

Abstract
Neuronal voltage‐gated potassium channels KV7.2/KV7.3 are sensitive to small‐molecule drugs such as flupirtine, even though physiological response occurs in the absence of ligands. Clinically, prolonged use of flupirtine as a pain medication is associated with rare cases of drug‐induced liver injury. Thus, safety concerns prevent a broader use of this non‐opioid and non‐steroidal analgesic in therapeutic areas with unmet medical needs such as hyperactive bladder or neonatal seizures. With the goal of studying influences of chemical structure on activity and toxicity of flupirtine, we explored modifications of the benzylamino bridge and the substitution pattern in both rings of flupirtine. Among twelve derivatives, four novel thioether derivatives showed the desired activity in cellular assays and may serve as leads for safer KV channel openers.
Keywords: medicinal chemistry, ion channels, oxidation, structure-activity relationships, sulfides
Flupirtine (1) is a non‐opioid and non‐steroidal, centrally acting analgesic with a unique mode of action. The analgesic effect of 1 is believed to be associated with the opening of hetero‐tetrameric voltage‐gated potassium channels, consisting of the subunits KV7.2 and KV7.3, which are encoded by KCNQ2 and KCNQ3, leading to membrane potential stabilization and decreased excitability.1 The KV7.2/KV7.3 channel generates M‐currents that control the subthreshold excitability of the cell membrane, therefore, drugs that stabilize the open state of KV7.2/KV7.3 channels could be used in a broad range of CNS diseases that are characterized by neuronal over‐activity, including pain, stress, anxiety and epilepsy. While 1 is regarded safe in short‐term use for acute pain, a recent clinical study provided indirect evidence that 1 can be oxidized in healthy volunteers to unstable ortho‐ or para‐azaquinone diimines 3 a and/or b (Figure 1) as reactive metabolites.2 While neither compound 3 a nor 3 b could be identified directly, their formation was deduced from presence of cysteine metabolites in biological fluids,2 formed by reactions of the reactive intermediates with glutathione to yield adducts such as 4. While it is not known whether reactive electrophiles 3 a or 3 b are the causative agent for drug‐induced liver injury (DILI), safety issues lead to a termination of a clinical study with 1 in hyperactive bladder in 2013.3
Figure 1.
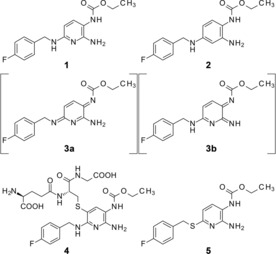
Structure of KV channel openers 1 and 2, elusive metabolites 3 a and b, product 4 of phase II drug metabolism, and direct thio‐analogue 5 of 1 used for theoretical comparison in Figure 2 B).
Although 1 was approved for use as a centrally acting analgesic in a number of European countries until 2018, it was not approved as an anticonvulsant. Two recent animal studies however have demonstrated that 1 terminates seizures in neonatal mammals effectively, and that a combination of 1 and diazepam is superior to diazepam alone.4 Because the closely‐related KV channel opener retigabine (2) had been used as antiepileptic, until it was also withdrawn from the market in 2017, this additional indication for 1 would seem a logical endeavor. To date, treatment options for severe cases of status epilepticus are scarce.
Follow‐up substance 2 marketed as Trobalt® has been taken of the market by GSK, last year. One of the main reasons was a blueish tissue discoloration.5 While the exact structure of the blue dye could not be resolved, it is most likely that the formation of polymers of 2 is the cause for this adverse effect. Based on earlier findings that 1 and its metabolites can only be traced to a limited extent in excretes, we speculate that diimines such as 3 a and b could form insoluble polymers in vivo, as well. This hypothesis is supported by the report, that DILI caused by 1 is not influenced by typical polymorphisms of metabolic enzymes but associated to Human leucocyte antigen (HLA) genotype.6 It seems possible, that polymerization in vivo triggers an immune response that could lead to the observed hepatotoxicity. Consequently, the search for flupirtine analogues with a better safety profile seems worthwhile.
