

Abstract
Candidatus Poribacteria is a little-known bacterial phylum, previously characterized by partial genomes from a single sponge host, but never isolated in culture. We have reconstructed multiple genome sequences from four different sponge genera and compared them to recently reported, uncharacterized Poribacteria genomes from the open ocean, discovering shared and unique functional characteristics. Two distinct, habitat-linked taxonomic lineages were identified, designated Entoporibacteria (sponge-associated) and Pelagiporibacteria (free-living). These lineages differed in flagellar motility and chemotaxis genes unique to Pelagiporibacteria, and highly expanded families of restriction endonucleases, DNA methylases, transposases, CRISPR repeats, and toxin–antitoxin gene pairs in Entoporibacteria. Both lineages shared pathways for facultative anaerobic metabolism, denitrification, fermentation, organosulfur compound utilization, type IV pili, cellulosomes, and bacterial proteosomes. Unexpectedly, many features characteristic of eukaryotic host association were also shared, including genes encoding the synthesis of eukaryotic-like cell adhesion molecules, extracellular matrix digestive enzymes, phosphoinositol-linked membrane glycolipids, and exopolysaccharide capsules. Complete Poribacteria 16S rRNA gene sequences were found to contain multiple mismatches to “universal” 16S rRNA gene primer sets, substantiating concerns about potential amplification failures in previous studies. A newly designed primer set corrects these mismatches, enabling more accurate assessment of Poribacteria abundance in diverse marine habitats where it may have previously been overlooked.
Subject terms: Microbiology, Microbial ecology
Introduction
Candidatus Poribacteria were first identified in the marine sponge Aplysina aerophoba more than 14 years ago [1], but have never been successfully isolated in laboratory culture. It is not known whether the relationship of Poribacteria with their hosts is mutualistic, commensal, or parasitic, although vertical transmission has been demonstrated throughout all host reproductive stages [2, 3]. Significant sequence divergence between Poribacteria and their nearest sister groups have made confident taxonomic placement challenging. Poribacteria form a deep-branching, monophyletic clade alternatively proposed, with limited bootstrap support, as most closely related to the Planctomycetes-Verrucomicrobia-Chlamyidiae superphylum [1], Hydrogenedentes [4], Spirochaetes [5], and Acidobacteria [6].
Cellular structure and potential metabolic capabilities of Poribacteria have been inferred from microscopic observations [1, 7], partial single-cell amplified genomes [8, 9], metatranscriptomic recruitment to these genomes [7], and taxonomic binning of metagenome-assembled contigs [5]. Pathway reconstructions from these sources have suggested a heterotrophic, primarily aerobic lifestyle that includes glycolysis, oxidative phosphorylation, and autotrophic carbon fixation via the Wood–Ljungdahl pathway. A diverse set of carbohydrate degrading enzymes and abundant sulfatases have been interpreted to enable the digestion of sponge host extracellular matrix [8], supported by in situ fluorescence hybridization and electron microscope images demonstrating the localization of ovoid-shaped Poribacteria cells embedded within Aplysina aerophoba mesohyl tissues [1, 7].
Several investigators have observed the presence of enclosed micro-compartments within Poribacteria cells. These structures were originally described as DNA-containing, membrane-bound nuclear bodies, based on fluorescence in situ hybridization and immuno-gold staining [1, 10], but this claim was later disputed based on transmission electron microscopy and correlative light and electron microscopy results [7]. In the absence of cultured cells for laboratory verification, the function of these compartments currently remains unresolved.
Ultrastructure and molecular characterization studies to date have relied heavily on samples obtained from a single host, Aplysina aerophoba, but Poribacteria-related 16S rRNA genes have also been detected in numerous other sponge genera, including Agelas, Astrosclera, Dactylospongia, Geodia, Ircinia, Plaktoris, Pseudoceratina, Rhabdastrella, Theonella, Vaceletia, and Xestospongia [2, 11–19]. Closely related 16S rRNA sequences have also been observed, albeit at much lower levels, in corals, seawater, and marine sediment samples [2, 3, 19–22].
Phylogenetic trees constructed from partial 16S rRNA sequences suggest that sponge-associated Poribacteria may fall into four [16, 23] or five [24] distinct subclasses, but no correlations have been observed between these classes and sponge host taxonomy or geographical location. Several studies analyzing sponge-associated microbial communities with specifically targeted 16S rRNA gene primers observed Poribacteria-related sequences at relative abundances of 20–30% [1, 12, 25–27]. Other studies, using only broader “universal” primers, found much lower relative abundances in samples from the same sponge host species [19, 28]. These differences may reflect natural biological variation, but could also be the result of reduced sensitivity due to amplification primer mismatches [1, 9, 12, 29], raising concerns that historical surveys relying exclusively on unsuitable primers may have systematically under-reported Poribacteria abundance.
A set of 2631 metagenome-assembled genomes from the Tara Oceans project, reconstructed from multiple samples of varying depths and filter sizes, were found to include thirteen putative Poribacteria-related genomes [6]. Metagenomic assembly has also identified two Poribacteria-related contig bins in the particulate fraction of deep-sea hydrothermal vent plumes, with estimated read abundances in some samples approaching 1.25% of the microbial community [20]. The extent to which the functional capabilities of these open ocean Poribacteria might resemble those of their sponge-associated relatives is unknown. Detailed evolutionary relationships between genomes from different habitats also remain to be determined.
In this study, metagenomic assembly techniques were used to reconstruct eight new high quality Poribacteria genomes from four different Verongid sponge genera, collected at distant geographic locations. These genomes have been compared with previously reported sponge-associated sequences as well as Poribacteria-related genomes from the open ocean, to determine their taxonomic relationships and explore shared versus unique functional activities associated with their collective pangenomic repertoire.
Materials and methods
Sample collection and processing
Sponge specimens, collection dates, and locations are described in Supplementary Figure 1. DNA was extracted from frozen whole sponge tissue by lysis at 55 °C for 30 min in 4 M guanidine thiocyanate, 2% sarkosyl, 50 mM EDTA, 40 μg/ml proteinase K, and 15% β-mercaptoethanol, followed by Mini‐Beadbeater‐8 (BioSpec Products, USA) homogenization for 20 s with 0.1 mm silica beads, extraction with one volume of phenol:chloroform:isoamyl alcohol (25:24:1), and cleaning with the Quick-gDNA MiniPrep kit (Zymo Research, USA).
16S rRNA analysis
V4 region amplification was performed using the Illumina two-reaction strategy [30] with Q5 polymerase (NEB, USA). Amplifications were performed using two different initial primer sets; 515FB-806RB (Fwd:GTGYCAGCMGCCGCGGTAA; Rev:GGACTACNVGGGTWTCTAAT) [31] and 515Fsp-806Rsp (Fwd:GTGCCAGCAGCYGCGGTAA; Rev:GGACTASCGGGGTATCTAAT), modified to eliminate Poribacteria-specific mismatches. First stage amplifications were performed in triplicate, with an initial 30 s denaturation at 98 °C, followed by 25 cycles of 10 s at 98 °C, 30 s at 54 °C, 20 s at 72 °C, and final extension 2 min at 72 °C. Barcoding reactions were performed on 5 µl pooled aliquots of each sample with 8 amplification cycles at annealing temperature 60 °C. Equimolar concentrations of dual-barcoded amplicons were sequenced using Illumina’s MiSeq platform to obtain 2 × 300 bp reads (UC Davis DNA Technologies Core).
MiSeq reads were trimmed with Trimmomatic version 0.35 [32] using the settings SLIDINGWINDOW:4:5, MINLEN:100. Paired reads were further processed in Qiime version 2017.12.0 [33] for denoising, primer trimming, read-pair merging, and non-ribosomal sequence filtering, using the DADA2 workflow [34]. Taxonomies were assigned using the scikit-learn naive bayes classifier [35] and SILVA database release 128 [36], supplemented with Poribacteria 16S rRNA gene sequences from this study. Taxonomies and count tables were imported into R using phyloseq version 1.20.0 [37], and normalized using the cumulative-sum scaling method from MetagenomeSeq version 1.1216 [38].
Metagenomic sequencing, assembly, and annotation
Metagenomic DNA libraries were constructed using TruSeq Nano kits (Illumina, San Diego CA) to obtain 150 bp paired-end reads using the Illumina HiSeq 2500 platform in Rapid Run mode. Paired-end reads were quality filtered and trimmed using Trimmomatic version 0.35 [32], with the following parameters: adapter-read alignment settings 2:30:10, LEADING:3, TRAILING:15, HEADCROP:15, SLIDINGWINDOW:4:15, MINLEN:115. Preliminary guide assemblies were created using IDBA-UD version 1.1.3 set to default parameters [39]. Input reads were mapped to scaffolds from these preliminary assemblies using the end-to-end option of Bowtie2, version 2.2.7 [40]. Coverage depth was calculated using the idxstats module of samtools version 1.3 [41]. Preliminary scaffolds were grouped into bins based on percent GC, nucleotide composition, assembly depth of coverage, and taxonomic assignment by DarkHorse version 2.0 [42, 43], as previously described [44]. Read subsets from scaffold bins identified as potentially belonging to Poribacteria were re-assembled using Celera Assembler version 8.3 [45], configured with merSize = 17, utgGenomeSize = 5 Mb and utgErrorRate = 0.01.
