003) Small molecule inhibitors of the RNA-dependent protein kinase. Biochem. Biophys. Res. Commun. ,308, 50–57. [DOI] [PubMed] [Google Scholar]
- Karaman M.W., et al. (2008) A quantitative analysis of kinase inhibitor selectivity. Nat. Biotechnol. ,26, 127–132. [DOI] [PubMed] [Google Scholar]
- Kinnings S.L., Jackson R.M. (2009) Binding site similarity analysis for the functional classification of the protein kinase family. J. Chem. Inf. Model. ,49, 318–329. [DOI] [PubMed] [Google Scholar]
- Kuhn D., et al. (2006) From the similarity analysis of protein cavities to the functional classification of protein families using cavbase. J. Mol. Biol. ,359, 1023–1044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu T., Altman R.B. (2011) Using multiple microenvironments to find similar ligand-binding sites: application to kinase inhibitor binding. PLoS Comput. Biol. ,7, e1002326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo Y.C., et al. (2016) 3D chemical similarity networks for structure-based target prediction and scaffold hopping. ACS Chem. Biol. ,11, 2244–2253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo Y.C., et al. (2017) Computational cell cycle profiling of cancer cells for prioritizing FDA-approved drugs with repurposing potential. Sci. Rep. ,7, 11261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo Y.C., et al. (2015) Large-scale chemical similarity networks for target profiling of compounds identified in cell-based chemical screens. PLoS Comput. Biol. ,11, e1004153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manning G., et al. (2002) The protein kinase complement of the human genome. Science ,298, 1912–1934. [DOI] [PubMed] [Google Scholar]
- Momcilovic M., et al. (2017) Targeted inhibition of EGFR and glutaminase induces metabolic crisis in EGFR mutant lung cancer. Cell. Rep. ,18, 601–610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel R.Y., Doerksen R.J. (2010) Protein kinase-inhibitor database: structural variability of and inhibitor interactions with the protein kinase P-loop. J. Proteome Res. ,9, 4433–4442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patterson H., et al. (2014) Protein kinase inhibitors in the treatment of inflammatory and autoimmune diseases. Clin. Exp. Immunol. ,176, 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosnet O., et al. (1991) Murine Flt3, a gene encoding a novel tyrosine kinase receptor of the PDGFR/CSF1R family. Oncogene ,6, 1641–1650. [PubMed] [Google Scholar]
- Sali A., Blundell T.L. (1993) Comparative protein modelling by satisfaction of spatial restraints. J. Mol. Biol. ,234, 779–815. [DOI] [PubMed] [Google Scholar]
- Santio N.M., et al. (2016) The PIM1 kinase promotes prostate cancer cell migration and adhesion via multiple signalling pathways. Exp. Cell. Res. ,342, 113–124. [DOI] [PubMed] [Google Scholar]
- Sciabola S., et al. (2008) Predicting kinase selectivity profiles using Free-Wilson QSAR analysis. J. Chem. Inf. Model. ,48, 1851–1867. [DOI] [PubMed] [Google Scholar]
- Sheridan R.P., et al. (2009) QSAR models for predicting the similarity in binding profiles for pairs of protein kinases and the variation of models between experimental data sets. J. Chem. Inf. Model. ,49, 1974–1985. [DOI] [PubMed] [Google Scholar]
- Sohl C.D., et al. (2015) Illuminating the molecular mechanisms of tyrosine kinase inhibitor resistance for the FGFR1 gatekeeper mutation: the Achilles’ heel of targeted therapy. ACS Chem. Biol. ,10, 1319–1329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stumpfe D., et al. (2014) Advancing the activity cliff concept, part II. F1000Res ,3, 75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian G., Sud M. (2010) Computational modeling of kinase inhibitor selectivity. ACS Med. Chem. Lett. ,1, 395–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swords R., et al. (2011) The Pim kinases: new targets for drug development. Curr. Drug Targets ,12, 2059–2066. [DOI] [PubMed] [Google Scholar]
- Tabernero J. (2007) The role of VEGF and EGFR inhibition: implications for combining anti-VEGF and anti-EGFR agents. Mol. Cancer Res. ,5, 203–220. [DOI] [PubMed] [Google Scholar]
- Verweij J., et al. (2004) Progression-free survival in gastrointestinal stromal tumours with high-dose imatinib: randomised trial. Lancet ,364, 1127–1134. [DOI] [PubMed] [Google Scholar]
- Vin H., et al. (2013) BRAF inhibitors suppress apoptosis through off-target inhibition of JNK signaling. Elife ,2, e00969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zahler S., et al. (2007) Inverse in silico screening for identification of kinase inhibitor targets. Chem. Biol. ,14, 1207–1214. [DOI] [PubMed] [Google Scholar]
- Zhao G., et al. (2011) A novel, selective inhibitor of fibroblast growth factor receptors that shows a potent broad spectrum of antitumor activity in several tumor xenograft models. Mol. Cancer Ther. ,10, 2200–2210. [DOI] [PubMed] [Google Scholar]
- Zhou W., et al. (2015) High resolution prediction of calcium-binding sites in 3D protein structures using FEATURE. J. Chem. Inf. Model. ,55, 1663–1672. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.