The tendency of 1 to form 3 a or 3 b or polymers may be attributed to electronic features such as oxidation potentials. By calculating the molecular orbitals of flupirtine (Figure 2) one finds that the highest occupied molecular orbital (HOMO) and HOMO‐1 are localized around the pyridine ring. This explains the oxidation susceptibility of the heterocyclic ring to form azaquinone diimine metabolites. The same calculation for deazathio‐flupirtine 5 showed that the HOMO orbital will shift from pyridine to the sulfur atom. This flupirtine analogue 5 is equally active as 1 but non‐toxic in vitro and was thus selected as a starting point for the synthesis of more potent flupirtine analogues.7 Our experiments on the oxidation of thio‐analogues to sulfoxides with a stoichiometric equivalent of m‐chloroperbenzoic acid also confirm this computational result. In order to alter these unfavorable electronic features of 1, we synthesized flupirtine analogues with modifications that should avoid the formation of azaquinone diimines and polymers in vivo.
Figure 2.
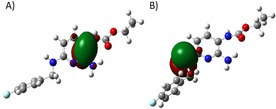
A) HOMO of flupirtine (1); B) HOMO of equally active but putatively non‐toxic lead 5.
To plan the modifications of 1, we divided its chemical structure into four regions; the 4‐fluorobenzyl moiety, the secondary amine linker, the primary amine substituent, and the carbamate group. All analogues reported here have a sulfur containing functional group in place of the secondary amine linker. As shown in the HOMO calculation, the placement of this sulfur atom is envisioned to hinder the possible formation of azaquinone metabolites like 3 a. Compounds 9 b–9 g and compounds 10 b–10 c have other rings or different substitution patterns in place of the 4‐fluorobenzyl group (Table 1). Compounds 9 g‐9 h and 10 d, on the other hand, have a methyl or methyl amine moiety instead of a primary amine. This modification, owning to the absence of primary amine, may result in analogues with little tendency to form metabolites like 3 b. In addition, the ethyl carbamate of 1 was replaced with fluoro‐substituted phenyl or benzyl groups to give compounds 9 a, 9 g and 10 a, respectively. The rational for this bulky, lipophilic modification comes from the recent patent literature on this class of compounds.8
Table 1.
Residues R1–R3 in intermediates 8 a–h, thioethers 9 a–d,f–h and thioester 9 e, and sulfoxides 10 a–d
| Entry | R1 | R2 | R3 | Yield[a] [%] |
|---|---|---|---|---|
| 8 a | NH2 | 4‐Fluorobenzyl | 85 | |
| 8 b | NH2 | 4‐Phenylbenzyl | 90 | |
| 8 c | NH2 | 3,5‐Dimethoxybenzyl | 76 | |
| 8 d | NH2 | 2‐Pyridylmethyl | 88 | |
| 8 e | NH2 | 4‐Fluorbenzoyl | 82 | |
| 8 f | NH2 | Piperidylethyl | 87 | |
| 8 g | NHCH3 | Benzyl | 89 | |
| 8 h | CH3 | 4‐Fluorobenzyl | 66 | |
| 9 a | NH2 | 4‐Fluorobenzyl | 3,4‐Difluorophenyl | 35 |
| 9 b | NH2 | 4‐Phenylbenzyl | Ethoxy | 38 |
| 9 c | NH2 | 3,5‐Dimethoxybenzyl | Ethoxy | 52 |
| 9 d | NH2 | 2‐Pyridylmethyl | Ethoxy | 30 |
| 9 e | NH2 | 4‐Fluorbenzoyl | Ethoxy | 28 |
| 9 f | NH2 | Piperidylethyl | Ethoxy | 32 |
| 9 g | NHCH3 | Benzyl | 3,5‐Difluorobenzyl | 8 |
| 9 h | CH3 | 4‐Fluorobenzyl | Ethoxy | 84 |
| 10 a | NH2 | 4‐Fluorobenzyl | 3,4‐Difluorobenzyl | 30 |
| 10 b | NH2 | 4‐Phenylbenzyl | Ethoxy | 88 |
| 10 c | NH2 | 3,5‐Dimethoxybenzyl | Ethoxy | 18 |
| 10 d | CH3 | 4‐Fluorobenzyl | Ethoxy | 25 |
[a] Isolated yield.