Previously reported Poribacteria single-cell genomes were downloaded from IMG-MER [46]. Poribacteria-related genomes assembled from the Aplysina aerophoba metagenome [5] and the Tara Oceans projects [6] were downloaded from the Genbank WGS sequence database. Corresponding metadata for Tara Oceans sequences were retrieved from supplementary online sources [47, 48]. Ab initio gene predictions and functional descriptions were obtained using Prokka version 1.12 [49] for genomes lacking publicly available annotation data, using default program settings. Poribacteria metagenome-assembled genomes generated in this study were also annotated at IMG-MER [46]. CRISPR repeat regions were identified using the CRISPR Repeat Tool software, version 1.2 [50]. Assembly bin quality was assessed using CheckM version 1.07 with the default set of bacterial marker genes [51]. Potentially over-represented protein functional families were identified using Hidden Markov Models (HMMs) from the Pfam-A version 32 [52] and TIGRFAM release 15 [53] databases. In cases where models with overlapping functional activities matched the same target protein (e.g., restriction endonucleases), only the HMM with the highest bitscore was included in quantitative tallies, so that no protein was counted more than once.
Phylogenetic placement and protein family clustering
Poribacteria-related 16S rRNA gene sequences were downloaded from SILVA database release 132 [36] and aligned with sequences extracted from assembled Poribacteria genomes using the SILVA Incremental Aligner (SINA) version 1.2.11 [54]. Concatenated alignments of 28 highly conserved single-copy genes were created using MUSCLE version 3.8.31 [55]. Phylogenetic trees were constructed using FastTree version 2.1.8 [56] and visualized using FigTree version 1.4.3 [57].
Average amino acid identity (AAI) and average nucleic acid identity (ANI) calculations were performed using the online ANI/AAI-Matrix Genome-based distance matrix calculator [58]. ANI scores below 75% were excluded from the analysis, as they have been shown to be unreliable [59]. Protein family clusters for predicted proteins were obtained using ProteinOrtho version 5.16b [60], excluding assembled genomes estimated to be <50% complete. Venn diagrams were produced using the venneuler module of the R software package, version 3.4.0 [61]. and EulerAPE version 3.0 [62].
Relative abundance measurements
Trimmed, quality-filtered reads from each sponge metagenomic sample were randomly down-sampled to 50,000 read subsets, then translated into all six frames using the EMBOSS 6.0 transeq tool [63]. Predicted proteins from each random subset were analyzed using DarkHorse version 2.0 [42, 43] with a filter threshold setting of 0.01 to find taxonomic classifications for database matches. Because none of the previously published single cell or metagenome-assembled Poribacteria genomes were included in Genbank nr as of January 2018, the DarkHorse reference database was customized to include these sequences as a supplement, along with the eight newly assembled Poribacteria genomes described in this study.
Database deposition Information
All sequence data associated with this study have been deposited under NCBI BioProject ID PRJNA433267 and the Joint Genome Institute Integrated Microbial Genomes and Microbiomes (IMG/M) resource [46] (Supplementary Table S1).
Results and discussion
Genome assembly and quality assessment
All available host-associated Poribacteria genomes prior to this study were obtained from a single sponge species, Aplysina aerophoba, collected from the Adriatic Sea, including five single-cell genomes (SAGs) and two metagenome-assembled genomes (MAGs). The current study has expanded the host range of Poribacteria-like genomes to encompass eight new MAGs from four additional sponge genera, Agelas, Dysidea, Melophlus, and Pseudoceratina, collected from geographically distant sites in the Atlantic and Pacific oceans (Table 1).
Table 1.
Assembled genome properties. Asterisks indicate genomes excluded from functional analyses due to incompleteness. Abbreviations: SAG, single-cell genome; MAG metagenomic assembly genome. Additional properties of assembled genomes are provided in Supplementary Table S1
| Assembly name | DNA source | Seq type | Num scf | Total length | pct GC | N50 | Max 16S length | CheckM pct comp | CheckM Contam | References | Tax group |
|---|---|---|---|---|---|---|---|---|---|---|---|
| PCPOR4 | Pseudoceratina sp. | MAG | 214 | 6,452,484 | 47.7 | 64,782 | 1554 | 96% | 0.0 | This study | G1 |
| PCPOR2 | Pseudoceratina sp. | MAG | 76 | 5,150,747 | 48.1 | 121,268 | 1541 | 95% | 5.2 | This study | G1 |
| DGPOR9 | Dysidea granulosa | MAG | 114 | 5,526,652 | 47.9 | 147,715 | 1555 | 95% | 14.7 | This study | G1 |
| PCPOR2b | Pseudoceratina sp. | MAG | 53 | 5,303,725 | 48.7 | 196,409 | 875 | 90% | 0.5 | This study | G4 |
| MSPOR6 | Melophlus sarasinorum | MAG | 50 | 4,328,976 | 38.2 | 108,750 | 887 | 88% | 0.0 | This study | G2 |
| PCPOR1 | Pseudoceratina sp. | MAG | 178 | 5,380,211 | 47.3 | 45,924 | 1554 | 88% | 0.0 | This study | G1 |
| AGPOR5 | Agelas tubulata | MAG | 213 | 4,351,829 | 53.9 | 28,430 | 1555 | 86% | 0.9 | This study | G2 |
| PCPOR2a | Pseudoceratina sp. | MAG | 141 | 4,568,562 | 43.1 | 52,945 | 0 | 79% | 0.0 | This study | G2 |
| MPMS | Aplysina aerophoba | MAG | 465 | 7,043,869 | 47.2 | 23,989 | 0 | 96% | 11.0 | [5] | G1 |
| MPMY | Aplysina aerophoba | MAG | 106 | 5,097,196 | 40.3 | 70,347 | 0 | 95% | 10.3 | [5] | G2 |
| WGA-3G | Aplysina aerophoba | SAG | 286 | 5,441,554 | 47.8 | 51,110 | 0 | 88% | 0.6 | [9] | G1 |
| WGA-4E | Aplysina aerophoba | SAG | 521 | 3,647,669 | 49.5 | 13,035 | 1554 | 59% | 1.3 | [4] | G4 |
| *WGA-4C | Aplysina aerophoba | SAG | 276 | 1,629,923 | 40.9 | 12,952 | 1545 | 30% | 0.8 | [4] | G2 |
| *WGA-4G | Aplysina aerophoba | SAG | 15 | 189,191 | 47.3 | 15,021 | 1546 | 7% | 0.0 | [4] | G1 |
| *WGA-A3 | Aplysina aerophoba | SAG | 157 | 414,219 | 48.7 | 4895 | 1326 | 0% | 0.0 | [4] | G1 |
| NP60 | Tara Oceans | MAG | 120 | 5,814,405 | 40.2 | 113,317 | 1518 | 98% | 4.5 | [6] | G6b |
| SP142 | Tara Oceans | MAG | 87 | 5,196,966 | 41.1 | 71,421 | 0 | 98% | 0.0 | [6] | G6a |
| RS423 | Tara Oceans | MAG | 166 | 5,326,059 | 40.4 | 59,315 | 1518 | 97% | 5.2 | [6] | G6b |
| MED599 | Tara Oceans | MAG | 217 | 4,811,953 | 40.2 | 28,937 | 1518 | 96% | 1.7 | [6] | G6b |
| SAT10 | Tara Oceans | MAG | 123 | 4,732,202 | 40.2 | 50,212 | 1518 | 96% | 3.5 | [6] | G6b |
| NAT79 | Tara Oceans | MAG | 122 | 4,242,771 | 40.4 | 44,479 | 1518 | 96% | 2.3 | [6] | G6b |
| SAT1451 | Tara Oceans | MAG | 160 | 4,963,030 | 42.0 | 39,891 | 1546 | 93% | 5.2 | [6] | G6a |
| NP41 | Tara Oceans | MAG | 243 | 5,344,530 | 43.6 | 28,426 | 1548 | 90% | 1.7 | [6] | G6a |
| NAT81 | Tara Oceans | MAG | 168 | 4,167,435 | 41.9 | 28,622 | 1546 | 90% | 1.9 | [6] | G6a |
| ARS61 | Tara Oceans | MAG | 457 | 5,339,246 | 66.6 | 12,287 | 1540 | 84% | 2.7 | [6] | Outgroup |
| NAT1 | Tara Oceans | MAG | 257 | 6,421,894 | 47.0 | 31,101 | 0 | 72% | 0.5 | [6] | G6c |
| ARS1035 | Tara Oceans | MAG | 187 | 3,967,361 | 42.0 | 28,336 | 1546 | 70% | 3.5 | [6] | G6a |
| ARS87 | Tara Oceans | MAG | 243 | 4,054,128 | 46.8 | 20,570 | 0 | 49% | 0.5 | [6] | G6c |
Thirteen MAGs from the Tara Oceans project have recently been classified as belonging to candidate phylum Poribacteria [6]. These 13 MAGs comprise <0.5% of the 2631 genomes described in the study, but cover a worldwide geographical distribution, with collection sites including the Red Sea, the Mediterranean Sea, and both Atlantic and Pacific oceans (Fig. 1).
Fig. 1.

Expanded global distribution of assembled Poribacteria genomes. SAGs single-cell assembled genomes, MAGs metagenomically assembled genomes. Detailed metadata and accession numbers for host-associated and Tara Oceans genomes are provided in Supplementary Tables S1-S2. Hydrothermal vent metadata were obtained from Anantharaman et al. [20]
Previously published tables mapping raw reads for assembled Tara oceans Poribacteria MAGS to underlying sample ids [48] were joined with metadata from the NCBI Short Read Archive [64] and an earlier publication documenting sample depths and filter sizes [47] to create Supplementary Table S2. Although multiple different filter sizes and sampling stations were included in the initial assembly pipeline, most of the final consensus sequences were reconstructed primarily with reads originating from a single source. Twelve of the 13 Poribacteria-related Tara Oceans genomes were assembled primarily from reads collected in deep chlorophyll maximum and mesopelagic zones, at depths ranging from 70 to 800 m. Ten of these genomes were predominantly derived from reads in the 0.8–5 µm filter fraction rather than the smaller 0.22–1.6 µm fraction typical of free-living bacteria. These data strongly suggest association with sinking particulate matter, consistent with the independent discovery of Poribacteria on particles collected from a deep hydrothermal vent environment [20].
Genomic characteristics and assembly quality metrics for Poribacteria genomes in the current study, including all standard parameters recommended in ref. [65], are presented in Table 1 and Supplementary Table S1. Two previously published MAGs (MPMS and MPMY) and DGPOR9 from the current study exceeded the maximum recommended CheckM “contamination” value of 10%, but were included in the study because they provided unique information unobtainable from other sources. MPMS and MPMY are the most complete available Poribacteria representatives from sponge family Aplysinidae, while DGPOR9 is the only representative from sponge host family Dysideidae, in which Poribacteria had not previously been reported. When duplicated proteins flagged by CheckM were tested in a blast search that also included all proteins in Genbank nr, they most closely matched other (non-self) Poribacteria sequences, suggesting the duplicated proteins were most likely co-assembled Poribacteria strain variants.
Sponge-associated Poribacteria genomes showed no evidence of symbiosis-related genome streamlining [66]. Average sizes for genome bins of sponge-associated Poribacteria reported as more than 90% complete by CheckM were actually slightly larger (5.4 ± 0.69 MB, n = 10) than those from the Tara Oceans dataset (5 ± 0.28 MB, n = 9), although this difference was not statistically significant (p-value = 0.18, two-tailed t-test). Average nucleotide compositions ranged from 40 to 50% GC, with the exception of one sponge-associated genome at 53.9% GC and one Tara Oceans genome at 66.6% GC. Estimated completeness for MAGs was generally higher than for SAGs, but some MAGs also had higher duplication levels for single-copy marker genes.
Taxonomic relationships and subgroups
Taxonomic relationships between Poribacteria genomes were analyzed using four different, complementary techniques: 16S rRNA gene trees, concatenated multi-locus gene trees, average amino acid identity (AAI), and average nucleotide identity (ANI). These independent approaches were especially valuable in compensating for unequal completeness among the genomes being analyzed. Phylogenetic trees based on 16S rRNA genes have the advantage of allowing comparison with publicly available sequences where no other genomic data are available; however, many Poribacteria assemblies, even some that are otherwise nearly complete, lack full-length 16S rRNA gene sequences (Table 1). Alternatively, 22 of the assembled Poribacteria genomes contain a complete set of 28 conserved, single-copy marker genes (Supplementary Table S3), which were used to construct a concatenated multi-locus tree. AAI and ANI clustering enable placement of incomplete genomes lacking 16S rRNA and/or single-copy marker genes, although no bootstrap support values can be inferred using these metrics.
All classification methods agreed in clustering the Tara Oceans MAGs together in a single, well-supported monophyletic clade, with the exception of genome ARS61, an outgroup to both open ocean and sponge-associated Poribacteria (Figs. 2, 3). The dissimilarity between ARS61 and all other Poribacteria-related sequences is consistent with its previously published placement as an outgroup in a much broader multi-locus tree [6], as well as its highly atypical nucleotide composition (Table 1). Although ARS61 is more closely related to Poribacteria than to Planctomyces outgroup Rhodopirellula baltica, its placement inside phylum Poribacteria cannot be confidently confirmed until additional genomes from suitably related sister phyla become available.
Fig. 2.
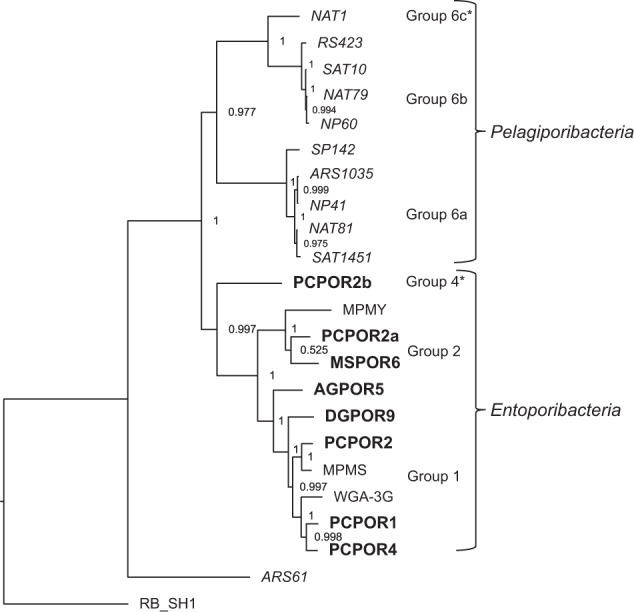
Concatenated multi-locus Poribacteria tree. Bolded names indicate sequences generated by the current study. Host abbreviations: Agelas tubulata, AG; Dysidea granulosa, DG; Melophlus sarasinorum, MS; Pseudoceratina sp., PC; Aplysina aerophoba, WGA MPMY, and MPMS. Tara Oceans genomes are italicized, with geographic abbreviations ARS, Arabian Sea; RS, Red Sea; MED Mediterranean Sea; NAT, North Atlantic; SAT, South Atlantic; NP, North Pacific; SP, South Pacific. Outgroup abbreviation RB_SH1 indicates Rhodopirellula baltica strain SH1. Asterisks highlight divisions supported by additional genomes in 16S rRNA trees (Supplementary Figure S2) and AAI distance clustering (Fig. 3). Supplementary Table S3 lists the 28 genes used to construct this tree, which were all present in all genomes shown
Fig. 3.

Percent average amino acid identity (AAI) shared between Poribacteria genomes. AAI values include all assembled Poribacteria genomes classified as >50% complete by CheckM (Table 1; [51]). Abbreviation RB_SH1 indicates Rhodopirellula baltica strain SH1. Cladogram was constructed using AAI values to create a distance matrix [58]. Areas colored in darker shades represent closer evolutionary relationships
16S rRNA gene phylogenetic analysis placed all sponge-associated Poribacteria genomes into previously classified subgroups 1, 2, or 4, with no representatives in subgroups 3 or 5 (Supplementary Figure S2). Searches of the NCBI, SILVA, and IMG-MER databases for additional Poribacteria-related sequences retrieved Tara Oceans entries from earlier, less complete metagenomic assemblies from North Atlantic, Peruvian Coast, and Red Sea samples (CEVJ01037068, CETA01044412, and CENY01011605), but none from 16S rRNA amplicon studies at these same locations. One additional Tara Oceans-related 16S rRNA sequence (JYMV01042177) was recovered from a hydrothermal vent plume sampling project [20], but this sequence was also obtained through metagenomic assembly rather than 16S rRNA gene amplification. No Poribacteria-related sequences were detected in an earlier 16S rRNA gene study of the same environment by the same authors [67]. The unexpected absence of database matches from 16S rRNA amplification studies to environmental sequences from the open ocean may be linked to mismatch issues with commonly used “universal” primers, as discussed below.
The names Entoporibacteria and Pelagiporibacteria have been selected to represent genomes from sponge-associated and open oceans clades, respectively. Relative taxonomic distances between Entoporibacteria and Pelagiporibacteria genomes were estimated based on pairwise comparisons of 16S rRNA gene nucleotide identity, ANI, and AAI (Fig. 3, Supplementary Figure S3a, b). AAI scores (49–53%) and 16 S rRNA gene identities (87–93%) suggest that Entoporibacteria and Pelagiporibacteria fall within ranges recently proposed to represent separate families (AAI) or orders (16S rRNA) [68]. Although some evidence suggests that AAI levels below 45% and 16S identities 73–85% might be characterized as phylum-level differences [59, 68, 69], confident assignment of higher level taxonomic categories typically requires comparisons of several closely related sister groups, an approach that is not currently feasible for Poribacteria.
Genomes with ANI scores > 95%, AAI scores > 65–95%, and 16S rRNA gene identities > 98.6% are most often classified as belonging to the same species, while those with AAI scores of 65–95%, or 16S rRNA gene identities > 95–98% are generally considered members of the same genus [68]. By these criteria, Pelagiporibacteria subgroups 6a, 6b, and 6c and Entoporibacteria subgroups 1 and 2 each represent separate genera. The hydrothermal vent particle-associated 16S rRNA gene sequence JYMV01042177 (Lau Basin; 2000 m depth) was 97.4% identical to Tara Oceans genome ARS1035 (Arabian Sea; 600 m depth), suggesting membership in the same genus. The identification of JYMV01042177 on a 43,681 bp contig containing 26 predicted protein sequences with 80–95% amino acid identity to other group 6a genomes corroborates this close relationship. AAI scores and 16S rRNA gene identities for Entoporibacteria subgroup 4 support its classification as an independent family, suggesting that Entoporibacteria and Pelagiporibacteria most likely represent different orders within phylum Poribacteria.