In an initial test set of systematically alkylated flupirtine derivatives reported earlier, EC50 values for their KV7.2/KV7.3 channel opening activity correlated with oxidation potentials.9 In order to evaluate this proposed connection, we followed a similar approach to increase stability towards oxidation. By replacement of the secondary amino group connecting the two aromatic moieties in 1 by a thioether or thioester group in 9 a–h, we aimed to alter the oxidation pathway of the molecule while retaining or even improving biological activity.
The synthesis of most of the compounds was commenced with the amination of 2,6‐dichloro‐3‐nitropyridine yielding intermediates 6 a or b (Scheme 1). The only exceptions were the syntheses of compounds 7 c, 8 h, 9 h and 10 d, which instead started with 6‐bromo‐2‐methyl‐3‐nitropyridine (6 c). The halogen substituents in 6 a–c were unambiguously replaced by a thiol group. Subsequently, thioether analogues were synthesized by straightforward nucleophilic attack of the thiol group on reactants with different cyclic structures in β‐position. Following the thioether synthesis, the 3‐nitro group in 8 a‐‐h was reduced to a primary amine with various classical reducing agents and then acylated to give the corresponding carbamate 9 b–f, and h or amide analogues 9 a and g in yields varying between 8 and 84 %.
Scheme 1.
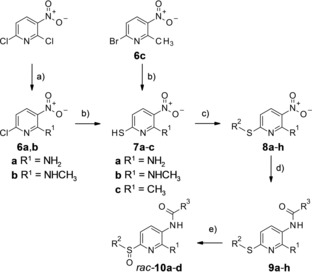
Synthesis of compounds 8 a–h, 9 a–h and 10 a–d (for residues R1, R2, R3 in 8 a–10 d consult Table 1). a) for 6 a: aq. NH3 (25 %), 2‐propanol, 35 °C, 5 days; for 6 b, methylamine, triethylamine, acetonitrile, 0 °C 10 min, then rt, 30 min; b) Na2S ⋅ 9 H2O, S8, NaOH, ethanol, reflux, 3–6 h; c) for 8 a–d and 8 f–h: alkylating agents, aq. KOH (10 %), DMF, rt, 1 h; for 8 e: 4‐fluorobenzoyl chloride, triethylamine, 2‐propanol, reflux, overnight; d) for 9 b–f, 9 h: SnCl2 ⋅ 2 H2O, absolute ethanol, 70 °C, argon, overnight ‐ 48 h, then triethylamine, acylating agent, 40 °C, 3 h ‐ overnight; for 9 a: iron powder, NH4Cl, 4 : 1 ethanol/water, 100 °C, 1 h, then triethylamine, acylating agent, 0 °C, 1.5 h; for 9 g: iron powder, NH4Cl, 4 : 1 ethanol/water, 100 °C, 2 h, then acylating agent, HATU, 40 °C, overnight e) m‐chloroperbenzoic acid, dichloromethane, 0 °C, 2 h.
The important anthelmintic albendazole, belonging to chemical family of alkylarylthioethers, is rapidly oxidized to albendazole sulfoxide and subsequently to albendazole sulfone (not shown).11 Albendazole sulfoxide is considered to be the therapeutically active form of the drug, even so re‐reduction inside the parasites contributes significantly to the mode of action. However, the role of albendazole metabolism for the toxicity in humans is unknown. In the case of 9 a and b, we similarly anticipate that the main reaction products of oxidation might be sulfoxides 10 a and b, respectively (Scheme 1). In order to investigate the toxicity of these putative metabolites along with their parent compounds, we prepared the sulfoxides 10 a–d. For sulfoxidation, sulfides 9 a–d were treated with almost a stoichiometric equivalent (1.1 equivalents) of m‐chloroperbenzoic acid in an ice cold water bath as described.12
After structural confirmation by NMR and MS experiments, the compounds were studied electrochemically by measuring the oxidation potential with cyclic voltammetry. KV7.2/KV7.3 channel opening activity were measured by a fluorescence‐based cellular thallium flux assay as described previously.9 To assess for possible hepatotoxicity, the synthesized analogues were evaluated with both a transgenic mouse hepatocyte (TAMH) and human hepatoma (HEP‐G2) cell line by using the MTT‐assay.
The closely related deazathio‐flupirtine analogues 9 a and b are as active as the marketed drug 1. The less similar compound 9 g is even markedly more potent and effective (Figure 3).
Figure 3.
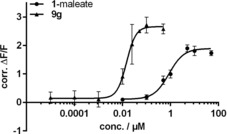
Concentration‐response curves of 1 (as maleate salt) and 9 g obtained with fluorescence‐based thallium‐flux KV7.2/3 channel‐opening assay; determined after 30 min exposure; data were normalized to control (1 % DMSO); values are the mean±SD (n≥3); EC50 values calculated from log(concentration)‐response curves which fit a four‐parameter logistic equation.
Thioester 9 e was inactive in the tested concentration range, which shows that a sulfide bridge is the much better bioisosteric surrogate for the amino bridge than a thioester in its thionoester form. The products of the chemical oxidation experiments demonstrated that formation of reactive diimines does not occur but instead the oxidative reactivity is shifted towards relatively benign S‐oxidation, at least in the sulfide series, as was anticipated. Oxidation is generally hampered in comparison to 1, as the anodic peak potentials (E pa) are higher. The resulting oxidation products, namely sulfoxides and sulfones, are putative metabolites that could also form from metabolic oxidation. Therefore, their cell toxicity is of interest and selected derivatives (10 a–d) were thus evaluated for in vitro hepatotoxicity in hepatocellular models with that use the TAMH and HEP‐G2 cell lines.13
Except for compounds 1, 9 a, c, and d, LD50 values after 24 h could not be determined due to a lack of aqueous solubility. To gain another quantitative indicator of toxicity, LD25 values after 48 h, which could be determined at lower concentrations where water solubility was not an issue, were determined for the most promising compounds. The LD25 for highly potent and effective 9 g is 6±3 and 4±4 μM in the TAMH and HEP‐G2 cell lines, respectively, and thus considerably lower than for 1 (Table 2). However, this increase in cell toxicity is more than compensated by the superior KV7.2/3 channel opening activity of 9 g compared to 1, yielding better safety indices of 400 versus 112 and 267 versus 81 in the TAMH and HEP‐G2 cell lines, respectively. Because 9 g has the highest logD 7.4 within this series of compounds, lipophilicity but not oxidizability or resulting reactivity might be important for the underlying mechanisms of action and toxicity.
Table 2.
Anodic peak potentials (E pa) of flupirtine (1) and derivatives 9 a–h and 10 a–d, EC50 and Emax values towards KV7.2/3 channels in HEK293 cells and LD50 (24 h exposure) and LD25 values (48 h exposure) in TAMH cells and HEP‐G2 cells as well as toxicity/activity ratios.
| Entry | E pa [a] [mV] | logD 7.4 | EC50 [b][μM] | Emax [%] | LD50 [c] [μM] | LD25 [μM][c] | Tox./Act.[c] | LD50 [d] [μM] | LD25 [μM][d] | Tox./Act.[d] |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 350 | 2.96 | 0.918±0.099[e] | 100 | 487±51 | 103±47 | 112 | 547±111 | 74±40 | 81 |
| 9 a | 442 | 3.90 | 0.26±0.082 | 100±23 | >30 | 13±04 | 50 | 8±3 | 4±1 | 15 |
| 9 b | 452 | 4.34 | 0.253±0.042 | 69±9 | >63 | 14±13 | 55 | >250 | n.d. [f] | n.d. [f] |
| 9 c | 450 | 3.61 | >10 | – | >250 | n.d. [f] | n.d. [f] | 134±22 | n.d. [f] | n.d. [f] |
| 9 d | 499 | 2.62 | >10 | – | >1000 | n.d. [f] | n.d. [f] | 831±149 | n.d. [f] | n.d. [f] |
| 9 e | 573 | 3.00 | >10 | – | >125 | n.d. [f] | n.d. [f] | >250 | n.d. [f] | n.d. [f] |
| 9 f | 573 | 2.04 | >10 | – | >500 | n.d. [f] | n.d. [f] | >500 | n.d. [f] | n.d. [f] |
| 9 g | 631 | 4.27 | 0.015±0.002 | 147±9 | >7.5 | 6±03 | 400 | >10 | 4±4 | 267 |
| 9 h | 855 | 3.85 | 0.269±0.031 | 129±3 | >10 | >10 | ‐ | >30 | 25±16 | 93 |
| 10 a | 628 | 3.24 | >10 | – | >100 | n.d. [f] | n.d. [f] | >125 | n.d. [f] | n.d. [f] |
| 10 b | 654 | 3.72 | >10 | – | >63 | n.d. [f] | n.d. [f] | >63 | n.d. [f] | n.d. [f] |
| 10 c | 442 | 2.81 | >10 | – | >500 | n.d. [f] | n.d. [f] | >500 | n.d. [f] | n.d. [f] |
| 10 d | n.o.[g] | 3.02 | >10 | – | >500 | n.d. [f] | n.d. [f] | >500 | n.d. [f] | n.d. [f] |
[a] Determined with 1.0 mM compound in 100 mM TRIS‐buffer (pH 7.4); [b] EC50‐ and LD50‐values are means and standard deviations of 4–5 independent determinations; [c] determined using TAMH cells; [d] determined using HEP‐G2 cells; [e] flupirtine maleate salt was used; [f] not determined; [g] non‐oxidizable.