Shared functional activities
A total of 106,793 predicted proteins from Entoporibacteria and Pelagiporibacteria draft genomes were processed using ProteinOrtho to yield 10,654 family clusters. Approximately one third of these protein families were shared in both Entoporibacteria and Pelagiporibacteria genomes (Fig. 4). Shared protein family percentages within subgroups of these major lineages are shown in Supplementary Figure S4.
Fig. 4.
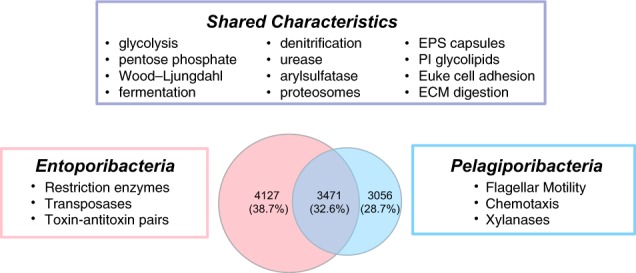
Shared versus unique protein family functions in Poribacteria. Shared families are defined as ProteinOrtho clusters found in two or more genomes from each different lineage. Specific functional characteristics attributed to Entoporibacteria and Pelagiporibacteria were found in all members of their respective groups. EPS exopolysaccharide, PI phosphatidyl inositol, Euke eukaryotic, ECM extracellular matrix. More detailed information on shared versus lineage-specific functional gene families is provided in Supplemetary Table S4
Predicted functions of protein families shared between Entoporibacteria and Pelagiporibacteria encompass most features of central metabolism previously described in Aplysina host-associated SAGs and MAGs, including complete pathways for glycolysis, oxidative phosphorylation, the tricarboxylic acid cycle, oxidative and non-oxidative branches of the pentose phosphate pathway, and carbon fixation via the Wood–Ljungdahl pathway (Supplementary Table S4). Entoporibacteria and Pelagiporibacteria genomes also shared predicted genes and pathways for complex carbohydrate degradation, assimilatory sulfate reduction, denitrification, urea degradation, propane/butane-diol utilization, and fermentation of citrate, lactate, and pyruvate, suggesting facultative adaptation to microaerophilic or anaerobic conditions. Both groups also encoded conserved Gram-negative outer membrane and periplasm components, translocases, type II protein secretion systems, protein families related to bacterial microcompartment and gas-vesicle shell formation (pfam00936 and pfam12732), and synthetic pathways for branched chain fatty acids, peptidoglycans, lipopolysaccharides, sterols, and vitamin cofactors (biotin, thiamine, and cobalamin).
The combined analysis of multiple genomes, including many that are nearly complete, enabled identification of some shared protein functions not previously described in Poribacteria. These include genes encoding exopolysaccharide capsule biosynthesis and assembly; bacterial proteosome/Pup mediation of protein turnover; cellulosome anchoring of extracellular enzyme complexes through cohesin and dockerin domains; metabolism of the osmolytes ectoine and glycine betaine; competence proteins involved in DNA uptake, type IV pili and plasmid transfer functions; and phage defense through CRISPR/Cas systems, restriction endonucleases, and nucleic acid modification methylases.
Several protein families previously suggested as potentially adaptive to a host-associated lifestyle [5, 8, 9] were unexpectedly abundant in Pelagiporibacteria genomes, with some averaging more than 10 copies per genome (Fig. 5). These include predicted membrane adhesion factors such as concanavalin A-like lectins and immunoglobin-like, fibronectin, leucine-rich repeat, and pleckstrin homology domains. Additional shared protein clusters included not only glycosaminoglycan degradation enzymes (for example heparinases), but also multiple neuraminidases, ceramidases, and chitinases.
Fig. 5.
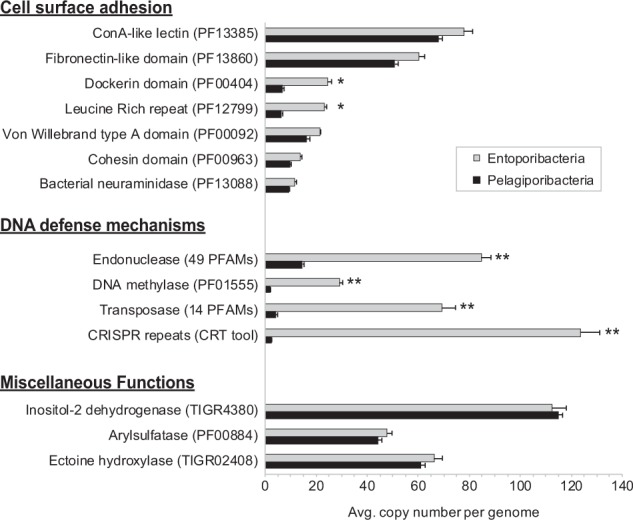
Highly over-represented gene families. Functional families shown averaged 10 or more copies per genome in either Entoporibacteria, Pelagiporibacteria, or both. Asterisks indicate statistically significant differences, as defined by two-tailed Student’s t-test. *p-values < 0.01, **p-values < 0.001
Both Entoporibacteria and Pelagiporibacteria encode pathways utilizing glycosylated phosphotidyl inositols, normally found in eukaryotes and archaea but not in bacteria outside of phylum Actinobacteria [70]. Pathogenic Mycobacteria use cell membrane glycolipids anchored by myo-inositol lipid head groups to mediate interactions with their terrestrial hosts [71]. Shared Poribacteria protein families included not only multiple variants of previously described degradative enzymes like myo-inositol 2-dehydrogenase and scyllo-inositol 2-dehydrogenase [8], but also newly predicted synthetic pathway components, including myo-inositol-1-phosphate synthase, phosphatidyl inositol mannoside acyltransferase, CDP-diacylglycerol-inositol 3-phosphatidyltransferase, and inosose isomerase. Additionally, Pelagiporibacteria genomes from groups 6b and 6c encoded Mycobacteria-like tuberculostearic acid methyltransferases. Both Entoporibacteria and Pelagiporibacteria genomes shared conserved gene families encoding mycothiol synthase and mycothiol S-conjugate amidase. Mycothiol, which can act as a substitute for glutathione, is noted for its ability to defend pathogenic Actinobacteria against toxic oxygen radicals produced by mammalian phagocytes [72].
Sporulation pathway genes have not previously been described in Poribacteria, but were discovered among shared protein families identified by the current study. These genes are of particular interest with respect to the controversial intracellular compartments previously reported in microscopic images [1, 7]. Although the function of these structures in Poribacteria is currently unknown, they bear a striking visual resemblance to transmission electron micrographs of endospores in dividing cells of Lactobacillus brevis [73]. Protein family clusters annotated as stage II sporulation proteins E and M, inner spore coat protein H, SpoIIAA-like anti-anti-sigma regulatory factor, SpoIVB peptidase S55, Spore protein SP21, and spore cortex peptidoglycan biosynthesis regulator SpoVE were found in all Poribacteria clades. It is possible that these genes were inherited from an ancient spore-forming ancestor and later re-purposed to perform other cellular functions, but this scenario is inconsistent with their high degree of conservation within the Poribacteria group. Comparative studies of known endospore-forming bacteria have identified several hundred conserved genes preferentially expressed during sporulation [74], but potential completeness of this pathway in Poribacteria is difficult to determine due to large evolutionary distances from well-studied reference genomes.
Predicted functional differences
The most obvious differences between Entoporibacteria and Pelagiporibacteria genomes were the exclusive presence of complete flagellar biosynthesis, assembly and methyl-accepting chemotaxis pathways in Pelagiporibacteria. The absence of these pathways in Entoporibacteria is consistent with a previously reported lack of flagella in microscopic observations [1, 7] and the absence of motility genes in single-cell genomes [9]. Pelagiporibacteria also contained unique beta-1,4-xylanases, potentially useful in breaking down refractory carbon from algal cell walls, as well as cryptochrome-like enzymes annotated as deoxyribodipyrimidine and (6–4) photo-lyases that were not present in any Entoporibacteria genomes. These latter enzymes, encoding ultraviolet light-induced DNA dimer repair, should not be essential at the sampling depths associated with the majority of reads used to construct Pelagiporibacteria genomes, but could be retained to support adaptive flexibility for living closer to the ocean surface.
Entoporibacteria genomes contained multiple toxin–antitoxin gene families that were absent from Pelagiporibacteria, including type II pairs MazE/MazF, ParDE, RelBE, HicA-HicB, BrnA/BrT, Phd/YefM, as well as the type IV AbiE system. Toxin–antitoxin modules often control transcriptional and translational regulation, causing apoptotic self-destruction if a toxin is expressed without co-expression of its cognate antitoxin. Originally discovered for their role in plasmid retention, toxin–antitoxin systems have recently been shown promote the creation and maintenance of “persister” populations, able to survive environmental stresses such as phage infection, antibiotic challenge, and host immune response through temporary dormancy (reviewed in ref. [75]).
Over-represented gene families
Potentially adaptive genomic characteristics can sometimes be inferred by quantitative expansion of functionally characterized gene families, beyond the simple presence or absence of individual proteins. Highly over-represented gene families in Entoporibacteria and Pelagiporibacteria are compared in Fig. 5. Copy numbers for families associated with cell surface adhesion were consistently enriched in Entoporibacteria, especially leucine-rich repeat and dockerin domains. Dockerins have been shown to pair with cohesin domains in the assembly of extracellular compartments called cellulosomes, anchoring fibronectin-domain containing, polysaccharide-degrading enzyme complexes in terrestrial bacteria [76]. Although cellulosomes are not commonly found outside phylum Firmicutes or in marine bacteria, they have been recently reported in candidate phylum Marinimicrobia MAGs from an oxygen minimum zone, where they are proposed to participate in recycling of high molecular weight carbon compounds [77].
Arylsulfatases are highly abundant in both Entoporibacteria and Pelagiporibacteria genomes, but determining the extent to which these enzymes participate in degradative versus synthetic pathways is difficult without experimental verification. Like many known complex polysaccharide-degrading bacteria, Poribacteria genomes contain abundant glycoside hydrolases and polysaccharide lyases. Some Poribacteria sulfatases may be acting in concert with these enzymes to enable the conversion of complex, sulfated polysaccharides into simpler monosaccharides and oligosaccharides that can be fed into other pathways for energy production [78] and/or nutrient acquisition. This metabolic strategy is consistent with the discovery of Pelagiporibacteria in sulfur-rich, black smoker hydrothermal vent plumes [20].
Poribacteria arylsulfatases may also have a role in tailoring sulfated polysaccharide compounds synthesized for extracellular surface display. This interpretation is supported by conserved operon structures in multiple Entoporibacteria genomes where arylsulfatases occur in tandem repeats adjacent to predicted lipopolysaccharide glycosyltransferase and capsular assembly proteins, along with multiple sulfotransferases, glycotransferases, and leucine-rich repeat proteins (Fig. 6). Gene neighborhoods for capsular assembly proteins in Pelagiporibacteria genomes are much less conserved, and do not include arylsulfatases or sulfotransferases.
Fig. 6.
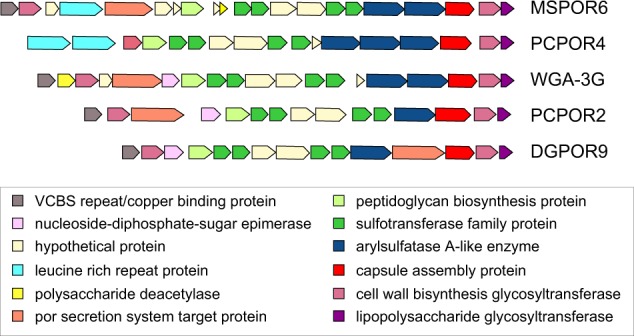
Conserved Entoporibacteria extracellular polysaccharide capsule assembly operon. Functional assignments for predicted proteins are based on the IMG-MER annotation pipeline [46]
A large number of Poribacteria arylsulfatases are predicted to encode enzymes with choline sulfatase activity, a key component of both synthetic and degradative pathways for glycine betaine. This compound is used as a compatible solute to counter osmotic dehydration stress in bacteria and some archaea [79]. Some Poribacteria choline sulfatases are located adjacent to predicted ectoine hydroxylase family proteins, which may be involved in metabolism of the osmolyte hydroxyectoine [80]. The presence of predicted transporters for both betaine and ectoine combined with the absence of canonical synthetic operons for these compounds suggests their potential use as nutritional resources when not required for osmoregulation.
DNA defense-related gene families for restriction endonucleases and DNA methylases were dramatically increased in Entoporibacteria (Fig. 5), consistent with previous observations of enrichment in pooled Aplysina aerophoba sponge metagenomes containing Poribacteria [5]. These results suggest potentially greater exposure to phage predation, further supported by elevated numbers of transposases and CRISPR repeat domains. Increased DNA defense-related gene abundance is consistent with the greatly expanded repertoire of type II toxin–antitoxin pairs in Entoporibacteria, potentially creating persister cells capable of surviving population-wide viral sweeps. The recovery of five completely independent Entoporibacteria genomes from a single Pseudoceratina sp. sponge sample (PCPOR1, PCPOR2, PCPOR2a, PCPOR2b, and PCPOR4) also supports a model of environmentally selective genome modification pressure.
Relative abundance of Poribacteria in sponge hosts
Bacterial community abundance measurements based on 16S rRNA genes are known to over-report taxonomic groups with higher rRNA gene copy numbers, while measurements based on unamplified metagenomic reads can be impacted by differing genome sizes. Both methods are susceptible to artifacts arising from database incompleteness, preventing classification of novel species. These biases cannot readily be corrected in environmental samples containing unidentified taxa, where 16S rRNA gene copy number and genome sizes for many community members are unknown. To address these potential limitations, sponge microbial community abundances for Poribacteria were analyzed by comparing results utilizing both techniques.
Measurements of Poribacteria relative abundance based on 16S rRNA gene analysis were approximately fivefold higher with newly designed primer set 515Fsp/806Rsp compared to “universal” V4 region primer set 515FB/806RB (Fig. 7). To maintain a total value of 100%, relative abundances for non-Poribacteria taxonomic groups were correspondingly reduced (Supplementary Figure S5). It was not possible to determine whether differences in non-Poribacteria abundance might be due to discriminatory bias by one primer set or the other, because the true, unbiased taxonomic composition of these natural samples is unknown. For sponge samples from Pseudoceratina, Dysidea, and Melophlus, Poribacteria abundance gains with the new primers were more than threefold larger than corresponding losses in any other individual taxonomic group. The gain in Poribacteria abundance with primer set 515Fsp/806Rsp was not larger than corresponding losses in other taxa for the Agelas sample, perhaps linked to low Poribacteria abundance.
Fig. 7.
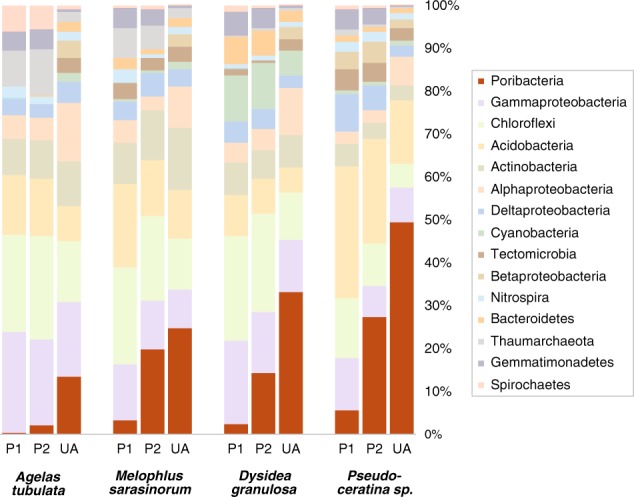
Relative abundance of Poribacteria in sponge microbial communities. P1 16S primer set 515FB/806RB (Earth Microbiome Project), P2 16S primer set 515Fsp/806Rsp (sponge-poribacteria adjusted), UA unamplified metagenomic reads, mapped to database protein sequences using the DarkHorse algorithm [42]
Impaired Poribacteria detection with primer set 515FB/806RB was further corroborated by relative abundance values that were 8- to 10-fold lower than those obtained using unamplified metagenomic reads for samples from Pseudoceratina, Dysidea, and Melophlus, versus only 1.5- to 1.8-fold lower for these same samples using primer set 515Fsp/806Rsp. In the Agelas sample, Poribacteria abundances with the 515FB/806RB primers were 37-fold lower than metagenomic estimates, versus 6-fold lower with primer set 515Fsp/806Rsp. These results are consistent with previously reported non-linearity of primer bias effects [81, 82], which may be exaggerated at lower relative abundance levels.
The 515FB and 806RB primers each contain nucleotide mismatches to some (806RB) or all (515FB) 16S rRNA genes from assembled Poribacteria genomes (Supplementary Figure S6a–c). These mismatches apparently hindered, but did not completely abolish amplification in the four sponge samples tested here, perhaps reflecting clade-specific alignment differences to primer 806RB (Supplementary Figure S6a) that might allow some Poribacteria strains to be more easily detected than others. All known mismatches have been corrected in primer pair 515Fsp/806Rsp, but many other widely used primers, including historical favorite 27F [83], contain two or more mismatches to Poribacteria 16S rRNA genes, making successful amplification highly unlikely (Supplementary Figure S6a).
Potential environmental adaptations
This work has shown that environmental habitats define the division of Poribacteria into two taxonomically distinct lineages, designated as Entoporibacteria, associated with marine sponge tissues, and Pelagiporibacteria from the open ocean. The availability of multiple genomes from both lineages has not only enabled recognition of shared versus unique gene families, metabolic pathways, and conserved operon structures, but also the discovery of potentially adaptive quantitative differences in functional capacities between the two groups.
Enzymes associated with the hydrolysis of sulfated glycan polymers from eukaryotic tissue and/or particulate organic matter are abundant features in both Entoporibacteria and Pelagiporibacteria genomes. Suitable substrates should be present in seawater filtrates collected by sponge hosts, as well as sinking particles in the open ocean [84]. Pelagiporibacteria genomes encode flagellar structures and chemotaxis machinery for sensing and responding to environmental chemical gradients, but Entoporibacteria have lost this capability. They may compensate for their immotility by embedding themselves in host tissues via extracellular matrix degrading enzymes, potentially using the resulting products for heterotrophic growth, and simultaneously reducing the risk of being washed away by host seawater pumping action. Phosphoinositol-linked glycolipids and capsular exopolysaccharides can create impermeable membrane barriers that mimic eukaryotic tissue surface chemistry [85], potentially allowing Entoporibacteria to avoid recognition by host phagocytes. Although all sponge specimens analyzed in this study appeared healthy at the time of collection, it is not known whether Poribacteria elicit any negative consequences for their hosts. Sponge-associated Poribacteria may act as commensal symbionts, or possibly opportunistic pathogens under appropriate circumstances.
Non-motile symbiotic microbes associated with sessile, benthic organisms may face additional challenges in escaping viral predators and dispersing to new locations in the event of host death. Evidence for the toll of phage predation on Entoporibacteria includes the coexistence of multiple closely related strains within the same host, along with greatly elevated numbers of transposases and CRISPR repeats within their genomes. Genomic evidence suggests that phage predation pressure may be addressed through the expansion of restriction endonuclease gene families and the acquisition of multiple toxin–antitoxin pairs enabling possible self-initiated reduction in population sizes, as well as the creation of inactive cells with a persister phenotype to enhance survival during viral blooms.
Many marine sponges can reproduce by tissue fragmentation [86–88], enabling embedded or tightly adhering microbiome components to be passed vertically during reproduction and dispersal. Dispersal of both Entoporibacteria and Pelagiporibacteria to new environments might also be facilitated by the formation of endospores, accumulation of osmoprotective ectoines and glycine betaines, and the formation of capsules to protect cells from environmental stresses. Microbial species inhabiting the marine sponge Haliclona have been proposed to use sporulation as a mechanism to survive ingestion by host phagocytes, which subsequently redistribute the engulfed bacteria to additional sites within the same host [89]. Whether or not actual spores are formed, resistance to eukaryotic digestive processes could provide an additional dispersal mechanism in the event that Poribacteria-containing tissues or marine particles are ingested by fish or invertebrate grazers.
Entoporibacteria and Pelagiporibacteria genomes both contain a large number of factors typically linked to a eukaryotic host-associated lifestyle. These include enhanced adhesion to eukaryotic cell surfaces; digestion of host tissue via heparinases [90], neuraminidases [91], ceramidases [92], and chitinases [93]; the incorporation of phosphoinositol-linked glycolipids into cell membranes [94]; the formation of exopolysaccharide capsules containing host cell surface-mimicking domains [85, 95]; the production of mycothiol to undermine oxidative host defenses [72]; and the targeting of pupylated substrates to bacterial proteosomes, previously shown to be a mechanism for inactivating lysosomal enzymes after engulfment by host macrophages [96]. The suggestion that much of the sinking particulate matter in the open ocean originates from marine eukaryotic hosts [84], offers an intriguing possible explanation for the unexpected abundance of functional activities characteristic of host association in Pelagiporibacteria genomes. Multiple resistance mechanisms to protect cells from ingestion by particle grazers may further contribute to survival and dispersal of Pelagiporibacteria in the water column. The presence of Pelagiporibacteria in hydrothermal vent plumes 2000 m deep suggests these organisms may be capable of adapting to a wide range of different depths.
Poribacteria may be more abundant in marine environments than previously appreciated. Historical studies relying exclusively on mismatched primers such as 27F and 515FB/806RB to quantify Poribacteria in environmental samples may have failed to detect these organisms due to amplification bias. These findings are particularly relevant given on-going collaborative efforts to catalog marine microbial diversity, including the Earth Microbiome Project [97] and the Global Sponge Microbiome Project [98]. More sensitive and accurate determination of Poribacteria abundance and distribution in marine habitats will undoubtedly provide new opportunities to assess their contributions to microbial ecosystem functions in places where they may have been previously overlooked.
Electronic supplementary material
Acknowledgements
We thank Dr. Michelle Schorn (Scripps Institution of Oceanography) and Dr. Eduardo Esquenazi and the Sirenas collection team (Sirenas Marine Discovery LLC) for sponge sample collection assistance. This work was supported by grants from NSF (MCB-1149552) to EEA and NSF (OCE-1313747) and NIEHS (P01-ES021921) to BSM and EEA.
Compliance with ethical standards
Conflict of interest
The authors declare that they have no conflict of interest.
Electronic supplementary material
The online version of this article (10.1038/s41396-018-0292-9) contains supplementary material, which is available to authorized users.
References
- 1.Fieseler L, Horn M, Wagner M, Hentschel U. Discovery of the novel candidate phylum “Poribacteria” in marine sponges. Appl Environ Microbiol. 2004;70:3724–32. doi: 10.1128/AEM.70.6.3724-3732.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Schmitt S, Angermeier H, Schiller R, Lindquist N, Hentschel U. Molecular microbial diversity survey of sponge reproductive stages and mechanistic insights into vertical transmission of microbial symbionts. Appl Environ Microbiol. 2008;74:7694–708. doi: 10.1128/AEM.00878-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Webster NS, Taylor MW, Behnam F, Lucker S, Rattei T, Whalan S, et al. Deep sequencing reveals exceptional diversity and modes of transmission for bacterial sponge symbionts. Environ Microbiol. 2010;12:2070–82. doi: 10.1111/j.1462-2920.2009.02065.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kamke J, Rinke C, Schwientek P, Mavromatis K, Ivanova N, Sczyrba A, et al. The candidate phylum Poribacteria by single-cell genomics: new insights into phylogeny, cell-compartmentation, eukaryote-like repeat proteins, and other genomic features. PLoS ONE. 2014;9:e87353. doi: 10.1371/journal.pone.0087353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Slaby BM, Hackl T, Horn H, Bayer K, Hentschel U. Metagenomic binning of a marine sponge microbiome reveals unity in defense but metabolic specialization. ISME J. 2017;11:2465–78. doi: 10.1038/ismej.2017.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tully BJ, Graham ED, Heidelberg JF. The reconstruction of 2631 draft metagenome-assembled genomes from the global oceans. Sci Data. 2018;5:170203. doi: 10.1038/sdata.2017.203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jahn MT, Markert SM, Ryu T, Ravasi T, Stigloher C, Hentschel U, et al. Shedding light on cell compartmentation in the candidate phylum Poribacteria by high resolution visualisation and transcriptional profiling. Sci Rep. 2016;6:35860. doi: 10.1038/srep35860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kamke J, Sczyrba A, Ivanova N, Schwientek P, Rinke C, Mavromatis K, et al. Single-cell genomics reveals complex carbohydrate degradation patterns in poribacterial symbionts of marine sponges. ISME J. 2013;7:2287–300. doi: 10.1038/ismej.2013.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Siegl A, Kamke J, Hochmuth T, Piel J, Richter M, Liang C, et al. Single-cell genomics reveals the lifestyle of Poribacteria, a candidate phylum symbiotically associated with marine sponges. ISME J. 2011;5:61–70. doi: 10.1038/ismej.2010.95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fuerst JA, Webb RI, Garson MJ, Hardy L, hm R. Membrane-bounded nuclear bodies in a diverse range of microbial symbionts of Great Barrier Reef sponges. Mem Qld Mus. 1999;44:193–203. [Google Scholar]
- 11.Erwin PM, Pita L, Lopez-Legentil S, Turon X. Stability of sponge-associated bacteria over large seasonal shifts in temperature and irradiance. Appl Environ Microbiol. 2012;78:7358–68. doi: 10.1128/AEM.02035-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gao ZM, Zhou GW, Huang H, Wang Y. The cyanobacteria-dominated sponge Dactylospongia elegans in the South China Sea: prokaryotic community and metagenomic insights. Front Microbiol. 2017;8:1387. doi: 10.3389/fmicb.2017.01387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jensen S, Fortunato SA, Hoffmann F, Hoem S, Rapp HT, Ovreas L, et al. The relative abundance and transcriptional activity of marine sponge-associated microorganisms emphasizing groups involved in sulfur cycle. Microb Ecol. 2017;73:668–76. doi: 10.1007/s00248-016-0836-3. [DOI] [PubMed] [Google Scholar]
- 14.Karlinska-Batres K, Worheide G. Phylogenetic diversity and community structure of the symbionts associated with the coralline sponge Astrosclera willeyana of the Great Barrier Reef. Microb Ecol. 2013;65:740–52. doi: 10.1007/s00248-013-0212-5. [DOI] [PubMed] [Google Scholar]
- 15.Karlinska-Batres K, Worheide G. Microbial diversity in the coralline sponge Vaceletia crypta. Antonie Van Leeuwenhoek. 2013;103:1041–56. doi: 10.1007/s10482-013-9884-6. [DOI] [PubMed] [Google Scholar]
- 16.Lafi FF, Fuerst JA, Fieseler L, Engels C, Goh WW, Hentschel U. Widespread distribution of poribacteria in demospongiae. Appl Environ Microbiol. 2009;75:5695–9. doi: 10.1128/AEM.00035-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mohamed NM, Saito K, Tal Y, Hill RT. Diversity of aerobic and anaerobic ammonia-oxidizing bacteria in marine sponges. ISME J. 2010;4:38–48. doi: 10.1038/ismej.2009.84. [DOI] [PubMed] [Google Scholar]
- 18.Montalvo NF, Hill RT. Sponge-associated bacteria are strictly maintained in two closely related but geographically distant sponge hosts. Appl Environ Microbiol. 2011;77:7207–16. doi: 10.1128/AEM.05285-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Thomas T, Moitinho-Silva L, Lurgi M, Bjork JR, Easson C, Astudillo-Garcia C, et al. Diversity, structure and convergent evolution of the global sponge microbiome. Nat Commun. 2016;7:11870. doi: 10.1038/ncomms11870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Anantharaman K, Breier JA, Dick GJ. Metagenomic resolution of microbial functions in deep-sea hydrothermal plumes across the Eastern Lau Spreading Center. ISME J. 2016;10:225–39. doi: 10.1038/ismej.2015.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pham VD, Konstantinidis KT, Palden T, DeLong EF. Phylogenetic analyses of ribosomal DNA-containing bacterioplankton genome fragments from a 4000 m vertical profile in the North Pacific Subtropical Gyre. Environ Microbiol. 2008;10:2313–30. doi: 10.1111/j.1462-2920.2008.01657.x. [DOI] [PubMed] [Google Scholar]
- 22.Taylor MW, Tsai P, Simister RL, Deines P, Botte E, Ericson G, et al. ‘Sponge-specific’ bacteria are widespread (but rare) in diverse marine environments. ISME J. 2013;7:438–43. doi: 10.1038/ismej.2012.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hardoim CC, Cox CJ, Peixoto RS, Rosado AS, Costa R, van Elsas JD. Diversity of the candidate phylum Poribacteria in the marine sponge Aplysina fulva. Braz J Microbiol. 2013;44:329–34. doi: 10.1590/S1517-83822013000100048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Steinert G, Gutleben J, Atikana A, Wijffels RH, Smidt H, Sipkema D. Coexistence of poribacterial phylotypes among geographically widespread and phylogenetically divergent sponge hosts. Environ Microbiol Rep. 2018;10:80–91. [DOI] [PubMed]
- 25.Germer J, Cerveau N, Jackson DJ. The holo-transcriptome of a calcified early branching metazoan. Front Mar Sci. 2017;4:81.
- 26.Bayer K, Kamke J, Hentschel U. Quantification of bacterial and archaeal symbionts in high and low microbial abundance sponges using real-time PCR. FEMS Microbiol Ecol. 2014;89:679–90. doi: 10.1111/1574-6941.12369. [DOI] [PubMed] [Google Scholar]
- 27.Moitinho-Silva L, Bayer K, Cannistraci CV, Giles EC, Ryu T, Seridi L, et al. Specificity and transcriptional activity of microbiota associated with low and high microbial abundance sponges from the Red Sea. Mol Ecol. 2014;23:1348–63. doi: 10.1111/mec.12365. [DOI] [PubMed] [Google Scholar]
- 28.Hentschel U, Hopke J, Horn M, Friedrich AB, Wagner M, Hacker J, et al. Molecular evidence for a uniform microbial community in sponges from different oceans. Appl Environ Microbiol. 2002;68:4431–40. doi: 10.1128/AEM.68.9.4431-4440.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Eloe-Fadrosh EA, Ivanova NN, Woyke T, Kyrpides NC. Metagenomics uncovers gaps in amplicon-based detection of microbial diversity. Nat Microbiol. 2016;1:15032. doi: 10.1038/nmicrobiol.2015.32. [DOI] [PubMed] [Google Scholar]
- 30.Illumina (2013). 16S metagenomic sequencing library preparation guide, version 15044223B https://support.illumina.com/downloads/16s_metagenomic_sequencing_library_preparation.html.
- 31.Earth_Microbiome_Project (2016). Earth microbiome project protocols and standards: 16S Illumina amplicon protocol http://press.igsb.anl.gov/earthmicrobiome/protocols-and-standards/16s/.
- 32.Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30:2114–20. doi: 10.1093/bioinformatics/btu170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK, et al. QIIME allows analysis of high-throughput community sequencing data. Nat Methods. 2010;7:335–6. doi: 10.1038/nmeth.f.303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Callahan BJ, McMurdie PJ, Rosen MJ, Han AW, Johnson AJ, Holmes SP. DADA2: High-resolution sample inference from Illumina amplicon data. Nat Methods. 2016;13:581–3. doi: 10.1038/nmeth.3869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pedregosa F, Varoquaux G, Gramfort A, Michel V, Thirion B, Grisel O, et al. Scikit-learn: Machine Learning in Python. J Mach Learn Res. 2011;12:2825–30. [Google Scholar]
- 36.Quast C, Pruesse E, Yilmaz P, Gerken J, Schweer T, Yarza P, et al. The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res. 2013;41:D590–6. doi: 10.1093/nar/gks1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.McMurdie PJ, Holmes S. phyloseq: an R package for reproducible interactive analysis and graphics of microbiome census data. PLoS ONE. 2013;8:e61217. doi: 10.1371/journal.pone.0061217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Paulson JN, Stine OC, Bravo HC, Pop M. Differential abundance analysis for microbial marker-gene surveys. Nat Methods. 2013;10:1200–2. doi: 10.1038/nmeth.2658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Peng Y, Leung HC, Yiu SM, Chin FY. IDBA-UD: a de novo assembler for single-cell and metagenomic sequencing data with highly uneven depth. Bioinformatics. 2012;28:1420–8. doi: 10.1093/bioinformatics/bts174. [DOI] [PubMed] [Google Scholar]
- 40.Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods. 2012;9:357–9. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078–9. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Podell S, Gaasterland T. DarkHorse: a method for genome-wide prediction of horizontal gene transfer. Genome Biol. 2007;8:R16. doi: 10.1186/gb-2007-8-2-r16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Podell S. DarkHorse2 GitHub repository. 2017 https://github.com/spodell/Darkhorse2.
- 44.Podell S, Ugalde JA, Narasingarao P, Banfield JF, Heidelberg KB, Allen EE. Assembly-driven community genomics of a hypersaline microbial ecosystem. PLoS ONE. 2013;8:e61692. doi: 10.1371/journal.pone.0061692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Myers EW, Sutton GG, Delcher AL, Dew IM, Fasulo DP, Flanigan MJ, et al. A whole-genome assembly of Drosophila. Science. 2000;287:2196–204. doi: 10.1126/science.287.5461.2196. [DOI] [PubMed] [Google Scholar]
- 46.Markowitz VM, Chen IM, Chu K, Szeto E, Palaniappan K, Grechkin Y, et al. IMG/M: the integrated metagenome data management and comparative analysis system. Nucleic Acids Res. 2012;40:D123–129. doi: 10.1093/nar/gkr975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sunagawa S, Coelho LP, Chaffron S, Kultima JR, Labadie K, Salazar G, et al. Ocean plankton. Structure and function of the global ocean microbiome. Science. 2015;348:1261359. doi: 10.1126/science.1261359. [DOI] [PubMed] [Google Scholar]
- 48.Tully BJ, Graham ED, Heidelberg JF. The reconstruction of 2631 draft metagenome-assembled genomes from the global oceans. figshare. Fileset. 10.6084/m9.figshare.5188273.v13. 2017.
- 49.Seemann T. Prokka: rapid prokaryotic genome annotation. Bioinformatics. 2014;30:2068–9. doi: 10.1093/bioinformatics/btu153. [DOI] [PubMed] [Google Scholar]
- 50.Bland C, Ramsey TL, Sabree F, Lowe M, Brown K, Kyrpides NC, et al. CRISPR recognition tool (CRT): a tool for automatic detection of clustered regularly interspaced palindromic repeats. BMC Bioinformatics. 2007;8:209. doi: 10.1186/1471-2105-8-209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Parks DH, Imelfort M, Skennerton CT, Hugenholtz P, Tyson GW. CheckM: assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 2015;25:1043–55. doi: 10.1101/gr.186072.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Finn RD, Coggill P, Eberhardt RY, Eddy SR, Mistry J, Mitchell AL, et al. The Pfam protein families database: towards a more sustainable future. Nucleic Acids Res. 2016;44:D279–85. doi: 10.1093/nar/gkv1344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Haft DH, Selengut JD, Richter RA, Harkins D, Basu MK, Beck E. TIGRFAMs and genome properties in 2013. Nucleic Acids Res. 2013;41:D387–395. doi: 10.1093/nar/gks1234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Pruesse E, Peplies J, Glockner FO. SINA: accurate high-throughput multiple sequence alignment of ribosomal RNA genes. Bioinformatics. 2012;28:1823–9. doi: 10.1093/bioinformatics/bts252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004;32:1792–7. doi: 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Price MN, Dehal PS, Arkin AP. FastTree 2—approximately maximum-likelihood trees for large alignments. PLoS ONE. 2010;5:e9490. doi: 10.1371/journal.pone.0009490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Rambaut A. Molecular evolution, phylogenetics and epidemiology. 2016 http://tree.bio.ed.ac.uk/software/figtree/.
- 58.Rodriguez-R L, Konstantinidis K. The enveomics collection: a toolbox for specialized analyses of microbial genomes and metagenomes. PeerJ Prepr. 2016;4:e1900v1. [Google Scholar]
- 59.Rodriguez-R LM, Konstantinos TK. Bypassing cultivation to identify bacterial species. Microbe. 2014;9:111–8. [Google Scholar]
- 60.Lechner M, Findeiss S, Steiner L, Marz M, Stadler PF, Prohaska SJ. Proteinortho: detection of (co-)orthologs in large-scale analysis. BMC Bioinformatics. 2011;12:124. doi: 10.1186/1471-2105-12-124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.R Core Team. R: a language and environment for statistical computing. Vienna, Austria: R Foundation for Statistical Computing; 2015.
- 62.Micallef L, Rodgers P. eulerAPE: drawing area-proportional 3-Venn diagrams using ellipses. PLoS ONE. 2014;9:e101717. doi: 10.1371/journal.pone.0101717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Rice P, Longden I, Bleasby A. EMBOSS: the European Molecular Biology Open Software Suite. Trends Genet. 2000;16:276–7. doi: 10.1016/S0168-9525(00)02024-2. [DOI] [PubMed] [Google Scholar]
- 64.Leinonen R, Sugawara H, Shumway M, International Nucleotide Sequence Database Collaboration. The sequence read archive. Nucleic Acids Res. 2011;39:D19–21. doi: 10.1093/nar/gkq1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Bowers RM, Kyrpides NC, Stepanauskas R, Harmon-Smith M, Doud D, Reddy TBK, et al. Minimum information about a single amplified genome (MISAG) and a metagenome-assembled genome (MIMAG) of bacteria and archaea. Nat Biotechnol. 2017;35:725–31. doi: 10.1038/nbt.3893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.McCutcheon JP, Moran NA. Extreme genome reduction in symbiotic bacteria. Nat Rev Microbiol. 2011;10:13–26. doi: 10.1038/nrmicro2670. [DOI] [PubMed] [Google Scholar]
- 67.Sheik CS, Anantharaman K, Breier JA, Sylvan JB, Edwards KJ, Dick GJ. Spatially resolved sampling reveals dynamic microbial communities in rising hydrothermal plumes across a back-arc basin. ISME J. 2015;9:1434–45. doi: 10.1038/ismej.2014.228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Konstantinidis KT, Rossello-Mora R, Amann R. Uncultivated microbes in need of their own taxonomy. ISME J. 2017;11:2399–406. doi: 10.1038/ismej.2017.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Stackebrandt E, Goebel BM. Taxonomic note: a place for DNA-DNA reassociation and 16S rRNA sequence analysis in the present species definition in bacteriology. Int J Syst Evolut Microbiol. 1994;44:846–9. doi: 10.1099/00207713-44-4-846. [DOI] [Google Scholar]
- 70.Morii H, Ogawa M, Fukuda K, Taniguchi H. Ubiquitous distribution of phosphatidylinositol phosphate synthase and archaetidylinositol phosphate synthase in Bacteria and Archaea, which contain inositol phospholipid. Biochem Biophys Res Commun. 2014;443:86–90. doi: 10.1016/j.bbrc.2013.11.054. [DOI] [PubMed] [Google Scholar]
- 71.Whitfield C, Szymanski CM, Aebi M. Eubacteria, Ch 21. Essentials of glycobiology, 3rd ed. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 2017. 823 p.
- 72.Fahey RC. Novel thiols of prokaryotes. Annu Rev Microbiol. 2001;55:333–56. doi: 10.1146/annurev.micro.55.1.333. [DOI] [PubMed] [Google Scholar]
- 73.Madigan MT. Brock biology of microorganisms. 13th ed. San Francisco: Benjamin Cummings; 2012. p. 521.
- 74.Galperin MY, Mekhedov SL, Puigbo P, Smirnov S, Wolf YI, Rigden DJ. Genomic determinants of sporulation in Bacilli and Clostridia: towards the minimal set of sporulation-specific genes. Environ Microbiol. 2012;14:2870–90. doi: 10.1111/j.1462-2920.2012.02841.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Harms A, Brodersen DE, Mitarai N, Gerdes K. Toxins, targets, and triggers: an overview of toxin-antitoxin biology. Mol Cell. 2018;70:768–84. [DOI] [PubMed]
- 76.Artzi L, Bayer EA, Morais S. Cellulosomes: bacterial nanomachines for dismantling plant polysaccharides. Nat Rev Microbiol. 2017;15:83–95. doi: 10.1038/nrmicro.2016.164. [DOI] [PubMed] [Google Scholar]
- 77.Bertagnolli AD, Padilla CC, Glass JB, Thamdrup B, Stewart FJ. Metabolic potential and in situ activity of marine Marinimicrobia bacteria in an anoxic water column. Environ Microbiol. 2017;19:4392–416. doi: 10.1111/1462-2920.13879. [DOI] [PubMed] [Google Scholar]
- 78.Helbert W. Marine polysaccharide sulfatases. Front Mar Sci. 2017;4:6. doi: 10.3389/fmars.2017.00006. [DOI] [Google Scholar]
- 79.Welsh DT. Ecological significance of compatible solute accumulation by micro-organisms: from single cells to global climate. FEMS Microbiol Rev. 2000;24:263–90. doi: 10.1111/j.1574-6976.2000.tb00542.x. [DOI] [PubMed] [Google Scholar]
- 80.Widderich N, Hoppner A, Pittelkow M, Heider J, Smits SH, Bremer E. Biochemical properties of ectoine hydroxylases from extremophiles and their wider taxonomic distribution among microorganisms. PLoS ONE. 2014;9:e93809. doi: 10.1371/journal.pone.0093809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Bru D, Martin-Laurent F, Philippot L. Quantification of the detrimental effect of a single primer-template mismatch by real-time PCR using the 16S rRNA gene as an example. Appl Environ Microbiol. 2008;74:1660–3. doi: 10.1128/AEM.02403-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Parada AE, Needham DM, Fuhrman JA. Every base matters: assessing small subunit rRNA primers for marine microbiomes with mock communities, time series and global field samples. Environ Microbiol. 2016;18:1403–14. doi: 10.1111/1462-2920.13023. [DOI] [PubMed] [Google Scholar]
- 83.Lane DJ. 16S/23S rRNA sequencing. In: Stackebrandt E, Goodfellow M, editors. Nucleic acid techniques in bacterial systematics. New York, NY: John Wiley and Sons; 1991. pp. 115–75. [Google Scholar]
- 84.Pelve EA, Fontanez KM, DeLong EF. Bacterial succession on sinking particles in the ocean’s interior. Front Microbiol. 2017;8:2269. doi: 10.3389/fmicb.2017.02269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Cress BF, Englaender JA, He W, Kasper D, Linhardt RJ, Koffas MA. Masquerading microbial pathogens: capsular polysaccharides mimic host-tissue molecules. FEMS Microbiol Rev. 2014;38:660–97. doi: 10.1111/1574-6976.12056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Ayling AL. Patterns of sexuality, asexual reproduction and recruitment in some subtidal marine demospongiae. Biol Bull. 1980;158:271–82. doi: 10.2307/1540854. [DOI] [Google Scholar]
- 87.Bergquist PR. Sponges. London: Hutchinson; 1978. [Google Scholar]
- 88.Wulff JL. Asexual fragmentation, genotype success, and population dynamics of erect branching sponges. J, Exp Mar Biol Ecol. 1991;149:227–47. doi: 10.1016/0022-0981(91)90047-Z. [DOI] [Google Scholar]
- 89.Phelan RW, O’Halloran JA, Kennedy J, Morrissey JP, Dobson AD, O’Gara F, et al. Diversity and bioactive potential of endospore-forming bacteria cultured from the marine sponge Haliclona simulans. J Appl Microbiol. 2012;112:65–78. doi: 10.1111/j.1365-2672.2011.05173.x. [DOI] [PubMed] [Google Scholar]
- 90.Dzvova N, Colmer-Hamood JA, Griswold JA, Hamood AN. Heparinase is essential for pseudomonas aeruginosa virulence during thermal injury and infection. Infect Immun. 2018;86:e00755-17. [DOI] [PMC free article] [PubMed]
- 91.Ghazaei C. Neuraminidase function in bacteria as a virulence factor. J PURE Appl Microbiol. 2013;7:1447–50. [Google Scholar]
- 92.Inoue H, Someno T, Kawada M, Ikeda D. Citric acid inhibits a bacterial ceramidase and alleviates atopic dermatitis in an animal model. J Antibiot. 2010;63:611–3. doi: 10.1038/ja.2010.91. [DOI] [PubMed] [Google Scholar]
- 93.Frederiksen RF, Paspaliari DK, Larsen T, Storgaard BG, Larsen MH, Ingmer H, et al. Bacterial chitinases and chitin-binding proteins as virulence factors. Microbiology. 2013;159:833–47. doi: 10.1099/mic.0.051839-0. [DOI] [PubMed] [Google Scholar]
- 94.Jackson M. The mycobacterial cell envelope-lipids. Cold Spring Harb Perspect Med. 2014;4:a021105. [DOI] [PMC free article] [PubMed]
- 95.Taylor CM, Roberts IS. Capsular polysaccharides and their role in virulence. Contrib Microbiol. 2005;12:55–66. doi: 10.1159/000081689. [DOI] [PubMed] [Google Scholar]
- 96.Becker SH, Darwin KH. Bacterial Proteasomes: Mechanistic and Functional Insights. Microbiol Mol Biol Rev. 2017;81:e00036-16. [DOI] [PMC free article] [PubMed]
- 97.Thompson LR, Sanders JG, McDonald D, Amir A, Ladau J, Locey KJ, et al. A communal catalogue reveals Earth’s multiscale microbial diversity. Nature. 2017;551:457–63. doi: 10.1038/551033a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Moitinho-Silva L, Nielsen S, Amir A, Gonzalez A, Ackermann GL, Cerrano C, et al. The sponge microbiome project. GigaScience. 2017;6:1–7. [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.