Based on these findings, we conclude that the development of thio‐analogues of known drugs 1 and 2 may result in KV7.2/7.3 channel openers with more favorable therapeutic indices than flupirtine. Because they do not form reactive oxidation products in vitro they could even help to separate structure‐activity from structure‐toxicity relationships.
Experimental Section
N‐[6‐(Benzylthio)‐2‐(methylamino)pyridin‐3‐yl]‐2‐(3,5‐difluorophenyl)acetamide (9 g)
Compound 8 g (2.8 mmol, 771 mg), iron powder (28 mmol, 1.57 g) and ammonium chloride (28 mmol, 1.5 g) were suspended in 15 mL ethanol 20 %. The suspension was stirred at 100 °C for 2 hours, filtered over diatomaceous earth, and the filter washed with ethyl acetate. The filtrate was poured into water. The collected precipitate was washed with ethyl acetate. 2,6‐Dichlorophenyl acetic acid (2.8 mmol, 600 mg) and O‐(7‐azabenzotriazole‐1‐yl)‐N,N,N′,N′‐tetramethyluroniumhexafluorophosphate (HATU, 5.6 mmol, 2.1 g) were added and the mixture was stirred at 40 °C overnight. The product was separated using silica gel chromatography (solvent: ethyl acetate/hexane). The combined product containing fractions were evaporated to dryness. The residue was dissolved in ethanol and water was added to precipitate the product. Lavender colored solid (yield=8 %); purity 100 %; mp: 201–202 °C; 1H NMR (400 MHz, DMSO‐d6): δ=9.25 (s, 1H), 7.40 (m, 2H), 7.30 (m, 4H), 7.13 (m, 3H), 6.41 (d, J=7.8 Hz, 1H), 6.23 (d, J=4.6 Hz, 2H), 4.38 (s, 2H), 3.70 (s, 2H), 2.89 (d, J=4.6 Hz, 2H); 13C NMR (100 MHz, DMSO‐d6): δ=168.7, 163.4 (dd, J=13 Hz, J=244 Hz, 2 C), 153.2, 151.6, 140.4 (t, J=10 Hz, 1 C), 138.9, 133.2, 128.6 (2 C), 128.3 (2 C), 126.8, 115.1, 112.7 (dd, J=6 Hz, J=17 Hz, 2 C), 107.9, 102.2 (t, J=26 Hz, 1 C), 41.8, 33.3, 27.9; IR: =3442, 3404, 3269, 1652, 1591, 1496, 1389, 1230, 1119, 991, 696 cm−1; HRMS‐ESI m/z [M−H]− calcd for C14H16N4O2S: 398.1144, found: 398.1157. For synthetic details of other compounds see supporting information.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
AS is thankful for support for the article processing charge from the DFG (German Research Foundation, 393148499) and the Open Access Publication Fund of the University of Greifswald. CB was supported by DFG grant LI 765/7‐1, KB by DFG BE 1287/6‐1.
A. S. Surur, K. Beirow, C. Bock, L. Schulig, M. K. Kindermann, A. Bodtke, W. Siegmund, P. J. Bednarski, A. Link, ChemistryOpen 2019, 8, 41.
Dedicated to Bernd Clement on the occasion of his 70th birthday and retirement as university professor
References
- 1. Gomis-Perez C., Soldovieri M. V., Malo C., Ambrosino P., Taglialatela M., Areso P., Villarroel A., Front. Mol. Neurosci. 2017, 10, 117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Scheuch E., Methling K., Bednarski P. J., Oswald S., Siegmund W., J. Pharm. Biomed. Anal. 2015, 102, 377–385. [DOI] [PubMed] [Google Scholar]
- 3. Douros A., Bronder E., Andersohn F., Klimpel A., Thomae M., Orzechowski H.-D., Kreutz R., Garbe E., Eur. J. Clin. Pharmacol. 2014, 70, 453–459; and (Erratum) 2015, 71, 387. [DOI] [PubMed] [Google Scholar]
- 4.
- 4a. Sampath D., Valdez R., White A. W., Raol Y. H., Neuropharmacology 2017, 123, 126–135; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4b. Zhang T., Todorovic M. S., Williamson J., Kapur J., Ann. Clin. Transl. Neurol. 2017, 4, 888–896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Shkolnik T. Garin, Feuerman H., Didkovsky E., Kaplan I., Bergman R., Pavlovsky L., Hodak E., JAMA Dermatol. 2014, 150, 984–989. [DOI] [PubMed] [Google Scholar]
- 6.
- 6a. Siegmund W., Modeß C., Scheuch E., Methling K., Keiser M., Nassif A., Rosskopf D., Bednarski P. J., Borlak J., Terhaag B., Br. J. Clin. Pharmacol. 2015, 79, 501–513; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6b. Nicoletti P., Werk A. N., Sawle A., Shen Y., Urban T. J., Coulthard S. A., Bjornsson E. S., Cascorbi I., Floratos A., Stammschulte T., Gundert-Remy U., Nelson M. R., Aithal G. P., Daly A. K., Pharmacogenet. Genomics, 2016, 26, 218–224. [DOI] [PubMed] [Google Scholar]
- 7.
- 7a.DE 102018212006.4;
- 7b. Bock C., Beirow K., Surur A. S., Schulig L., Bodtke A., Bednarski P. J., Link A., Org. Biomol. Chem. 2018, 16, 8695–8699. [DOI] [PubMed] [Google Scholar]
- 8. Bock C., Link A., Fut. Med. Chem. 2019, 11, in press. [DOI] [PubMed] [Google Scholar]
- 9. Lemmerhirt C. J., Rombach M., Bodtke A., Bednarski P. J. and Link A., ChemMedChem. 2015, 10, 368–379. [DOI] [PubMed] [Google Scholar]
- 10. Bottoni A., Calvaresi M., Ciogli A., Cosimelli B., Mazzeo G., Pissani L., Severi E., Spinelli D., Superchi S., Adv. Synth. Catal. 2013, 355, 191–202. [Google Scholar]
- 11. Turner J. D., Sharma R., Jayoussi G. Al, Tyrer H. E., Gamble J., Hayward L., Priestley R. S., Murphy E. A., Davies J., Waterhouse D., Cook D. A. N., Clare R. H., Cassidy A., Steven A., Johnston K. L., McCall J., Ford L., Hemingway J., Ward S. A., Taylor M. J., Proc. Natl. Acad. Sci. USA 2017, 114, E9712-E9721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.
- 12a. Barrese V., Miceli F., Soldovieri M. V., Ambrosino P., Iannotti F. A., Cilio M. R., Taglialatela M., Clin. Pharmacol. 2010, 2, 225–236; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12b. Surur A. S., Schulig L., Link A., Arch. Pharm. Chem. Life Sci. 2019, 352: e1800248.. [DOI] [PubMed] [Google Scholar]
- 13.
- 13a. Van Summeren A., Renes J., Bouwman F. G., Noben J.-P., Delft J. H. M. van, Kleinjans J. C. S., Mariman E. C. M., Toxicol. Sci. 2011, 120, 109–122; [DOI] [PubMed] [Google Scholar]
- 13b. Davis M., Stamper B. D., BioMed Res. Int. 2016, DOI: 10.1155/2016/4780872. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary