

Abstract
Centrosome amplification is a hallmark of human cancers that can trigger cancer cell invasion. To survive, cancer cells cluster amplified extra centrosomes and achieve pseudobipolar division. Here, we set out to prevent clustering of extra centrosomes. Tubulin, by interacting with the centrosomal protein CPAP, negatively regulates CPAP‐dependent peri‐centriolar material recruitment, and concurrently microtubule nucleation. Screening for compounds that perturb CPAP–tubulin interaction led to the identification of CCB02, which selectively binds at the CPAP binding site of tubulin. Genetic and chemical perturbation of CPAP–tubulin interaction activates extra centrosomes to nucleate enhanced numbers of microtubules prior to mitosis. This causes cells to undergo centrosome de‐clustering, prolonged multipolar mitosis, and cell death. 3D‐organotypic invasion assays reveal that CCB02 has broad anti‐invasive activity in various cancer models, including tyrosine kinase inhibitor (TKI)‐resistant EGFR‐mutant non‐small‐cell lung cancers. Thus, we have identified a vulnerability of cancer cells to activation of extra centrosomes, which may serve as a global approach to target various tumors, including drug‐resistant cancers exhibiting high incidence of centrosome amplification.
Keywords: CCB02, centrosome activation, centrosome clustering, centrosomes, CPAP‐tubulin module
Subject Categories: Cancer; Cell Adhesion, Polarity & Cytoskeleton; Cell Cycle
Introduction
Centrosomes are the major microtubule organizing centers (MTOC) of mammalian cells (Zheng et al, 1995; Nigg, 2004; Nigg & Raff, 2009). Each centrosome consists of a pair of centrioles surrounded by peri‐centriolar material (PCM), from which spindle and astral microtubules emanate (Bettencourt‐Dias & Glover, 2007; Conduit et al, 2010). In healthy cells, strict regulation of centrosome duplication ensures the formation of only two functional centrosomes, which assemble bipolar spindles to avoid chromosomal aberrations in mitosis. In contrast, many cancer cells harbor extra centrosomes accompanied by chromosomal instability (Nigg, 2002). Thus, centrosome amplification and its structural aberrations is a hallmark of human cancers and has direct consequences on chromosomal instability and cancer cell invasion (Ganem et al, 2009; Godinho et al, 2014; Ganier et al, 2018; Marteil et al, 2018).
Intriguingly, centrosome loss in normal cells leads to irreversible cell cycle arrest, whereas in cancer cells, proliferation can still continue (Wong et al, 2015). This raises the possibility that cancer cells with extra centrosomes are fundamentally different from normal cells and use extra centrosomes for the benefit of cellular invasion. Consequently, there is a need to identify unique centrosomal properties of cancer cells, which can be exploited for conceptually new strategies to counteract cancer cell proliferation.
Amplified extra centrosomes should in theory cause multipolar mitosis, leading cancer cells to undergo mitotic catastrophe and cell death. However, cells with extra centrosomes achieve a pseudobipolar spindle via centrosome clustering, a key mechanism by which cancer cells cluster their extra centrosomes to circumvent mitotic catastrophe (Basto et al, 2008; Kwon et al, 2008; Leber et al, 2010; Pannu et al, 2015; Chavali et al, 2016). Indeed, when cells failed to cluster extra centrosomes in mitosis, they have been shown to undergo multipolar divisions and enter apoptosis (Ganem et al, 2009; Leber et al, 2010; Kawamura et al, 2013; Marthiens et al, 2013; Mason et al, 2014). Thus, inhibiting centrosome clustering to induce multipolar divisions has been proposed as a strategy to counteract tumors with high incidences of centrosome amplification (Ogden et al, 2012). An allosteric inhibitor of HSET, a kinesin motor protein required for centrosome clustering in mitosis, in fact induced multipolar spindles in cells containing extra centrosomes (Watts et al, 2013). Accordingly, direct interaction between the PCM protein CEP215 and HSET was shown to be required for centrosome clustering, highlighting an essential role for PCM in centrosome clustering (Chavali et al, 2016).
While these studies reveal the advantages of targeting centrosome clustering in cancer cells, how extra centrosomes can be manipulated to prevent them from clustering remains largely unknown. Microtubule nucleation by centrosomal PCM is spatiotemporally regulated to be minimal during interphase and increases as cells enter mitosis (Conduit et al, 2010, 2014; Avidor‐Reiss & Gopalakrishnan, 2013). Accordingly, compared to metaphase centrosomes, interphase centrosomes remain relatively inactive displaying reduced to moderate level of microtubule nucleation (Piehl et al, 2004; Pannu et al, 2014; Sabino et al, 2015). Thus, we wondered that activating extra centrosomes to nucleate an enhanced level of microtubules before they cluster in mitosis could potentially lead to centrosome declustering. Although this rationale may differ from current view of centrosome‐declustering mechanisms (Kwon et al, 2008; Fielding et al, 2011; Kramer et al, 2011; Watts et al, 2013), it may represent as one of the alternative mechanisms linking microtubule‐nucleating activity and centrosome declustering.
Our studies in Drosophila have shown that cytoplasmic‐free tubulin negatively regulates the microtubule‐nucleating activity of centrosomes through its direct interaction with Sas‐4 (CPAP in humans; Gopalakrishnan et al, 2012). Mutated Sas‐4, which cannot interact with tubulin, activates interphase centrosomes to nucleate robust microtubules by recruiting increasing amounts of PCM proteins (Gopalakrishnan et al, 2012). Thus, to induce extra centrosomes to nucleate an enhanced level of microtubules prior to mitosis, we developed a proof‐of‐principle experiment in cells with extra centrosomes by genetically perturbing CPAP–tubulin interaction. Based on this experiment, we established AlphaScreen, a proximity‐based protein–protein interaction assay that identified CCB02, a selective inhibitor of CPAP–tubulin interaction. Nuclear magnetic resonance (NMR) experiments and cellular pull‐down assays have identified CCB02 as a tubulin binder that competes for the CPAP binding site of β‐tubulin, a previously uncharacterized site that has not been occupied by known tubulin binders. CCB02 but not conventional tubulin binders activated extra centrosomes to nucleate an enhanced level of microtubules prior to mitosis and prevented them from clustering. Finally, via mouse xenograft experiments, we found that CCB02 has an anti‐tumor activity.
Results
CPAP–tubulin interaction as a target to prevent proliferation of cells with extra centrosomes
To identify unique centrosomal properties that can be targeted for cancer‐selective chemotherapy, we analyzed centrosomes in a spectrum of cancer cell lines. In contrast to normal cells containing only a pair of centrosomes, the tested cancer cell lines invariably displayed extra centrosomes (Appendix Fig S1Ai and ii). Highly increased centrosome numbers were observed in TKI‐resistant non‐small‐cell lung cancer cell lines (NSCLC) carrying somatic and activating mutations in EGFR (H1975T790M and HCC827‐GR; Engelman et al, 2007; Guo et al, 2008; Pagliarini et al, 2015; Ahsan, 2016). Extra centrosomes cluster during interphase and mitosis (Gergely & Basto, 2008; Kwon et al, 2008; Leber et al, 2010; Pannu et al, 2014). Interestingly, we noticed that clustered interphase centrosomes in these cancer cells remained inactive with reduced microtubule nucleation (Pannu et al, 2014; Appendix Fig S1Aiii). Thus, we sought to identify a mechanism that could activate extra centrosomes to nucleate enhanced microtubules prior to mitosis. We hypothesized that such a manipulation as one of the mechanisms that may perturb clustering of extra centrosomes in metaphase leading to multipolar spindles and eventual mitotic catastrophe (Appendix Fig S1B and C).
We previously showed that perturbing cytoplasmic tubulin from interacting with the centrosomal protein Sas‐4 (the Drosophila homologue of CPAP) could activate interphase centrosomes to nucleate an elevated level of microtubules by recruiting increasing amounts of PCM proteins (Gopalakrishnan et al, 2012). Our recent CPAP‐tubulin crystal structure revealed that CPAP binds β‐tubulin at the microtubule outer surface via its conserved PN2‐3 C‐terminal loop–helix, thereby forming a high‐affinity complex requiring CPAP's Phe375 (Sharma et al, 2016; Zheng et al, 2016). Importantly, this CPAP binding site of tubulin is not occupied by so‐far‐studied tubulin binders (Pryor et al, 2002; Prota et al, 2013).
Building on these studies, we introduced CPAPF375A, a mutant with significantly reduced tubulin interaction into MCF10A cells, which were engineered to amplify centrosomes via doxycycline‐induced overexpression of Plk4 (Godinho et al, 2014). For clarity, we term CPAPF375A as CPAPΔT, a CPAP variant with diminished interaction with tubulin.
To test whether introducing CPAP‐WT or CPAPΔT activates extra centrosomes to nucleate enhanced levels of microtubules, we performed live cell imaging experiments to identify events occurring during centrosome clustering or declustering in real time. Expressing CPAP‐WT or CPAPΔT in two centrosomes‐containing MCF10A cells (without doxycycline induction; −Dox, two centrosomes) did not cause centrosome amplification or delay in mitosis (Fig 1Ai and ii, and Movie EV1A and B).
Figure 1. CPAP–tubulin interaction is a cancer target.
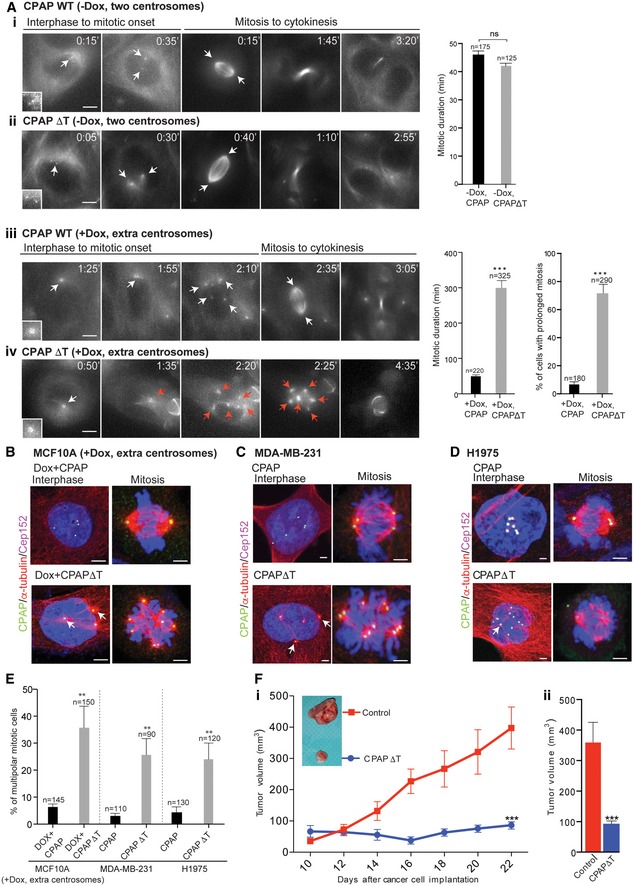
-
AProof‐of‐principle experiment. Time‐lapse images show overexpression of Myc‐tagged CPAP‐WT (i) or CPAPΔT (ii) has no effect on two centrosome‐containing MCF10A cells (−Dox, two centrosomes). The cells were imaged from interphase to cytokinesis. Bar graph at right quantifies mitotic duration of cells overexpressing CPAP‐WT and CPAPΔT. We define mitotic duration as time consumed from the onset of cell rounding until cytokinesis. The number of cells (n) analyzed in each condition is indicated at the top of each bar. Number of independent experiments (N), (N) = 3. Error bars, mean ± SEM. Unpaired t‐test. CPAP‐WT‐ (iii) or CPAPΔT‐expressing (iv) extra centrosome‐containing MCF10A cells (+Dox, extra centrosomes). Note compared to CPAP‐WT‐expressing centrosomes (iii) CPAPΔT‐expressing centrosomes nucleate enhanced levels of microtubule asters already at prophase‐like stage (iv). White arrows indicate centrosomal dots. Red arrows indicate centrosomes nucleating microtubule asters causing multipolar spindles. Scale bar, 2 μm. Bar graph at right quantifies mitotic duration and percentage of cells exhibiting prolonged mitosis in cells expressing CPAP‐WT and CPAPΔT. (N) = 3. Error bars, mean ± SEM. Unpaired t‐test ***P < 0.0001.
-
B–DFixed cell images showing the expression of CPAP‐WT (top panel) and CPAPΔT (bottom panel) in extra centrosome‐containing MCF10A (+Dox, extra centrosomes), MDA‐MB‐231, and H1975T790M cells. Note, in CPAP‐WT‐expressing cells, clustered extra centrosomes remain inactive with no or reduced microtubule nucleation. On the other hand, CPAPΔT expression enhances microtubule nucleation in extra centrosomes (arrows) and preventing them from clustering at G2‐like interphase and mitosis. Cells were stained with CPAP (GFP), microtubules (α‐tubulin, red), Cep152 (magenta). Scale bar, 2 μm.
-
EBar graph quantifies percentages of mitotic cells exhibiting multipolar spindles upon CPAP‐WT or CPAPΔT expression. (N) = 3. Error bars, mean ± SEM. Unpaired t‐test **P < 0.001.
-
FSubcutaneous xenograft tumor volume measurements of MDA‐MB‐231 cells expressing CPAP‐WT (red) as control or CPAPΔT (blue) in nude mice. (i) Bar graph at right shows total tumor volume reduction at day 22. (ii) Each condition used four mice each containing 4 xenograft tumors. Error bars, mean ± SEM with n = 16 for CPAP‐WT and n = 16 for CPAPΔT. Unpaired t‐test ***P < 0.0001.
We then monitored extra centrosomes‐containing MCF10A cells (with doxycycline induction; +Dox, extra centrosomes) expressing CPAP‐WT or CPAPΔT. In CPAP‐WT‐expressing cells, clustered centrosomes initially dispersed with a minimal microtubule nucleation, which appears to be G2 phase (from 1:55 to 2:10th minutes of Movie EV1C). These dispersed centrosomes eventually re‐clustered to form bipolar metaphase (from 2:10 to 2:35th minutes of Movie EV1C). We speculated that activating these dispersed extra centrosomes to nucleate an enhanced level of microtubules could prevent them from re‐clustering. CPAPΔT expression indeed caused an enhanced level of microtubules at the dispersed state of G2 (from 1:35 to 2:20th minutes of Movie EV1C). As a result, these cells failed to re‐cluster centrosomes and consumed much longer time in mitosis leading to multipolar metaphase (from 2:20 to 3:35th minutes of Movie EV1C). Overall, CPAPΔT‐expressing cells resided in mitosis ~ 4 times longer than control cells and apparently underwent cell death (Fig 1Aiii and iv, Appendix Fig S2A, Movie EV1C and D). We observed similar effects of multipolar mitosis when we introduced CPAPΔT into extra centrosome‐containing breast cancer (MDA‐MB‐231) and NSCLC (H1975T790M) cells (Fig 1B–E).
Finally, to test whether perturbing CPAP–tubulin interaction could prevent in vivo growth of cancer cells, we subcutaneously implanted CPAPΔT‐carrying MDA‐MB‐231 cells and observed a significant decrease of in vivo growth of breast cancer xenografts (Fig 1Fi and ii, and Appendix Fig S2Bi and ii). Together, these proof‐of‐principle experiments suggest that the CPAP–tubulin interaction is a target to prevent cancer cell proliferation.
Identification of CCB02, a specific inhibitor of CPAP–tubulin interaction
In order to identify a small molecule that can perturb CPAP–tubulin interaction, we initiated a high‐throughput compound screen based on the AlphaScreen assay technology (Schorpp et al, 2013). A library of 25,000 compounds was tested for their ability to disrupt the interaction between the conserved PN2‐3 domain of CPAP (amino acids 319–394) and free tubulin (Appendix Fig S3A). The PN2‐3 domain of CPAP harbors a tubulin‐binding site and interacts with tubulin to form a non‐polymerizable 1:1 complex (Hsu et al, 2008; Cormier et al, 2009). To exclude frequent hitters, we applied an algorithm, which resulted in a total of 49 initial hits (Schorpp et al, 2013; Table EV1, and Appendix Fig S3B and C). Subsequent cell‐based assays evaluating centrosome‐declustering activity further confirmed HTS1 as a compound that perturbs CPAP–tubulin interaction (Appendix Fig S3D and E). To increase the solubility of HTS1, we performed structural optimization, replacing the alkylamino residue at the C‐3 position of the benzo[b][1,6]naphthyridine system with a methoxy group. This led to CCB02, which inhibits CPAP‐tubulin interaction with an IC50 value of 0.689 μM as estimated by our AlphaScreen assay (Fig EV1A and B). This finding is further supported by our PN2‐3 CPAP‐GST pull‐down assay in the presence of CCB02. We noticed that CCB02 could inhibit CPAP–tubulin interaction with an approximate IC50 value of 0.441 μM (Fig EV1Ci and ii). Finally, a similar PN2‐3 CPAP‐GST pull‐down assay using cellular extract in the presence of CCB02 revealed that CCB02 could perturb interaction between CPAP and cellular tubulin (Fig EV1Ciii).
Figure EV1. Synthesis and characterization of CCB02.
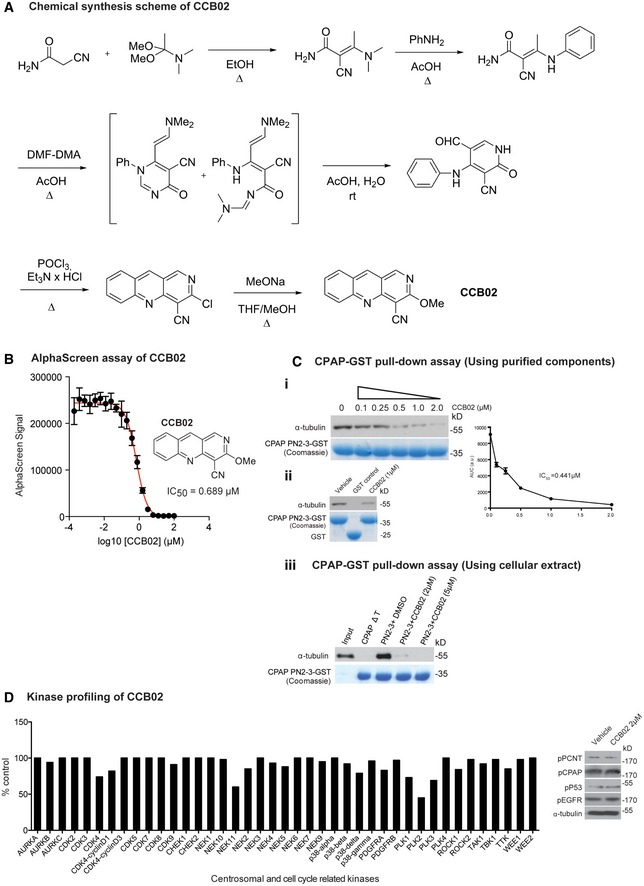
- Chemical synthesis of CCB02.
- CCB02 the derivative of HTS1 exhibits a dose‐dependent reduction of CPAP‐tubulin AlphaScreen signal. Error bars indicate data from a triplicate determination.
- CPAP‐GST pull‐down assay to support the findings of AlphaScreen assay. This CPAP‐GST pull‐down assay uses purified components of CPAP PN2‐3 and tubulin. Semi‐quantitative Western blot shows that CCB02 perturbs CPAP PN2‐3–tubulin interaction in a dose‐dependent manner with an approximately estimated IC50 value of 0.441 μM. The bait protein GST‐tagged PN2‐3 domain of CPAP is shown in Coomassie (bottom panel). The results derived from at least three independent experiments for intensity calculations (i). CPAP PN2‐3‐GST but not GST alone interacts with tubulin. CCB02 (1 μM) perturbs CPAP PN2‐3–tubulin interaction. Purified GST‐tagged PN2‐3 domains of CPAP and GST are shown in Coomassie (bottom panel) (ii). Similar CPAP‐GST pull‐down assay using cell extracts to show CCB02 prevents cellular tubulin binding to GST‐tagged PN2‐3 domain of CPAP. CCB02 at 2 and 5 μM (top panel Western blot) prevents binding of cellular tubulin to PN2‐3 (iii). We used CPAPΔT, a variant that will not interact with tubulin as a negative control. Addition of DMSO instead of CCB02 is a positive control.
- To evaluate the specificity, CCB02 was profiled against a broad panel of human protein kinases, including cancer‐relevant mutant kinases. Of the 469 kinases tested, CCB02 showed no significant inhibitory activity against family of centrosomal and other cell cycle‐related kinases even at higher concentration of CCB02 (5 μM). Note that our cell‐based experiments used concentration ranging from 1 to 2 μM. Experimental values are average of three independent experiments. (N) = 3. The kinases that represent < 10% of inhibitory activity compared to control are considered significant. Based on this, CCB02 does not seem to affect the activity of centrosomal and cell cycle‐related kinases. The complete result of in vitro kinases profiling is given in Table EV2 in the article. Western blot at right panel: Cell extracts treated with 2 μM of CCB02 were analyzed for phosphorylated substrates such as p‐PCNT, p‐CPAP, p‐P53, and p‐EGFR that are phosphorylated by PLK1, Aurora A, CDK2 (other like, CHK1 or CHK2 or ATM or ATR) and EGFR, respectively. Treatment with CCB02 does not alter the phosphorylation status of these proteins, indicating that the mechanism of CCB02 is not through inhibiting any of these cell cycle‐ or centrosome‐related kinase activities.
To exclude the off‐target effects of CCB02 on kinases, we screened a panel of kinases and determined that CCB02 does not significantly inhibit the tested kinases, which include cell cycle‐ and centrosome‐related kinases (Table EV2 and Fig EV1D). To further validate that CCB02 does not affect the tested cell cycle‐ and centrosome‐related kinase activities in cells, we performed Western blots using phospho‐specific antibodies that recognize substrates phosphorylated by Aurora A, Plk1, Plk2, CDK2, and CHK1. We identified that CCB02 does not affect these kinase activities (Fig EV1D, right panel).
CCB02 binds at the CPAP binding site of β‐tubulin to perturb CPAP–tubulin interaction
To dissect how CCB02 perturbs CPAP–tubulin interaction, we performed 1D‐1H NMR spectroscopy of CCB02 in the presence of tubulin and identified CCB02 as a tubulin binder (Fig 2A). INPHARMA experiments were then performed to identify the binding site of CCB02 using a CPAP‐derived peptide (residues 375–386), which binds to the microtubule outer surface on β‐tubulin with K D = 3.56 μM for tubulin (Sanchez‐Pedregal et al, 2005; Orts et al, 2009). To obtain comparable binding affinities of the CPAP peptide and CCB02 to tubulin, we used CCB02.1, a CCB02 derivative that prevents CPAP–tubulin interaction with an IC50 value of 6.94 μM (Appendix Fig S4A). We then generated NOESY spectra of a mixture of CPAP peptide (400 μM), CCB02.1 (200 μM), and tubulin (6.5 μM) at different mixing times (40, 70, 100, 150 ms) in D2O. Importantly, we ensured that tubulin at 6.5 μM used in these NMR analyses is folded properly and not forming aggregates (For details, see Appendix Fig S4B).
Figure 2. CCB02 binds tubulin, perturbing CPAP–tubulin interaction.
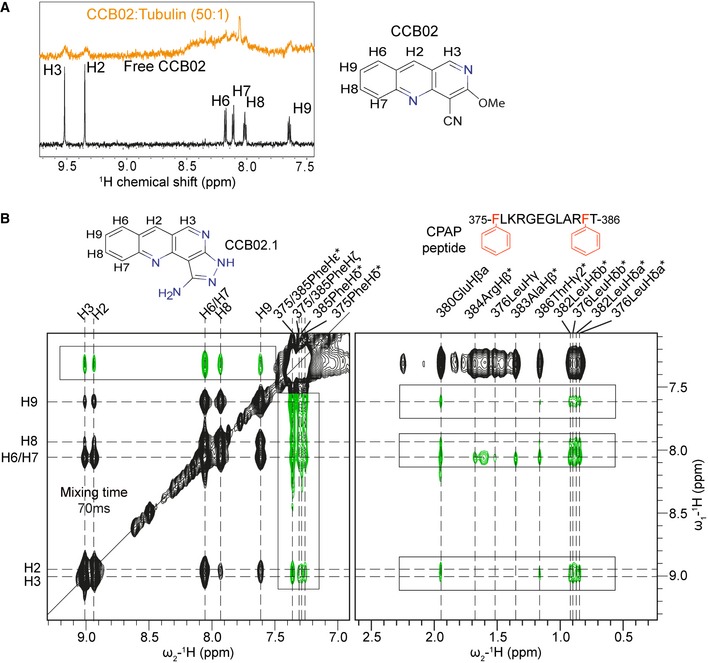
- 1H NMR spectra of free CCB02 (black) and bound to tubulin in 50:1 ratio (orange). Broadening of CCB02 proton peaks indicates its binding ability to tubulin.
- NOESY spectrum recorded for a solution comprising tubulin (6.5 μM), CPAP peptide (400 μM), and CCB02.1 (200 μM) with 70 ms mixing time. The signals colored in green and (highlighted by boxes) correspond to inter‐ligand trNOE signals arising due to protein‐mediated spin diffusion (INPHARMA NOEs), while the black peaks represent intra‐ligand trNOE signals.
We observed 10 intermolecular NOE peaks with a mixing time of 40 ms, while 50 peaks were observed at 70 ms (Fig 2B). Plotting NOE build‐up curves of six non‐overlapping intermolecular NOE peaks revealed a damped parabolic‐shaped curve, characteristic of inter‐ligand NOE cross peaks arising from protein‐mediated spin diffusion. This observation suggests that the CPAP peptide and CCB02.1 have the same binding site on tubulin (Fig 2B and Appendix Fig S4C). The NOESY spectra showed strong inter‐ligand NOEs from the H6/H7 protons of CCB02.1 to the aromatic side chain of Phe385 on the CPAP peptide although weaker NOEs were observed throughout the peptide at higher mixing times (Fig 2B and Appendix Fig S4D). In a control experiment, no intermolecular NOEs were observed when the CPAP peptide and CCB02.1 were mixed at a 1:1 ratio in the absence of tubulin.
In silico docking models combined with the NMR data suggest that CCB02.1 can occupy both the Phe385/Phe375 binding pockets on tubulin, with preference for the Phe385 pocket, which occupies the microtubule outer surface of β‐tubulin (Appendix Fig S5A). Finally, we performed isothermal titration calorimetry (ITC) to validate specific interaction between CCB02 and tubulin. Under our optimized condition, we were able to capture a titration curve (light blue curve, Appendix Fig S5B) that displayed a fitted binding K D of 2.2 μM with ΔH of −6.1 kcal/mol and ΔS of 4.7 cal/mol/deg, which showed a binding trend for specific interaction between CCB02 and tubulin.
Taken together, these results indicate that CCB02 is a novel tubulin binder whose binding site overlaps with the CPAP peptide at the microtubule outer surface of β‐tubulin (Sharma et al, 2016; Zheng et al, 2016). Importantly, some of the known conventional tubulin binders do not occupy this CCB02 binding site (Appendix Fig S5C; Pryor et al, 2002; Ravelli et al, 2004; Gigant et al, 2005; Lu et al, 2012; Prota et al, 2013). Via this mode of tubulin binding, CCB02 could perturb CPAP binding to tubulin in cells. To evaluate whether the major binding partner of CCB02 is cellular tubulin, we performed a CCB02‐Biotin pull‐down assay using cellular extracts and identified that CCB02 pulls down cellular tubulin (Appendix Fig S5Di–iii). Indeed, mass spectrometric analysis of CCB02 complexes identified tubulin as the most significant binding partner of CCB02 (Appendix Fig S5E). In summary, these experiments substantiate that CCB02 is a specific tubulin binder in cells.
CCB02‐mediated inhibition of CPAP–tubulin interaction impairs proliferation of cells with centrosome amplification
We then tested CCB02's effect on a spectrum of cancer cells exhibiting extra centrosomes compared to normal cells containing two centrosomes. A 72‐hr exposure of CCB02 prevented cancer cell proliferation with IC50 values between 0.86 and 2.9 μM (Fig EV2A). Importantly, correlating the percentages of extra centrosomes‐containing cancer cells to their respective IC50 values revealed that cells with higher percentages of extra centrosomes have inversely proportional IC50 values for CCB02 (Fig EV2B). If this were true, we would expect that a long‐term CCB02 treatment would selectively eliminate extra‐centrosome‐containing cells, but allowing the survival of two centrosomes‐containing cells. To test this aspect, we exposed MDA‐MB‐231, HCC827‐GR, Calu6 and Plk4‐overexpressing MCF10A cells to CCB02. After 14 days of exposure, we observed that the CCB02‐treated cultures mostly contained two centrosomes‐containing cells as compared to vehicle‐treated cultures, indicating that cells with higher levels of centrosome amplification are more sensitive to CCB02 (Fig 3A).
Figure EV2. Related to Fig 3: CCB02 impairs proliferation of extra centrosomes‐containing cells.
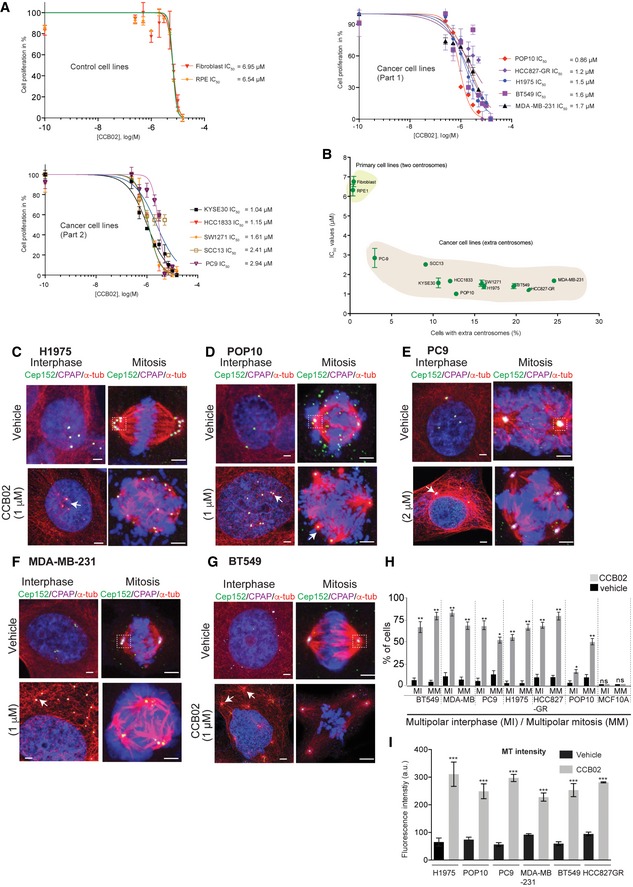
-
ADose–response curves of control (fibroblast and RPE) and cancer cell lines (separated as part 1 and 2) in response to CCB02 treatment. IC50 values were calculated after 72 h of treatment. MTS assay was used to calculate IC50 values (detailed description is given in the Materials and Methods section). Each data point is representative of three independent experiments. (N) = 3. Error bars, mean ± SEM.
-
BCorrelation of IC50 values to the percentage of cells containing extra centrosomes. An enhanced sensitivity of CCB02 is observed with cells harboring extra centrosomes. While cells with extra centrosomes segregate to the right side of the graph exhibiting lower IC50 values, two centrosome‐containing control cells segregate to the upper left side of the graph exhibiting higher IC50 values. Data represent mean ± SEM. (N) = 3. Pearson correlation coefficient was calculated for XY pairs (r = −0.86). ***P < 0.0001. Also refer Appendix Fig S1Aii for percentages of cells exhibiting extra centrosomes.
-
C–GCCB02 treatment prevents extra centrosomes of cancer cells from clustering by activating them to nucleate an enhanced level of microtubules prior to mitosis (Arrows). Activated centrosomes fail to cluster, instead of causing multipolar mitosis. H1975T790M, POP10, PC9, MDA‐MB‐231, and BT549 cancer cells exhibiting extra centrosomes were used. For cell‐based assays, CCB02 was used in the range of 1–2 µM concentrations depending on the estimated IC50 values for respective cell lines. All these cells were stained with Cep152 (green), CPAP (magenta), microtubules (α‐tubulin, red), and DNA (DAPI blue). Scale bar, 2 µm.
-
HBars show percentages of multipolar interphase (MI) and multipolar mitotic cells (MM) after 48 h of CCB02 treatment. Compared to vehicle treatment, CCB02 treatment causes multipolar spindles in both interphase and mitosis. (N) = 3 with total number of cells > 300 in each cell line. Error bars, mean ± SEM. Unpaired t‐test. *P < 0.01, **P < 0.001. Note that CCB02 does not affect two centrosome‐containing MCF10A wild‐type cells.
-
IBar graphs show MT intensities of interphase cells from the figure panels (C–G). Error bars, mean ± SEM. Two‐way ANOVA test. ***P < 0.0001.
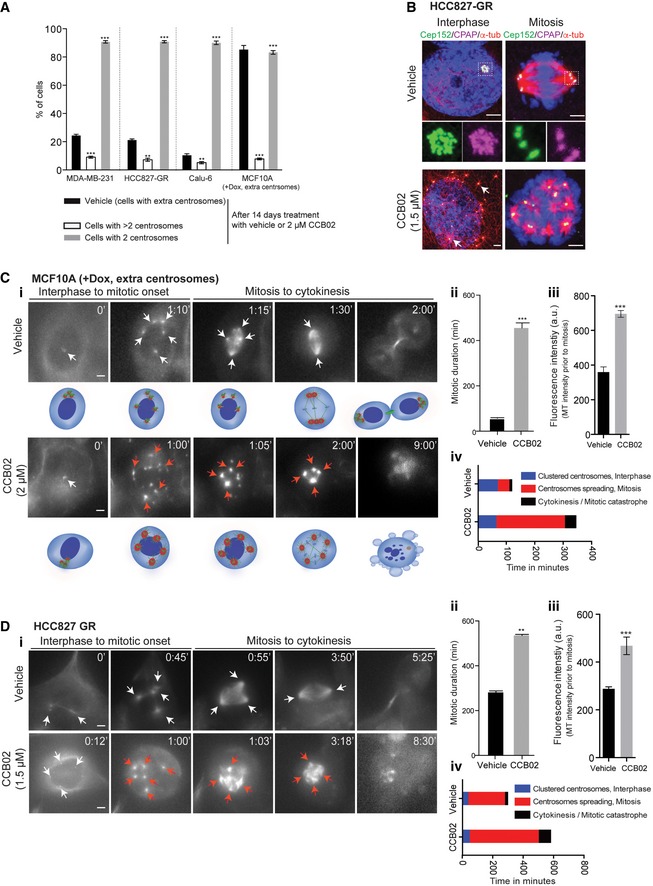
Figure 3. CCB02 prevents proliferation of cells with centrosome amplification.
-
AFourteen‐day CCB02 treatment of MDA‐MB‐231, HCC827‐GR, Calu‐6, and MCF10A (+Dox, extra centrosomes) mostly eliminated cells with extra centrosomes. At the end of the treatment, cells were fixed and stained for centrosomes. Bar graph shows the percentage of cells displaying two and more than two centrosomes. “Vehicle” at the bar graph indicates percentages of cells with extra centrosomes (also refer to Appendix Fig S1A). Note that percentages of cells containing more than two centrosomes are significantly reduced upon CCB02‐treatment (open bars). Data represent mean ± SEM of three independent experiments. (N) = 3, (n = 70). P‐values were obtained using unpaired t‐test **P < 0.001, ***P < 0.0001.
-
BCCB02 treatment prevents centrosome clustering in HCC827‐GR cells. Activated centrosomes (arrows) fail to cluster leading to multipolar mitosis. Cells were stained with Cep152 (green), CPAP (magenta), and microtubules (α‐tubulin, red). Scale bar, 2 μm, Insets, 0.5 μm.
-
C, DTime‐lapse images show the effect of CCB02‐mediated centrosome activation in cell cycle progression of MCF10A (+Dox, extra centrosomes) (C), HCC827‐GR (D) cells. White arrows indicate centrosome dots. Red arrows (in CCB02‐treated cells) indicate activated centrosomes with enhanced microtubule intensity compared to vehicle‐treated cells. Note, centrosome activation, declustering, and prolonged multipolar mitosis occur only with CCB02 treatment. Cartoons of a cell with extra centrosomes below to the panel (C) represent various events occurring upon vehicle or CCB02 treatment. Scale bar, 2 μm. Bar graphs at right to panel (C) and (D) quantify mitotic duration (ii), quantify microtubule intensity prior to mitosis (iii), and depict relative events occurring in cells treated with vehicle or CCB02 (iv). At least 100 cells were analyzed in each condition. (N) = 3. Error bars, mean ± SEM. Unpaired t‐test **P < 0.001, ***P < 0.0001.
We then analyzed the effect of CCB02 on extra centrosomes. Similar to genetic perturbation, chemical perturbation of CPAP‐tubulin interaction has also activated extra centrosomes to nucleate an enhanced level of microtubules prior to mitosis, resulting in the formation of multipolar spindles in mitosis (Appendix Fig S3B and Fig EV2C–I). To further verify that CCB02's action occurs prior to mitosis and to make sure that we score similar stages of cells between treatment and control groups, we profiled CCB02‐treated MDA‐MB‐231 cells using cyclin A staining, a bona fide G1‐S transition marker (Hochegger et al, 2008; Gabriel et al, 2016). In vehicle‐treated groups, cyclin A‐positive cells mostly exhibited clustered (G1/S phase) or dispersed (G2 phase) centrosomes with less or no microtubule nucleation. In contrast, CCB02‐treated cells exhibited declustered centrosomes with enhanced microtubule nucleation (Appendix Fig S6A and B).
To better capture CCB02‐mediated effects, we performed live imaging of CCB02‐treated MCF10A (+Dox, extra centrosomes) and HCC827‐GR cells. Similar to CPAPΔT expression, CCB02‐treatment prevented extra centrosomes from clustering leading to multipolar mitosis with an apparent cell death (Fig 3C and D, and Movie EV2A–D). Importantly, cells with extra centrosomes exhibited much longer mitotic duration than two centrosomes‐containing cells (Appendix Fig S6C and D).
We then tested whether CCB02‐induced mitotic delay causes spindle assembly checkpoint (SAC) activation. To do this, we performed experiments using antibodies against Bub1 and Mad1 proteins. These components are known to accumulate on unattached kinetochores (Shah & Cleveland, 2000; Kim et al, 2015; Musacchio, 2015). First, we verified that cells (regardless of two‐ or extra‐centrosome‐containing cells) normally showed an accumulation of Bub1 and Mad1 proteins in prophase (Fig EV3A, prophase panel). This is due to unaligned chromosomes to kinetochores at this stage of cell cycle (Johnson et al, 2004; Bolanos‐Garcia & Blundell, 2011). As also expected, bipolar and pseudobipolar metaphase cells did not show a detectable accumulation of these proteins (Fig EV3A, metaphase panel). We then analyzed CCB02‐treated MCF10A (−Dox, two centrosomes), MCF10A (+Dox, extra centrosomes), and MDA‐MB‐231 cells. Importantly, we have also used MCF10A (+Dox, extra centrosomes)‐expressing CPAPΔT as a control where CPAP–tubulin interaction is genetically perturbed. In both cases, we observed an accumulation of Bub1 and Mad1 proteins in prophase (Fig EV3B and C, prophase panels). However, in contrast to bipolar metaphase cells as observed in Fig EV3, these proteins are still accumulated in multipolar metaphase cells where CPAP‐tubulin interaction is perturbed either by CCB02 treatment or CPAPΔT overexpression (Fig EV3B and C, metaphase panels). These observations suggest that perturbing CPAP–tubulin interaction could activate the spindle assembly checkpoint in extra centrosome‐containing cells.
Figure EV3. Related to Fig 3: Inhibiting CPAP–tubulin interaction activates spindle assembly checkpoint in extra centrosomes‐containing cells.
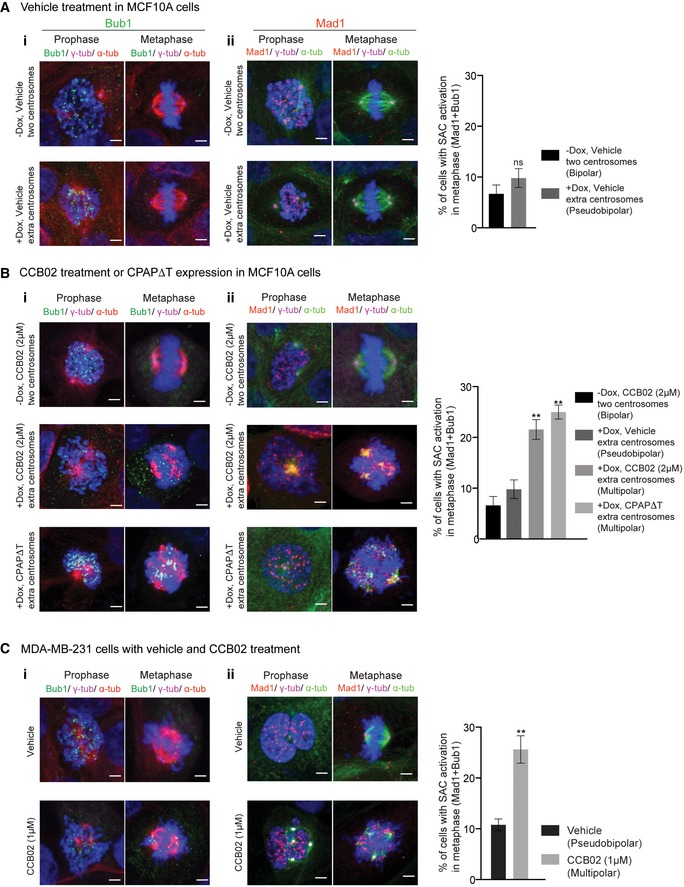
- Vehicle treatment. SAC components Bub1 (i, green) and Mad1 (ii, red) mostly accumulate in prophase but not in bipolar mitotic (two centrosomes‐containing) or pseudobipolar mitotic (extra centrosome containing) cells. MCF10A cells containing two centrosomes (−Dox, two centrosomes) and extra centrosome (+Dox, extra centrosomes) were used. Cells were stained for Bub1 (green) and Mad1 (red), γ‐tubulin (magenta), microtubules (α‐tubulin, red) and DNA (DAPI, blue). Scale bar, 2 μm. Bar diagram at right quantifies SAC activation only in metaphase cells (both bipolar and pseudobipolar). Data represent mean ± SEM of three independent experiments, (N) = 3 (n = 220 cells per condition). P‐values were obtained using one‐way ANOVA. **P < 0.001.
- CCB02 treatment or CPAPΔT overexpression. In contrast to bipolar metaphase cells (as observed above in panel A), Bub1 (i) and Mad1 (ii) proteins accumulated in multipolar metaphase cells where CPAP‐tubulin interaction is perturbed either by CCB02 treatment or CPAPΔT overexpression. MCF10A (−Dox, two centrosomes) and MCF10A (+Dox, extra centrosomes) cells were used. MCF10A cells (+Dox, extra centrosomes) expressing CPAPΔT were used as control where CPAP‐tubulin interaction is genetically perturbed. Bar diagram at right quantifies SAC activation only in metaphase cells (both bipolar and multipolar). Data represent mean ± SEM of three independent experiments (N) = 3 (n = 250 cell per condition). P‐values were obtained using one‐way ANOVA. **P < 0.001.
- SAC activation assay in MDA‐MB‐231 cells. Bar diagram at right quantifies SAC activation only in metaphase cells (both bipolar and multipolar). Data represent mean ± SEM of three independent experiments. (N) = 3 (n = 240 cells per condition). P‐values were obtained using one‐way ANOVA. **P < 0.001.
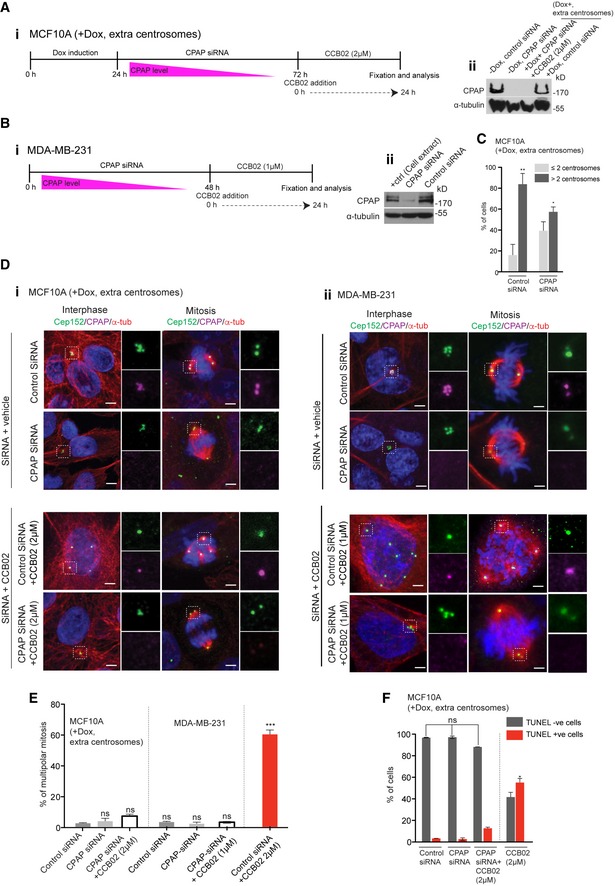
Finally to dissect the observed effect of CCB02 is CPAP dependent; we depleted CPAP in MDA‐MB‐231 and MCF10A cells harboring extra centrosomes for 48 h and analyzed a fraction of cells that still contained extra centrosomes (Fig 4A–C). We noticed that CPAP depletion did not prevent centrosome clustering (Fig 4D). We then treated these cells with CCB02 and identified that CCB02 treatment did not induce declustering of centrosomes or cell death (Fig 4E and F). These results suggest that CCB02‐treatment specifically impairs proliferation of extra‐centrosome‐containing cells and the effect induced by CCB02 is CPAP dependent.
Figure 4. siRNA depletion of CPAP abrogates the effects of CCB02 in extra centrosome‐containing cells.
-
A, B(i) Experimental strategy to test whether the effect of CCB02 is CPAP dependent in MCF10A cells containing extra centrosomes (+Dox, extra centrosomes) (A) and MDA‐MB‐231 cells (B). Cells were treated with CPAP siRNA and control (scramble) siRNA for 48 h. (ii) Western blots show depletion of CPAP when cells were treated with siRNA specific for CPAP.
-
CFractions of cells containing centrosomes after siRNA treatment. Forty‐eight hours of siRNA treatment still contained a fraction of cells with extra centrosomes that were analyzed in the following experiments. Bar graph shows the percentage of cells with extra centrosomes in MCF10A (+Dox, extra centrosomes) in the presence of control siRNA or CPAP siRNA. Data represent mean ± SEM of three independent experiments. (N) = 3, (n = 300 cells per condition). P‐values were obtained using ordinary one‐way ANOVA test *P < 0.01, **P < 0.001.
-
DEffect of CCB02 in the presence of control siRNA and CPAP siRNA in MCF10A (i) MDA‐MB‐231 cells (ii) CPAP depletion does not prevent centrosomes from clustering (second rows of i and ii). Note, in CPAP‐depleted cultures, centrosomes are marked with Cep152 (green) where CPAP (magenta) is detected faintly. In CPAP‐depleted cells, CCB02 does not prevent extra centrosomes from clustering (fourth rows of i and ii). Note CCB02 does prevent extra centrosomes from clustering in cultures treated with control siRNA (third rows of i and ii). Cells are stained for Cep152 (green), CPAP (magenta), microtubules (α‐tubulin, red), and DNA (DAPI, blue). Scale bar, 2 μm.
-
EBar graph shows the percentage of cells with multipolar mitosis. Higher percentage of multipolar mitotic cells is observed in control siRNA‐treated cultures. Bar graph on right shows the percentage of cells with multipolar mitosis. Data represent mean ± SEM of three independent experiments. (N) = 3, (n = 680 cells). P‐values were obtained using unpaired t‐test. ***P < 0.0001.
-
FBar graph shows the percentage of living (TUNEL‐negative) and dyeing (TUNEL‐positive) cells under various conditions tested. Higher percentage of TUNEL‐positive cells is observed in CCB02‐treated cultures. Data represent mean ± SEM of three independent experiments. (N) = 3 (n = 500 cells per condition). P‐values were obtained using ordinary one‐way ANOVA test *P < 0.01.
Inhibiting CPAP–tubulin interaction by CCB02 enhances PCM recruitment to centrosomes
We next sought to identify mechanisms by which CCB02 activates extra centrosomes for an enhanced microtubule nucleation. PCM recruitment to centrosomes is required for microtubule nucleation (Oegema et al, 1999; Nigg & Raff, 2009; Gopalakrishnan et al, 2011; Lee & Rhee, 2011). Enhanced microtubule nucleation of CCB02‐treated centrosomes prior to mitosis suggests that these centrosomes recruit enhanced levels of PCM. To test this, we estimated the amounts of Cep152, PCNT, and CDK5RAP2 recruitment to interphase centrosomes of CCB02‐treated two centrosomes‐containing MCF10A cells. Notably, both human and Drosophila CPAP interacts with these proteins to form the S‐CAP complex (Gopalakrishnan et al, 2011; Conduit et al, 2015; Chou et al, 2016). High‐resolution imaging and heat map intensity analyses revealed that interphase centrosomes recruit enhanced amounts of these proteins compared to vehicle‐treated cells (Fig 5A and B).
Figure 5. Chemical inhibition of CPAP–tubulin interaction causes interphase centrosomes to recruit enhanced levels of PCM .
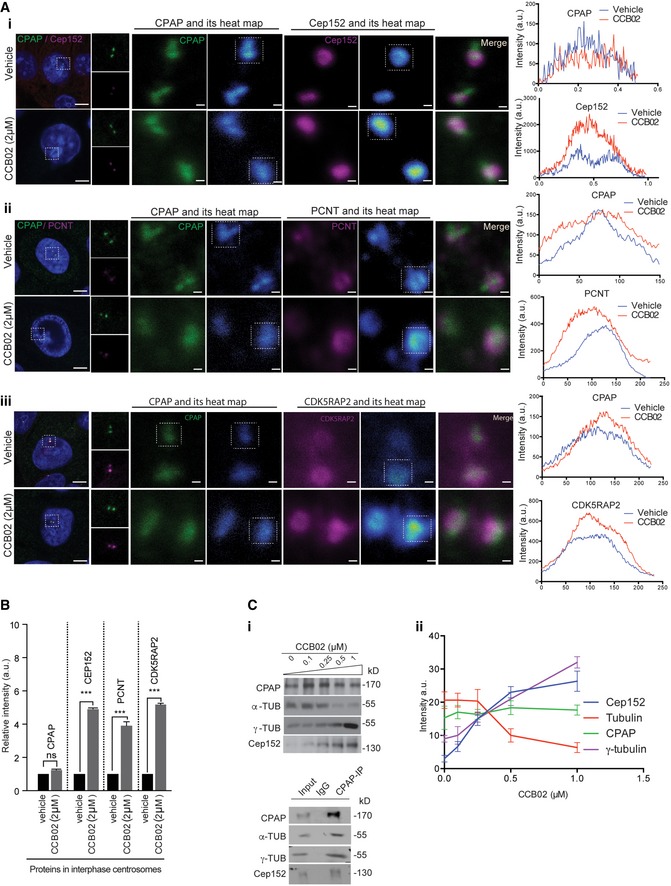
- Interphase centrosomes of CCB02‐treated two centrosomes‐containing MCF10A cells display an enhanced level of PCM proteins. CPAP (green) intensity remains same. Cells are stained for CPAP (green), PCM proteins (magenta) Cep152 (i), PCNT (ii) and CDK5RAP2 (iii), and microtubules (α‐tubulin, red). Scale bars, 2 μm for left panels and 0.2 μm for right panels. Heat map images show intensity saturation (blue) for CPAP, Cep152, PCNT, and CDK5RAP2 upon CCB02 treatment. Line graphs at right to each panel indicate the heat map intensity of single centrosomes of cells (given in boxes at heat map panels) treated with vehicle (blue line) or CCB02 (red line). Images were recorded with low laser excitation and high zoom factor.
- Bar graph quantifies relative intensity of CPAP‐interacting proteins in interphase centrosomes compared to vehicle treatment. At least 150 centrosomes (for vehicle) and 130 centrosomes (for CCB02) were analyzed in each condition. Error bars, mean ± SEM. (N) = 3. Ordinary two‐way ANOVA test ***P < 0.0001.
- Immunopurification of CPAP complexes from CCB02‐treated cell extracts. (i) CCB02 specifically prevents CPAP–tubulin interaction. This allows CPAP to bind an enhanced amount of Cep152 and γ‐tubulin. Control IP experiment is shown below. (ii) Plot showing PCM protein intensities co‐immunoprecipitated with CPAP at various concentrations of CCB02 treatment. CPAP amount does not change with CCB02 treatment. Data represent mean ± SEM. (N) = 3.
Furthermore, biochemically fractionated centrosomes from CCB02‐treated cells revealed the presence of elevated levels of CPAP‐interacting proteins (Appendix Fig S7A and B). Finally, we tested whether CCB02 could prevent tubulin binding to CPAP in cells and simultaneously enhance the ability of CPAP to bind its interacting proteins. To do this, we immunopurified CPAP complexes from cytoplasmic extracts of MCF10A cells in the presence of CCB02 and identified that CCB02 specifically perturbs tubulin binding to CPAP, thereby allowing CPAP to bind enhanced amounts of its interacting proteins (Fig 5C). We also observed a similar finding when we used cytoplasmic extracts prepared from interphase‐synchronized HeLa cells (Appendix Fig S7C).
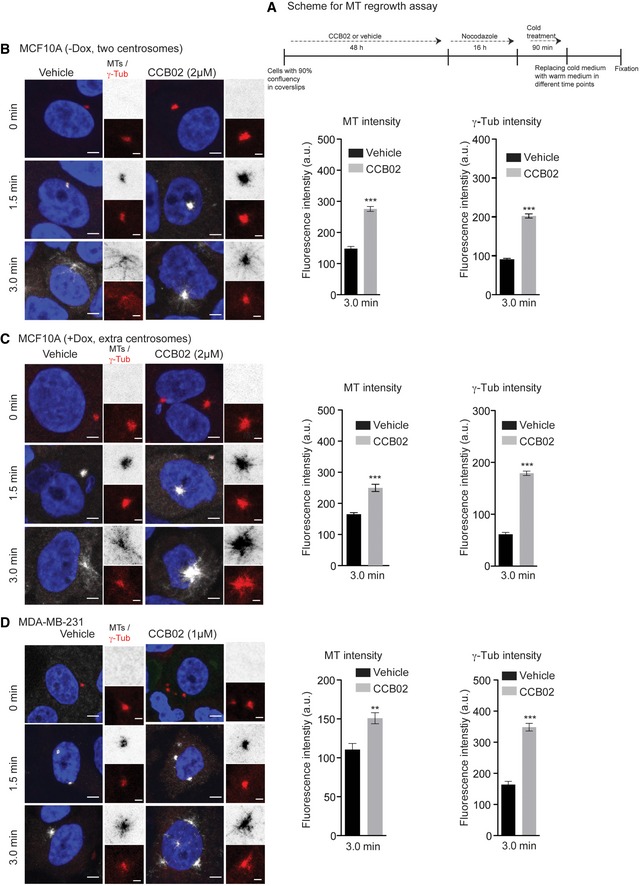
To validate that the enhanced recruitment PCM could cause microtubule nucleation, we performed a three‐time point microtubule regrowth assay with MCF10A (−Dox, two centrosomes), MCF10A (+Dox, extra centrosomes), and MDA‐MB‐231 cells (Sankaran et al, 2005; Choi et al, 2010; Fig 6). CCB02 treatment caused centrosomes to nucleate an enhanced level of microtubules already at 1.5 min after induction of regrowth. Importantly, these centrosomes recruited significantly higher levels of γ‐tubulin (Fig 6). This finding is in agreement with our live imaging experiments where CCB02‐treated cells display centrosomes with robust microtubule nucleation (Movie EV2B and D, and Fig 6).
Figure 6. CCB02 enhances microtubule nucleation activities of centrosomes.
-
AExperimental scheme of microtubule (MT) regrowth assay.
-
B–DMT regrowth assays at 0, 1.5, and 3.0 min using MCF10A (−Dox, two centrosomes), MCF10A (+Dox, extra centrosomes), and MDA‐MB‐231 cells. MT nucleation panel is shown in gray scale (inset images are inverted), and γ‐tubulin is shown in red. Note that in contrast to vehicle treatment, CCB02 treatment caused centrosomes to nucleate an enhanced level of microtubules already at 1.5 min after induction of regrowth with simultaneous increase in γ‐tubulin recruitment. All these cells were stained with γ‐tubulin (red), microtubules (α‐tubulin, gray), and DNA (DAPI blue). Scale bar, 2 μm and insets, 0.5 μm. Bar graphs show MT and γ‐tubulin intensities at 3 min after induction of MT regrowth. (N) = 3. At least 80 centrosomes were considered to calculate intensities from each cell line. Error bars, mean ± SEM. Unpaired t‐test **P < 0.001, ***P < 0.0001.
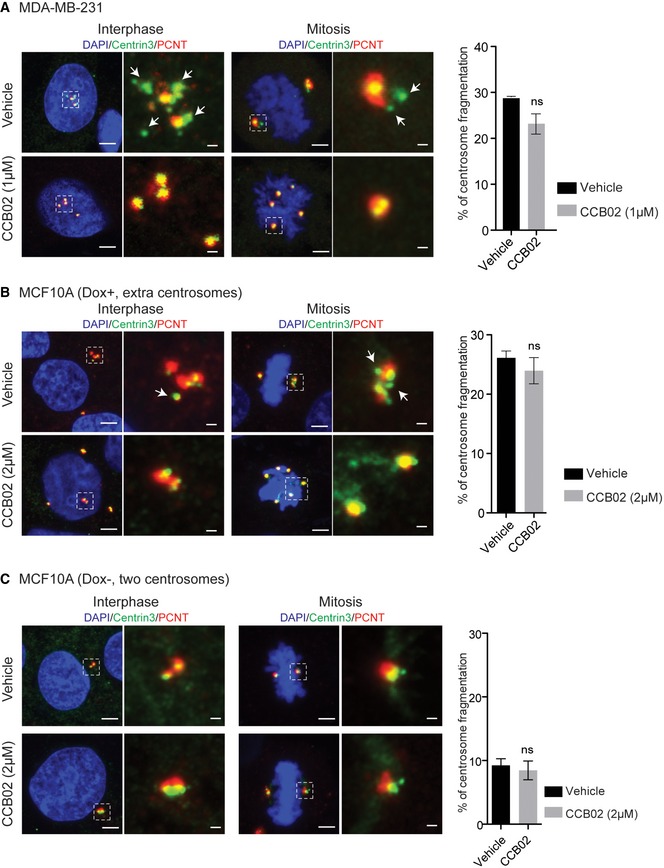
To exclude that the observed effects of CCB02 are not due to centrosome fragmentation, we analyzed MDA‐MB231 and MCF10A cells (+Dox, extra centrosomes) before and after CCB02 treatment. We did observe fragmented centrosomes in MDA‐MB231 and MCF10A cells (+Dox, extra centrosomes) cells as determined by PCNT‐negative centrin dots (centrin‐3; Kohlmaier et al, 2009; Lawo et al, 2012; Godinho & Pellman, 2014; Karki et al, 2017). The intact centrosomes are determined by centrin colocalization with PCNT. Importantly, we did not observe any increase in the frequencies of fragmented centrosomes indicating that CCB02 does not induce centrosome fragmentation (Fig EV4A–C). Together, these results suggest that chemical inhibition of the CPAP–tubulin interaction could enhance the recruitment of CPAP‐interacting proteins to interphase centrosomes. This finding is similar to what was observed when Sas‐4‐tubulin interaction was genetically perturbed in flies (Gopalakrishnan et al, 2012).
Figure EV4. Related to Fig 5: CCB02 does not induce centrosome fragmentation.
-
A, BFragmented centrosomes in extra centrosomes‐containing MDA‐MB231 and MCF10A cells (+Dox, extra centrosomes) before and after CCB02 treatment. Fragmented centrosomes in these cells are determined by PCNT‐negative centrin dots (centrin‐3). No increase in centrosome fragmentation is observed after CCB02 treatment.
-
CSimilar experiment was performed in two centrosome‐containing MCF10A cells. This was because, in centrosome‐amplified cells, extra centrosomes might mislead us in distinguishing them from fragmented centrosomes. No centrosome fragmentation is identified before and after CCB02 treatment. All these cells were stained for centrin‐3 (green) and PCNT (red), and DNA (DAPI, blue). Scale bar, 2 μm and insets, 0.5 μm. Bar diagrams at right quantify the findings. (N) = 3. At least 200 cells were used for quantifications from each cell line. Error bars, mean ± SEM. Unpaired t‐test.
Effects of CCB02 in cells differ from the effects of known tubulin‐binding agents
Most tubulin‐binding agents act on spindle microtubules and thus prevent mitosis non‐specifically (Kavallaris, 2010). To evaluate whether the effects of CCB02 are specific to CPAP–tubulin interaction and not due to general effects caused by known tubulin binders, we compared CCB02 to known tubulin binders such as taxol, bactallin III, docetaxel, and vinblastine. Except CCB02, neither of the tested tubulin binders could perturb CPAP–tubulin interaction, enhance PCM recruitment to interphase centrosomes, and prevent extra centrosomes from clustering (Fig 7A–C and Appendix Fig S8A–E). Finally, to test that CCB02 does not alter microtubule in vitro and live cells, we performed microtubule plus end‐tracking assay using GFP‐tagged EB1 and EB3, respectively. CCB02 at 1, 2, and 5 μM did not detectably influence various parameters of microtubule dynamics (Fig 7E and F, and Movies EV3 and EV4). Taken together, these results suggest that most effects of CCB02 differ from the effects of known tubulin‐binding agents.
Figure 7. CCB02 effects differ from the effects of known tubulin‐binding agents.
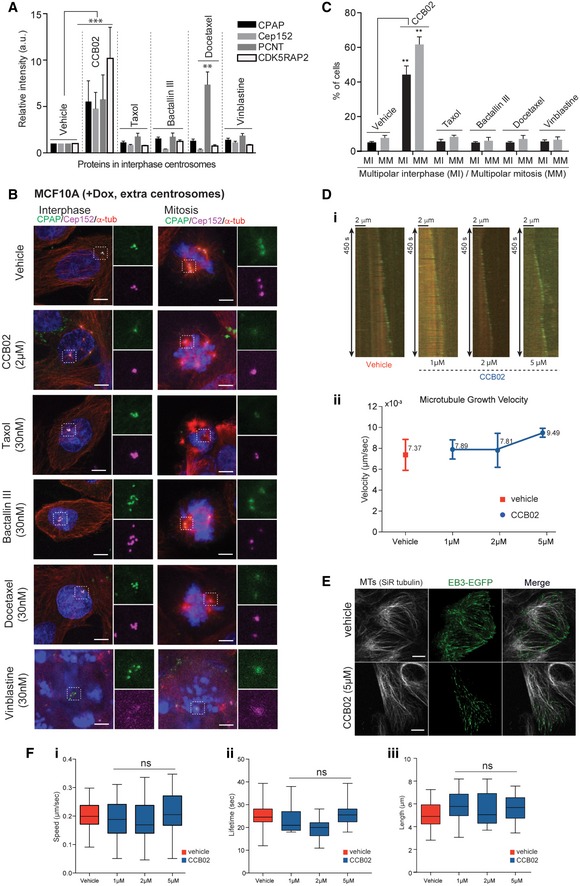
- Bar graph quantifies relative intensity of PCM proteins at interphase centrosomes of cells treated with vehicle, CCB02, and known tubulin binders. Two centrosomes containing MCF10A (−Dox, two centrosomes) cells were used. Elevated intensities of PCM proteins were detected only with CCB02 compared to vehicle‐treated cells. However, a slight increase in PCNT was observed with docetaxel treatment. Number of centrosomes tested, n = 100 (vehicle); n = 100 (for CCB02, taxol, bactallin III, docetaxel, and vinblastine). (N) = 3. Error bars, mean ± SEM. Ordinary two‐way ANOVA test **P < 0.001, ***P < 0.0001.
- CCB02 but not conventional tubulin binders (taxol, bactallin III, docetaxel, and vinblastine; concentrations used 30 nM) causes activation of extra centrosomes and prevents them from clustering. MCF10A cells with extra centrosomes (+Dox, extra centrosomes) were used. In contrast to CCB02‐treatment, interphase centrosomes of tubulin binder‐treated cells consistently display clustered centrosomes. Note, disrupted mitotic spindles were observed in tubulin binder‐treated cells due to their general toxicity to microtubules. Cells were stained with CPAP (green), Cep152 (magenta), and microtubules (α‐tubulin, red). Scale bar, 2 μm. Three independent experiments (N) = 3 with each at least 100 cells were examined per treatment.
- Bar graph quantifies percentages of cells exhibiting multipolar spindles in interphase and mitosis of vehicle, CCB02, and conventional tubulin binder‐treated cells. At least 150 cells were analyzed for each independent experiment (N) = 3. Error bars, mean ± SEM. Ordinary two‐way ANOVA test **P < 0.001.
- Kymographs (i) and averaged microtubule polymerization velocities mediated by vehicle and CCB02. Graph below represents the microtubule growth velocities mediated by vehicle (red) and CCB02 (blue) during microtubule end‐tracking assay (ii). CCB02 does not alter the microtubule growth although there is a slight increase in growth at 5 μM. At least 75 filaments were analyzed for each condition. The error bars represent mean ± SEM of three independent experiments (N) = 3.
- Snapshot images show the effect of CCB02 on microtubule dynamics in two centrosomes‐containing MCF10A cells. The microtubule was stained with SiR‐tubulin, and +end tip of microtubule was marked with EB3‐EGFP. A 5 μM CCB02 did not alter localization and dynamics of EB3 at the microtubules, indicating that the CCB02 does not affect microtubule dynamics (For details, refer to Movies EV4 and EV5). At least, 50 cells were analyzed for each condition. Scale bar, 2 μm.
- Box plots represent the speed (μm/s) (i), lifetime (s) (ii), and length (μm) (iii) of growing microtubule tracks in MCF10A cells treated with 1, 2, and 5 μM of CCB02. At least, 60 filaments were analyzed from each cell from a total of 30–40 cells. (N) = 3. Error bars, mean ± SD. Ordinary One‐way ANOVA test was performed. Boxes in all plots show the growing microtubule parameters such as speed (μm/s), lifetime (s), and length (μm/s). Horizontal lines in boxes show the median value. Whiskers at both extremes of boxes indicate minimum and maximum values.
CCB02 impairs the invasiveness of NSCLC cells in 3D‐organotypic cultures and has anti‐tumor activity in vivo
Centrosome amplification triggers cellular invasion in 3D cultures (Godinho et al, 2014; Ganier et al, 2018). To test whether CCB02‐mediated effects could impair the invasive behavior of NSCLC cells, we used 3D‐organotypic cultures of H1975T790M and HCC827‐GR exhibiting resistance to EGFR‐TKIs. In contrast to 5 μM erlotinib treatment, which is a known TKI, CCB02 at 5 μM was sufficient to prevent cellular invasion emerging from 3D spheres (Fig 8A, and Appendix Fig S8F and G). This could be due to the lack of proliferation and cellular death mediated by CCB02 treatment. Indeed, we noticed cell rounding, characteristics of prolonged mitotic arrest with concomitant cell death as revealed by activated caspase‐positive cells in H1975T790M spheroids (Appendix Fig S8G). As a result, CCB02‐treated spheroids did not grow further from its original size (Fig 8B). To corroborate this finding and further to test whether CCB02 treatment could prevent the migration of extra centrosome‐containing cells, we performed a wound‐healing assay using MDA‐MB‐231 cells (Wang et al, 2012). CCB02 treatment significantly inhibited the migration of cells as determined by prolonged duration in wound closure (Fig EV5A and B). Together, these data suggest that CCB02 has the ability to impair the invasion and migration behavior of cancer cells in vitro.
Figure 8. CCB02 treatment prevents invasive behavior of NSCLC in 3D organotypic spheroids.
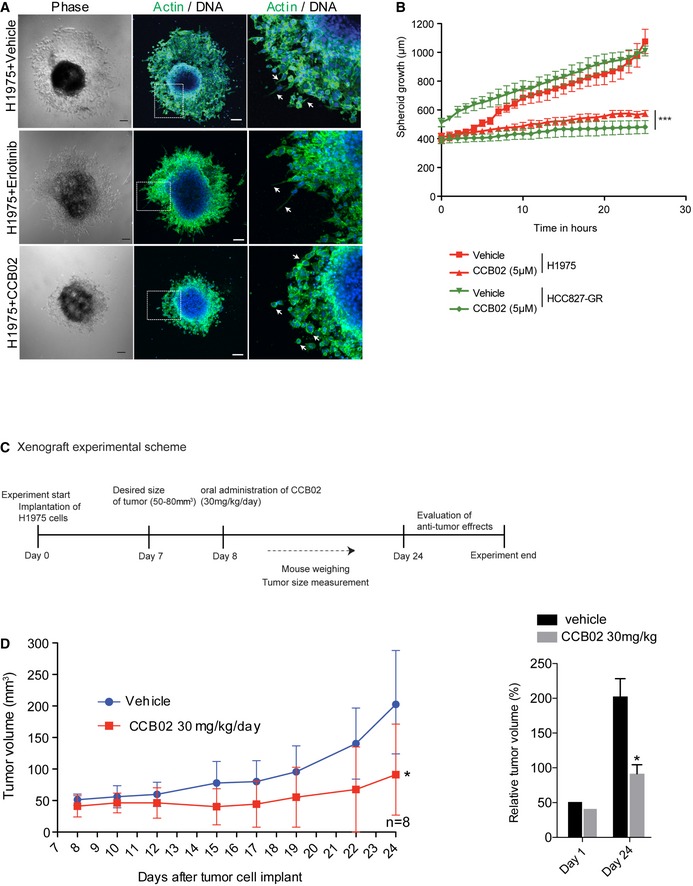
- Real‐time live imaging of NSCLC (H1975T790M) 3D spheroids with vehicle, 5 μM erlotinib, or 5 μM CCB02. At the end of live imaging, spheroids were fixed and stained for F‐actin (green). Arrow indicates invasive (vehicle‐treated) and non‐invasive (CCB02‐treated) structures in spheroids. Scale bar, 100 μm.
- Time series plot shows the spheroid growth rate measurements. CCB02 treatment prevents the spheroid growth rate by inhibiting the formation of invasive protrusions. More NSCLC spheroid experiments are given in Fig EV5A and B, and Movies EV5 and EV6. (N) = 3. At least 10 spheroids were measured for each condition. Error bars, mean ± SEM. Unpaired t‐test ***P < 0.0001.
- Experimental scheme of mouse xenograft using H1975T790M.
- Anti‐tumor activity of CCB02 in vivo. Left: Subcutaneous xenograft tumor volume measurements of H1975T790M (30 mg/kg of weight, daily) in nude mice treated with vehicle or CCB02. Right: Bar graph shows total tumor volume at day 1 and day 24 of vehicle‐ or CCB02‐treated xenograft. Error bars, mean ± SD with n = 8 for control vehicle and n = 8 for CCB02 treatment. Unpaired t‐test *P < 0.01.
Figure EV5. Related to Fig 8: CCB02 prevents MDA‐MB‐231 cell migration and induces multipolar mitosis in mouse xenografts.
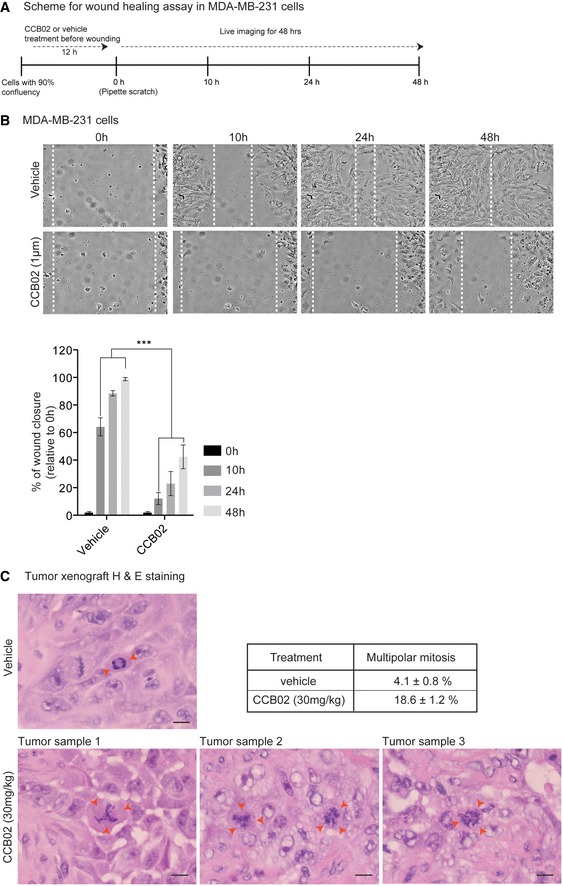
- Experimental scheme of wound‐healing assay using MDA‐MB‐231 cells.
- Snapshot of live cell images shows wound closure at various time points (0, 10, 24, and 48 h). Relative to vehicle treatment, CCB02 delays wound closure. Dashed lines mark cell‐free empty space. Scale bar, 100 μm. Bar diagrams at right quantify the percentage of relative wound closure. (N) = 3. Error bars, mean ± SEM. P‐values were obtained using two‐way ANOVA. ***P < 0.0001.
- Image shows H&E immunohistochemistry staining of representative xenograft tumor samples. In contrast to vehicle control xenograft tumors that show bipolar spindles (red arrows, top panel), CCB02 xenograft‐treated tumors show increased frequencies of multipolar mitotic cells, indicating mitotic catastrophe (red arrows, bottom panel). Scale bar, 10 μm. Table shows the percentage of multipolar mitotic cells. Data are represented as mean ± SEM. At least 350 mitotic cells were scored for vehicle and CCB02 treatment from independent xenograft tumors.
To evaluate whether CCB02 has anti‐tumor activity in vivo, we used nude mice bearing subcutaneous human lung (H1975T790M) tumor xenografts. We used two groups of mice bearing tumor volume > 100 mm3. We then delivered CCB02 (30 mg/kg/day) by oral gavage. At the end of the treatment, we measured the total tumor volume of CCB02‐treated groups and compared them with vehicle‐treated controls. We noticed a significant reduction in tumor growth rate in CCB02‐treated mice, indicating that CCB02 has an anti‐tumor activity (Fig 8C and D). Furthermore, immunohistochemistry analysis of tumor xenografts revealed that CCB02‐treated tumors harbor a significantly increased multipolar mitosis (Fig EV5C).
Discussion
Since centrosome amplification and clustering is a hallmark of cancer cells, perturbing centrosome clustering to induce mitotic catastrophe has been proposed as a selective strategy for tumors with a high incidence of centrosome amplification (Ogden et al, 2012; Chavali et al, 2014). Mechanisms to manipulate centrosomal activities in living cells have not been thoroughly investigated.
At interphase of the cell cycle, the centrosome contains a basal level of PCM (Piehl et al, 2004; Wiese & Zheng, 2006; Nigg & Stearns, 2011; Roostalu & Surrey, 2017). However, as cells enter into mitosis, centrosomes recruit PCM and increase in size. The process of PCM recruitment enabling centrosomes to nucleate robust microtubules in mitosis is termed as centrosome maturation (Conduit & Raff, 2010; Conduit et al, 2010, 2014). However, how the timing and amount of PCM recruitment is determined is yet to be critically analyzed.
Our earlier biochemical studies in Drosophila have identified that tubulin negatively affects Sas‐4's (in humans, it is CPAP) ability to form cytoplasmic protein complexes. Drosophila expressing a Sas‐4 variant that does not bind tubulin (Sas‐4‐ΔT) exhibited abnormal PCM recruitment (Zheng et al, 2016). In Sas‐4‐ΔT flies, the major PCM protein Cnn, normally detected only in mitotic centrosomes, was observed in interphase centrosomes, while mitotic centrosomes recruited at least twice the amount of Cnn as control centrosomes (Rusan & Peifer, 2007).10 These results suggested that tubulin present in wild‐type Sas‐4 complexes spatiotemporally regulates the amount of PCM recruitment; i.e., tubulin can function as a molecular switch in regulating Sas‐4‐/CPAP‐mediated PCM recruitment. From this, we hypothesized that perturbing CPAP–tubulin interaction could be used as a tool to enhance the microtubule‐nucleating activities of centrosomes prior to mitosis.
In this work, we tested the significance of perturbing CPAP‐tubulin interaction in cells, which harbor extra centrosomes. Indeed, our live imaging experiments revealed that CPAPΔT‐expressing cells exhibited an enhanced level of microtubule nucleation prior to mitosis starting from interphase itself. Importantly, the microtubule nucleation persisted over a longer period of time and prevented them from clustering leading to multipolar mitosis (Fig 1Aiv, Appendix S2A, and Movie EV1D).
To chemically inhibit CPAP–tubulin interaction, we identified CCB02, a selective inhibitor of CPAP–tubulin interaction, which caused similar effects as also caused by genetic perturbation of CPAP–tubulin interaction (Fig EV1). To dissect the mode of action, we identified that CCB02 binds β‐tubulin at the microtubule outer surface, thereby perturbing its interaction with CPAP. This enhances recruitment of CPAP‐interacting proteins to interphase centrosomes, which simultaneously activates centrosomes to nucleate an enhanced level of microtubules prior to mitosis (Fig 5 and Appendix Fig S6). Thus, the primary effect of perturbing CPAP–tubulin interaction is to activate centrosomes prior to mitosis.
When analyzing mitosis, we noticed that both genetic and chemical perturbation of CPAP–tubulin interaction specifically in extra centrosome‐containing cells causes prolonged activation of spindle assembly checkpoint (SAC; Fig EV3B and C). This could be due to at least two reasons. First, mitotic delay caused by centrosome‐declustering activity. Second, CCB02 as a tubulin binder could have an additional effect on spindle microtubules possibly via perturbing microtubule dynamics. In fact, such an effect has been demonstrated for griseofulvin, another tubulin binder that can prevent extra centrosomes from clustering (Rebacz et al, 2007; Raab et al, 2012; Ronnest et al, 2012). Interestingly, griseofulvin enhances Sas‐4 (in human CPAP)‐dependent PCM protein binding and recruitment (Gopalakrishnan et al, 2012). From this aspect, it appears that griseofulvin could also function through centrosome activation mechanism by elevating PCM recruitment.
The complete mechanisms of action of CCB02 remain unknown. Given the inherent nature of small molecules such as off‐target effects and cross‐reactivity, it is plausible that CCB02 can compete with microtubule‐binding proteins including kinesins such as HSET which has been shown to promote clustering of extra centrosomes (Kwon et al, 2008; Fielding et al, 2011). We could not exclude these additional possibilities that warrant for future experiments. However, based on our current data, CCB02's action is likely to be selective for extra centrosomes‐containing cells. Various experimental data support this notion. First, half‐maximal inhibition value (IC50) of CCB02 in various cancer cells ranges from 0.86 to 2.9 μM. Interestingly, PC9 cells which display a relatively weak inhibitory response (IC50 = 2.9 μM) harbor only a mild centrosome amplification. In contrast, cell lines with a high degree of centrosome amplification display strong inhibitory responses (Fig EV2A and B). Thus, the higher inhibitory response observed with these cells suggests that CCB02 is more potent in cancer cells with a high incidence of centrosome amplification.
EGFR activating mutations are some of the most common oncogenic driver mutations found in NSCLC patients, which reduces the overall survival rate of cancer patients. Although patients respond to initial EGFR‐TKI treatment, subsequent development of secondary resistance leads to treatment failure. Recent clinical and preclinical evidence suggests that combined approaches using multiple kinase inhibition or a combination of kinases and small‐molecule inhibitors of cell proliferation and cell migration can overcome secondary resistance (Brugger & Thomas, 2012). Interestingly, in contrast to erlotinib, we found CCB02 is able to perturb proliferation of EGFR‐mutant NSCLC (Fig 3B and Fig EV2C). Indeed, these cells exhibited an extreme increase in centrosome numbers. These findings suggest that developing a combination therapeutic strategy that uses centrosome‐activating agents could be beneficial in drug‐resistant cancers.
In conclusion, our work identifies a vulnerability of cancer cells to extra centrosomal activation providing a conceptually new strategy to specifically prevent cancer cell proliferation. Recent studies have linked extra centrosomes to tumor aggressiveness identifying differential functions of extra centrosomes in cancer cells (Ganem et al, 2009; Godinho et al, 2014; Wong et al, 2015). Our work provides mechanistic evidence that activating these extra centrosomes via enhanced PCM recruitment and microtubule nucleation may be a broadly useful tool to target cancers that exhibit extra centrosomes. The CPAP‐tubulin inhibitor CCB02 may not only serve as a useful tool to study centrosome functions in cells, but also a starting point for developing combinatorial treatment strategies, specifically when extra centrosomes indirectly contribute to a “bypass track” by which therapy‐resistant cancers develop.
Materials and Methods
Screening instruments
We used a HTS platform with an integrated instrumentation for plate and liquid handling. The screening was performed using a Sciclone G3 Liquid Handler from PerkinElmer (Waltham, MA, USA) with a Mitsubishi robotic arm (Mitsubishi Electric, RV‐3S11) and a Flexdrop dispenser (PerkinElmer, Waltham, MA, USA). The AlphaScreen assay was performed in white 384‐well Optiplates™ (PerkinElmer, 6007299). AlphaScreen signal was detected on the EnVision® Multilabel Reader (PerkinElmer, Waltham, MA, USA).
Compound library
The small‐molecule diversity set used in the CPAP‐tubulin HTS campaign was composed of compounds acquired from three providers, namely ChemDiv, Inc. (10,000 compounds), Enamine, Ltd. (10,000 compounds), and ChemBridge, Corp. (5,000 compounds). The purity of the compounds was > 90% as reported by the providers of the compounds. More detailed information on compound selection criteria is provided in Schorpp et al (2013).
AlphaScreen reagents
The AlphaScreen™ detection system (PerkinElmer, USA) used in this study consists of streptavidin donor beads and nickel chelate acceptor beads (AlphaScreen Histidine, Nickel Chelate Detection Kit, product #6760619C).
Assay development and screening of the CPAP‐tubulin AlphaScreen protein–protein interaction assay
Biotinylated tubulin (Tebu‐Bio, product number: 027T333P‐B) was captured by the streptavidin donor beads. His‐tagged PN2‐3 domain (aa 319–389) of CPAP that has tubulin‐binding region has been purified and bound with NiNTA Acceptor beads (Hsu et al, 2008; Zheng et al, 2016). All proteins and beads were diluted in assay buffer containing 1× PBS (pH 7.4), 0.5% bovine serum albumin (BSA) and 0.01% Tween‐20. Prior to performing the screening with 25,000 small molecules, the PPI assay was adapted to automation using a liquid handler and a compound transfer station (see instruments). The CPAP‐tubulin HTS campaign was performed in white 384‐well Optiplates™ as follows: (i) dispensation of 30 μl of 2× concentrated (20 nM, 10 nM final) biotinylated tubulin into white 384‐well plates using a robotic liquid handler (ii) transfer of 0.6 μl of compounds in DMSO (1 mM stock) or DMSO alone into each well using a compound transfer station with a nanoliter head yielding a final assay concentration of each compound of 10 μM and 1% v/v DMSO; for IC50 determination compounds were diluted in 100% DMSO (20 concentrations, 0.2 nM–100 μM final) (iii) dispensation of 10 μl of 6× concentrated (75 nM, 12.5 nM final) His‐CPAP or His‐CPAPF375A (CPAPΔT). CPAPΔT cannot interact with tubulin and was therefore included as a negative control to all assay plates; (iv) incubation of the plates for 1 h at room temperature; (v) addition of 10 μl of streptavidin donor and nickel chelate acceptor beads (30, 5 μg/ml final) followed by a further incubation for 1 h at room temperature in the dark; (vi) reading of the assay plates using laser excitation at 680 nm, with emission detected at 520–620 nm in an EnVision 2102 Multilabel Reader (PerkinElmer, USA). Subsequently, AlphaScreen and His‐tag frequent hitters (FH) were identified and could bioinformatically be excluded to create a final hit list (Schorpp et al, 2013). The quality and robustness of the assay, represented as Z′, were calculated.
Kinase screening assay
Kinase screening assay was performed commercially using KINOMEscan™ screening platform from DiscoverX. CCB02 was used at a concentration of 5,000 nM in duplicates. Kinase assay was performed as previously described (Fabian et al, 2005).
Cell culture
Unless and otherwise stated, all cancer cell lines were originally purchased from American Type Culture Collection (ATCC) and the German Resource Centre for Biological Material (DSMZ). Cell lines were cultivated at 37°C, 5% CO2, and 80–90% humidity with DMEM (#61965‐026, Gibco) or RPMI 1640 (#61870010, Gibco) containing 10% FBS (#P30‐19375, PAN Biotech), MEM (minimum non‐essential amino acids, #11140‐035, Gibco), 100 units/ml penicillin (#15140‐122, Gibco), and 100 units/ml streptomycin (#15140‐122, Gibco). All handling steps with the cell lines were done under a laminar flow. When the cells reached a density of about 90% the adherent cells were passaged beginning with removing the medium and a washing step with PBS (#18912014, Gibco). To detach the cells from the bottom of the 75 cm2/150 cm2 flask 0.5–2 ml of trypsin/EDTA (#25200056, Gibco) was added for 5 min at 37°C. The cells were then re‐suspended in at least 7 ml fresh medium and seeded at the desired density. Suspension cell lines were passaged by suitable dilution of the cell suspension. Cells were tested for mycoplasma using MycoAlert mycoplasma detection kit (#LT07‐418, Lonza).
Depletion of CPAP by siRNA
For depleting CPAP in cells, siRNA against 5′‐CCAAACAACUUCAUUCAUU‐3′ (Dharmacon siRNA, D‐010209‐02) CPAP was generated as previously described (Tang et al, 2009; Zhao et al, 2010; Zheng et al, 2014). Cells were treated with scramble and CPAP siRNA for 48 h. The depletion of CPAP was confirmed by Western blot and immunofluorescence.
Double thymidine block of HeLa cells
Double thymidine block of HeLa cells was performed as previously described (Harper, 2005). Briefly, cells were treated with 2 mM thymidine for 18 h. After this first thymidine block, cells were washed and released for 9 h. Second thymidine (2 mM) block was done for 15 h. Finally, cells released from the block and collected for G1/S stage of cell cycle.
Cell viability assay
Cell lines were plated as triplicates into sterile 96‐well plates at 5,000 cells/well densities for adherent cells. Every well was filled with 100 μl cell‐containing medium. To determine the cell number, improved Neubauer cell‐counting chamber (Marienfeld, Germany) was used. After 24 h of incubation of the plates at 37°C, CCB02 was added at increasing dosages, ranging from 0.1 to 15 μM together with a separate DMSO control. After 72–96 h, cell proliferation was performed colorimetrically by 3‐(4,5‐dimethylthiazol‐2‐yl)‐5‐(3‐carboxymethoxyphenyl)‐2‐(4‐sulphophenyl)‐2H‐tetrazolium, inner salt (MTS) assay, using 20 μl of CellTiter Aqueous One (#G3582, Promega) Solution reagent to each well. After 1–3 h of incubation at 37°C in a humidified, 5% CO2 atmosphere, the absorbance at 490 nm was recorded using a multimode plate reader (Mithras, Berthold Technologies, Germany) and the raw data were obtained. The IC50 (concentration needed to prevent cell proliferation by 50%) value was calculated using GraphPad Prism by plotting the percentage of cell survival as a function of drug concentration. Three independent experiments were performed per cell line.
3D organotypic culture system
Spheroid model was adapted from previously described methods (Friedrich et al, 2009). In short, 100 μl of culture medium containing 5,000 cells was added to an ultra‐low‐attachment 96‐well plate. The cells were supplemented with 2.5% of Matrigel (#356230, BD Biosciences) for compact spheroid formation (Ivascu & Kubbies, 2007). After 48–96 h, spheroids were transferred to a Labtek multi‐well plate (#154534, Thermo Scientific) or Ibidi μ‐Slide chamber plates (#80426 and #80286, Ibidi) containing polymerized Matrigel: collagen‐1 mixture in each well. For spheroid treatment, the supernatant medium on top of Matrigel: collagen‐1 mixture was replaced with vehicle or diluted drugs supplemented in a standard medium.
Wound‐healing assay
Wound‐healing assay was performed as previously described (Wang et al, 2012; Jonkman et al, 2014). In brief, MDA‐MB‐231 cells were grown as monolayer of cells to confluence in 2‐well ibidi chamber plate (#80286), and at experimental time point 0 h, a scratch was made in each well using a pipette tip (p10). The cells were washed twice with PBS before their subsequent incubation with culture medium in the presence of vehicle or CCB02 (1 μM). The cells were pretreated with CCB02 for 12 h before the scratch. In order to monitor cell migration in the scratched area, live cell imaging was performed using Leica DMI6000B widefield microscope with 10×/0.22. During live cell imaging, cells were maintained at 37°C with humidified CO2 (3–5%) using an enclosed temperature and CO2 controller. Images were collected at 0, 10, 24, and 48 h. All the captured images from each experiment were processed using Fiji/ImageJ.
2D and 3D indirect immunofluorescence microscopy
For 2D immunofluorescence, cells were grown in glass coverslips (18 mm, No. 1.5H, #0117580, Marienfeld) in multi‐well plates. Cancer cells were treated with CCB02 in a concentration range of 1–2 μM depending on the cell type. After vehicle or CCB02 treatment, cells were washed with PBS and fixed with ice‐cold methanol at −20°C for 10 min or 4% PFA. Fixed cells were blocked with 0.5% fish gelatin in PBS (wash buffer) for 10–30 min at room temperature or at 4°C overnight and then incubated with primary antibodies (centrosomal markers, as in figure legends) for 60 min at room temperature or at 4°C overnight. After incubation, cells were washed with wash buffer for three times and incubated with species‐specific secondary antibodies (#A11001, Alexa Fluor 488 anti‐mouse, #A11037, #A11008, Alexa Fluor 488 anti‐rabbit, Alexa Fluor 594 anti‐rabbit, #A11005 Alexa Fluro 594 anti‐mouse, #A21245, Alexa Fluor 647 anti‐rabbit, and #A21235, Alexa Fluro 647 anti‐mouse; Molecular Probes, Invitrogen) at 1:1,000 dilution for 60 min at room temperature. DAPI (1:1,000; #D21490, Invitrogen) was used to stain DNA along with last secondary antibody. Antibodies used include anti‐γ‐tubulin mouse (#T6657, Sigma), anti‐pericentrin rabbit (#ab4448, Abcam), anti‐tubulin, anti‐CPAP mouse (Zheng et al, 2014), anti‐Cep152 rabbit (Kind gift from Erich Nigg), anti‐centrin‐3 (#9E306, Novus Biologicals), and cyclin A2 (#ab16726, Abcam). Images were collected with Olympus FV1000 laser scanning confocal microscope using 405‐, 559‐, and 635‐nm laser light. 60× objective was used to collect images. Capture images were corrected for background, brightness, and contrast using Fiji/ImageJ (Schindelin et al, 2012) and Adobe Photoshop. For immunofluorescent quantifications, at least five image fields from three independent experiments with vehicle or compound treatment were used. Fiji was used to quantify the intensity. Statistical analysis was performed using ordinary two‐way ANOVA in GraphPad Prism version.
Staining of 3D spheroids was done after fixing the organoid with 4% PFA for 30 min at room temperature, followed by washing with PBS containing 30 mM glycine for three times 10 min each. After fixation, cells were permeabilized with 0.5% Triton X‐100 in PBS for 15 min at 4°C. Permeabilized cells were blocked with 0.5% fish gelatin in PBS (wash buffer) for 1 h at room temperature. Live actin dye (#R37110, Invitrogen) was used to stain actin filaments for 2 h at room temperature. After actin staining, cells were washed with wash buffer for 30 min for three times. DAPI (1:1,000 in wash buffer) was used to stain DNA for 30 min. 3D spheroids were imaged using 10×/0.4 plan Apo air objective on Olympus FV1000 laser confocal scanning microscope. Images were processed using Fiji/ImageJ and Adobe Photoshop.
Lentiviral production and transduction of target cells
Constitutive overexpression of GFP‐tagged CPAP‐WT and CPAPΔT lentiviral vectors was prepared using pSinEF2. The cloned vectors were packed into lentivirus using second‐generation packaging plasmids (pPMD, addgene #12259 and pPAX, addgene #12260). Briefly, GFP‐tagged CPAP vectors and packaging plasmids were transfected into HEK293TS cells using calcium chloride. After 16 h, medium was changed and the virus was collected after 8 h. The freshly collected virus was used to transduce target cells in 1:1 ratio for 72–96 h. For transient experiments, cells were immediately fixed after 48–72 h of transduction.
To generate inducible expression of CPAP‐WT and CPAPΔT (For spheroid experiment given in Fig 1), we used pLIX lenti inducible gateway cloning vector (addgene #41395). The target cells were infected with lentivirus containing pLIX‐CPAP‐WT and pLIX‐CPAPΔT and selected with puromycin (#ant‐pr‐1, InvivoGen). No individual clones were selected at any point rather all the selected cells were pooled to make a population. For the expression of the transgenes, all the cell lines generated were induced with 2 μg/ml of doxycycline (#D9891‐1G, Sigma) for 24–48 h. For live cell imaging, pcDNA CPAP‐Myc and CPAP‐ΔT versions were introduced transiently into MCF10A cells using TransIT‐X2 dynamic transfection reagent. These plasmids were kindly provided Dr. Tang TK (Tang et al, 2009).
Immunoprecipitations and Western blotting
Immunopurification of cytoplasmic CPAP complex was done as previously described (Gopalakrishnan et al, 2012). In brief, as previously described, cell extracts were prepared by lysing the cells using BRB80 buffer. The cell extracts were centrifuged 100,000 g for at least 60 min, and the high‐speed lysate (the supernatant) was used for further purifications. Protein G beads (#17061801, GE healthcare) were coated with anti‐CPAP antibody overnight at 4°C. The antibody‐coated beads and vehicle‐ or CCB02‐treated extracts were mixed and incubated at 4°C for 4 h. Then, it was washed with extract buffer containing 0.1% Triton X‐100, and then twice with cell extract buffer. For eluting the complexes, beads were boiled with 2× Laemmli buffer. Protein lysates were subjected to SDS–PAGE on 8 or 10% polyacrylamide gel, transferred onto nitrocellulose membranes, which were incubated with indicated primary antibodies, washed, and probed with HRP‐conjugated secondary antibodies [#G21040, goat mouse IgG (H + L) and #G21234, goat rabbit IgG (H + L)]. The band intensities were quantified from two independent experiments (n = 3, technical replicates) using Fiji. Pull‐down experiments were performed using GST‐tagged PN2‐3 domain of CPAP. In Brief, GST‐PN2‐3 was expressed in BL21 and we purified the protein using GST beads. Once the GST‐PN2‐3 bound to GST beads, we performed a pull‐down experiment using cellular extracts or using purified tubulin in the presence of different concentrations of CCB02. Porcine/bovine tubulin was purified as previously described (Zheng et al, 2016). Similarly, CCB02‐biotin pull‐down assay was performed using cellular extracts. Before this, the CCB02‐biotin was bound to strep‐Tactin sepharose resins (#2‐1201‐002, IBA, Germany) and washed several times with wash buffer (#2‐1003‐100, IBA, Germany) before cellular extracts were loaded.
Mass spectrometric analysis
The pull‐down proteins were on‐bead digested and processed for mass spectrometric analysis. Briefly, the beads were boiled in 10 μl of RapiGest™ at 95°C for 10 min followed by reduction of disulfide bridges by incubating with 5 mM TCEP for 30 min at RT, 750 rpm. The free cysteines were alkylated by adding 10 mM of chloroacetamide and incubating the vials for 30 min, 750 rpm, RT. Finally, the proteins were digested using 1 μg of trypsin (Promega) at 37°C, 750 rpm overnight. The protease activity was quenched by adding 20 μl of 10% formic acid (Sigma). The eluted peptides were desalted using C18 STAGE‐Tips and dried in SpeedVac. For LC‐MS analysis, each sample was reconstituted in 20 μl of 5% ACN and 0.1% formic acid and 5 μl was injected into the mass spectrometer. LC separation was carried on an Agilent 1100 nano‐flow LC system (Agilent Technologies). Buffer A was 0.1% formic acid in water. Buffer B was 95% acetonitrile, 0.1% formic acid in water. Injected peptides were loaded on an in‐house packed C18 trap column (1.5 cm, 360 μm outer diameter, 150 μm inner diameter, Reprosil‐Pur 120 Å, 5 μm, C18‐AQ, Dr. Maisch) at flow rate 10 μl/min and washed for 5 min with Buffer A. Peptide separation was done on an analytical C18 capillary column (15 cm, 360 μm outer diameter, 75 μm inner diameter, Reprosil‐Pur 120 Å, 5 μm, C18‐AQ, Dr. Maisch) at a flow rate of 300 nl/min with a gradient from 5–38% of Buffer B for 90 min. Eluting peptides were analyzed on a LTQ‐Orbitrap Velos hybrid mass spectrometer (Thermo Electron) in positive ion mode. The instrument was operated in a data‐dependent acquisition mode where the 30 most intense ions in the MS scan (m/z range from 350 to 1,600, resolution set to 60,000 at m/z 400) were selected for fragmentation by HCD mode. Automatic gain control target was set at 106 and 105 for MS1 and MS2, respectively. Sequenced precursors were put on an exclusion list for 30 s. The lock mass option (m/z 445.1200 was used for internal recalibration (Olsen et al, 2005).
The acquired RAW data were analyzed using MaxQuant software (Cox & Mann, 2008) version 1.5.2.8 based on Andromeda search engine (Cox et al, 2011). Each sample was given a unique name. Trypsin protease was selected, and LFQ option was highlighted. The human UniProt database (downloaded in December 2016; containing 20,129 reviewed entries) was used for identifying proteins. The protein and the peptide FDR was set to 0.01. The identified proteinGroup.txt file was processed and analyzed using Perseus (Hubner et al, 2010; Tyanova et al, 2016) version 1.5.5.3. Briefly, the logarithm of LFQ intensities of CCB02/lysate was analyzed by Student's t‐test (n = 3). For volcano plot, the obtained P‐values and logarithm of CCB02/lysate were plotted with S0 = 0.1, FDR = 0.05. The significant hits were categorized into various functional categories manually.
Centrosome fractionation
Discontinuous sucrose gradient (35–70%) was used to isolate centrosomes as described previously (Moritz et al, 1995; Gopalakrishnan et al, 2011, 2012). In brief, cells were treated with vehicle or CCB02 and lysed using BRB80 buffer containing 100 mM KCl. Lysed extracts were first centrifuged at 1,500 g for 20 min. The resulting supernatant was then layered on top of the discontinuous sucrose gradient of 35–70% prepared manually. After centrifugation at 243,000 g for 2 h at 4°C, fractions were collected and resolved using 8–10% SDS–PAGE. Proteins were transferred to nitrocellulose membrane and incubated with primary antibodies overnight at 4°C after blocking. Following this, membranes were incubated with species‐specific secondary antibody at room temperature for 1 h and developed using chemiluminescence (Thermo Scientific).
Long‐term time‐lapse imaging
Cells expressing inducible CPAPWT and CPAPF375A as monolayer cells (MCF10A with Plk4 overexpression) or spheroids (MDA‐MB‐231) were grown in Labtek multi‐well plate (#154534, Thermo Scientific) or Ibidi μ‐Slide chamber plates (#80426 and #80286, Ibidi) for 2D and 3D imaging. The microtubules (MTs) were stained with live SiR‐Tubulin (Spirochrome AG). Images were captured using Leica DMI6000B widefield microscope with 10×/0.22 or 20×/0.40 Objective. The microscope is equipped with Leica DFC365 FX camera, a high‐precision Pecon motorized stage, and Leica Adaptive focus control. During live cell imaging, cells were maintained at 37°C with humidified CO2 (3–5%) using an enclosed temperature and CO2 controller. All the captured images from each experiment were processed using Fiji/ImageJ.
NMR spectroscopy
All NMR spectra were recorded on an 800‐MHz Bruker spectrometer equipped with a TCI cryoprobe, in buffer containing 1.5 mM phosphate, 1.5 mM calcium, and sodium, 5% DMSO at pH 7 at 298 K. For NOESY spectra, the H2O buffer was exchanged to D2O buffer in the concentrator at 4°C. The NOESY spectra were collected with mixing times of 40, 70, 100, and 150 ms, processed with NMRPipe (Delaglio et al, 1995) and analyzed using CCPNmr analysis (Vranken et al, 2005).
Modeling by docking
CCB02.3 structure was built in Maestro 2012 version 9.3.5 (Schrödinger, LLC) and imported into UCSF Chimera after minimization (Pettersen et al, 2004). The docking poses of compound CCB02.3 with tubulin were generated using AutoDock Vina tool in UCSF Chimera (Trott & Olson, 2010). Docking was performed using grid box as the peptide‐binding interface on tubulin. Ten docking poses of CCB02.3 were generated, out of which eight accessed Phe385 binding pocket while two were docked into Phe375 pocket.
Isothermal titration calorimetry
Calorimetric experiments were conducted at 15°C with a MicroCal PEAQ‐ITC instrument (Malvern). Tubulin samples were dialyzed against the 1×BRB80 buffer (80 mM PIPES‐K, 1 mM MgCl2, 1 mM EGTA, pH 6.8, 0.5% DMSO) prior to titration. And the lyophilized compound CCB02 was solubilized using the dialysis buffer. For all ITC titration curves, 25 μM tubulin and 173 μΜ compound CCB02 were used. Protein concentration was determined by absorbance spectroscopy at 280 nm. Compound was quantified by weighing on a large scale. For fitting the ITC titration curve, acquired calorimetric titration data were analyzed using MicroCal PEAQ‐ITC Analysis Software using the One Set of Binding Sites fitting model.
Microtubule regrowth assay
The microtubule (MT) regrowth assay was performed as previously described (Sankaran et al, 2005; Choi et al, 2010). For microtubule regrowth assay, MDA‐MB‐231 and MCF10A (+Dox, extra centrosomes) cells were pretreated with vehicle or CCB02 for 72 h and then treated with nocodazole for 16 h. After this, cells were placed in ice with cold medium for 90 min to completely depolymerize the microtubules. MT regrowth was induced by replacing the cold medium with prewarmed medium at 37°C. At the indicated time points after the medium replacement, the cells were subsequently fixed with 4% PFA and stained for anti‐α‐tubulin and anti‐γ‐tubulin.
Microtubule end‐tracking assay
Microtubule polymerization assay was conducted following the previously reported method (Bieling et al, 2010). For preparation, the reaction chambers, microscope slides, and biotin‐coated coverslips were assembled using double‐sided tape. After blocking reaction chambers with 1% pluronic F127 and 0.5 mg/ml κ‐casein for 5 min, 50 μg/ml streptavidin was flowed in and incubate for 5 min. Microtubule seeds assembled from 50 μM tubulin mixed with 10% rhodamine‐labeled tubulin and 10% biotin‐labeled tubulin under 1 mM GMPCPP were then specifically attached to the functionalized surface by previously bound streptavidin. After washing the chamber with 1×BRB80 buffer, tubulin polymerizing was initiated by flowing in 1, 2 or 5 μM of CCB02 and taxol, 20 nM GFP‐tagged EB1, 15 μM tubulin, and 1.5 μM rhodamine‐labeled tubulin, which were diluted in oxygen scavenger system (50 mM glucose, 400 μg/ml glucose‐oxidase, 200 μg/ml catalase, and 4 mM DTT) supplied tubulin polymerization buffer (80 mM PIPES‐K, 150 mM KCl, 1 mM EGTA, 5 mM GTP, 4 mM MgCl2, pH 6.8). After immediately sealing the reaction chamber with candle wax, images were collected every 3 s using a TIRF (total internal reflection fluorescence) microscope (Nikon Eclipse Ti). ImageJ software (http://rsbweb.nih.gov/ij/) was used for kymograph presentation and image analysis.
Live EB3 imaging was performed using EB3‐EGFP constructs (kindly provided by Dr. Anna Akhmanova). Briefly, cells were grown in Ibidi μ‐Slide chamber plates (#80426 and #80286, Ibidi) and transiently transfected with EB3‐EGFP plasmids. After 24 h of transfection, cells were treated with DMSO vehicle or CCB02 (1, 2 and 5 μM) and EB3 dynamics was observed using laser scanning confocal microscope (Leica SP8, Leica Germany). The microtubules (MTs) were stained with live SiR‐Tubulin (Spirochrome AG). The images were recorded every 2 s. During live cell imaging, cells were maintained at 37°C with humidified CO2 (3–5%) using an enclosed temperature and CO2 controller. ImageJ software and MTrackJ plugin (http://rsbweb.nih.gov/ij/) was used for tracking and measuring EB3 and MT dynamics.
Animals
For mouse xenograft experiments, NMRI‐nu (RjOrl:NMRI‐Foxn1nu/Foxn1nu) female mice of 4–6 weeks old were used for experiments. All experiments and protocols were performed in accordance with the guidelines of the responsible national authority and approved by the local Governmental Committee for Animal Experimentation (license: 84‐02.04.2015.A541).
Breast cancer xenograft model
Proof‐of‐principle xenograft model: For each xenograft (n = 4), 5 × 106 MDA‐MB‐231 tumor cells expressing CPAP‐WT and CPAPΔT (a mutant version of CPAP that does not bind to tubulin) suspended in PBS were injected subcutaneously into the flank of male nude mice. Tumor size was monitored every second day by measuring perpendicular diameters. Tumor volumes were calculated by determination of the largest diameter and its perpendicular according to the following equation: tumor volume = a × (b 2/2), where a represents the largest diameter and b represents the perpendicular diameter. The experimenter was not blinded.
Lung cancer xenograft model
Lung cancer (H1975T790M) xenograft model: Immunodeficient NMRI‐nu (RjOrl:NMRI‐Foxn1nu/Foxn1nu) female mice from Harlan, the Netherlands, were delivered at the age of 4–6 weeks and were used for experiments after at least 1 week of quarantine. H1975 cells were cultivated at 37° C, 5% CO2, and 80–90% humidity with DMEM (#61965‐026, Gibco) containing 10% FBS (#P30‐19375, PAN Biotech), MEM (minimum non‐essential amino acids, #11140‐035, Gibco), 100 units/ml penicillin (#15140‐122, Gibco), and 100 units/ml streptomycin (#15140‐122, Gibco). H1975 cells (5 × 106 in 200 μl) were injected into 14 NMRI‐nu female mice. Animals and tumor implants were monitored daily until solid tumor growth was detectable in a sufficient number of animals. At randomization, the volume of growing tumors was determined. Animals fulfilling the randomization criteria (i.e., bearing tumors of 80–100 mm3) were then distributed into experimental groups (n = 8 for vehicle control and n = 8 for CCB02 treatment), aiming at comparable median and mean group tumor volumes of approximately 100–120 mm3. CCB02 was administered (30 mg/kg of weight, daily) by oral gavage to mice. Animals were routinely monitored at least twice daily. Animals were weighed thrice a week or daily if body weight losses in excess of 15% were recorded. Tumor volumes were determined by two‐dimensional measurement with a caliper on the day of randomization and then twice weekly according to the formula: (a × b 2) × 0.5 where a represents the largest and b the perpendicular tumor diameter of the tumor representing an idealized ellipsoid. Institutional guidelines were strictly followed while performing the animal experiments. All experiments are approved by the local authorities and are conducted according to the guidelines of the German Animal Welfare Act (Tierschutzgesetz). The experimenter was not blinded.
Statistical analysis
The statistical analyses were performed using GraphPad Prism version 5‐7. Statistical information including test results (P‐value), definition of center values as mean, and definition of error bars as standard error mean is indicated in the text or the figure legends. Different compound treatments per cell line were done in triplicate (N = 3) as independent experiments. IC50 values were calculated (N = 3–4 replicates per cell line) using dose–response curve. Western blot band intensities were calculated using Fiji. The intensity values from area under the curve (AUC) were used to calculate Western blot IC50 values. Dose–response inhibition model was used to calculate IC50 value. Unpaired Student's t‐test or ordinary one‐way or two‐way ANOVA was used to determine statistical significance of graphs (P < 0.01 vs. control was considered as significance). Error bars represent standard error mean.
Chemical synthesis of CCB02
Unless otherwise noted, all reagents were obtained from commercial sources and used without further purification. Technical grade solvents used for aqueous workup were distilled prior to use. Dry tetrahydrofuran and methanol were purchased from Acros. Reverse‐phase flash chromatography was performed on a Grace Reveleris® Prep Purification System using C18 40 μM cartridges. Analytical thin‐layer chromatography (TLC) was performed on silica (silica gel 60 F 254)‐coated plates. Compounds were detected by ultraviolet (UV) irradiation at 254 or 366 nm. HPLC‐UV/MS analysis was performed on a Waters X‐Bridge C18 (4.6 × 30 mm, 3.5 μm) column using a Dionex UltiMate 3000 HPLC system coupled with a Thermo Finnigan LCQ ultrafleet mass spectrometer (gradient: 5–95% of acetonitrile + 0.1% formic acid v/v in water + 0.1% formic acid v/v over 5‐min period, then hold 95% of acetonitrile + 0.1% formic acid v/v in water + 0.1% formic acid v/v for 1 min; flow rate: 1.1 ml/min; UV detection at 214 and 280 nm). Infrared (IR) spectra were recorded on a JASCO IR‐4100 (ATR). High‐resolution mass spectrometry (HRMS) measurements were performed on a Thermo Finnigan LTQ FT apparatus using an electrospray ionization (ESI) detector. NMR spectra were recorded at 303 K on a Bruker Avance III HD 400 (400 MHz) spectrometer. Chemical shifts are reported in parts per million (ppm) relative to residual d5‐DMSO (δH = 2.50 ppm) and d6‐DMSO (δC = 39.52 ppm). Splitting patterns are designated as s (singlet), d (doublet), t (triplet), td (triplet of doublets), ddd (doublet of doublets of doublets), m (multiplet), or bs (broad signal). The coupling constants (J) are reported in Hertz (Hz). 3‐Chlorobenzo[b][1,6]naphthyridine‐4‐carbonitrile was synthesized as described in Ivanov et al (2002), Ivanov et al (2004) and Yalysheva et al (1986).
CCB02 (3‐Methoxybenzo[b][1,6]naphthyridine‐4‐carbonitrile) was prepared from Chlorobenzo[b][1,6]naphthyridine‐4‐carbonitrile (Appendix Fig S5) according to the following procedure: A suspension of 3‐Chlorobenzo[b][1,6]naphthyridine‐4‐carbonitrile (400 mg, 1.67 mmol) in a mixture of absolute methanol (30 ml) and dry tetrahydrofuran (20 ml) was stirred at reflux, and 0.5 M solution of sodium methoxide in methanol (4.00 ml, 2.00 mmol, 1.2 eq) was added dropwise over 1 h. The resulting brown solution was kept at reflux for further 30 min, cooled, quenched with saturated aqueous solution of ammonium chloride (2 ml), and concentrated in vacuo. The residue was partitioned between water (10 ml) and methylene chloride (50 ml). The layers were separated, and the aqueous phase was extracted with methylene chloride (2 × 15 ml). The combined organic extract was washed with saturated aqueous ammonium chloride, dried over anhydrous sodium sulfate, filtered, and concentrated in vacuo. The residue was subjected to reverse‐phase flash chromatography (gradient: 0–45% acetonitrile in water v/v) to provide the title compound (210 mg, 0.89 mmol, 54%) as a yellow solid.
Thin‐layer chromatography (pentane: ethyl acetate, 2:1 v/v): Rf = 0.33; 1H NMR (400 MHz, d6‐DMSO) δ 9.71 (s, 1H, H‐1), 9.48 (s, 1H, H‐10), 8.26 (d, J = 8.4 Hz, 1H, H‐9), 8.15 (d, J = 8.8 Hz, 1H, H‐6), 8.04 (ddd, J = 8.6, 6.6, 1.5 Hz, 1H, H‐7), 7.70 (td, J = 6.8, 3.3 Hz, 1H, H‐8), 4.23 (s, 3H, OCH 3); 13C NMR (101 MHz, d6‐DMSO) δ 166.6 (C‐3), 160.4 (C‐1), 152.6 (C‐9a), 150.0 (C‐10a), 141.5 (C‐10), 135.2 (C‐7), 130.5 (C‐9), 128.8 (C‐6), 126.8 (C‐8), 126.3 (C‐4a), 118.3 (C‐5a), 115.2 (CN), 86.5 (C‐4), 56.1 (OCH3); IR (ATR) νmax (cm−1) 3044, 3019, 2958, 2894, 2847, 2224, 1605, 1557, 1512, 1466, 1411, 1331, 1285, 1181, 1140, 1107, 1041, 969, 800, 741, 613; ESI‐H RMS (m/z): [M + H]+ calcd. For C14H10N3O, 236.08184; found, 236.08194; LCMS (m/z): [M + H]+ 236, retention time 3.23 min.
Chemical synthesis of CCB02‐Biotin and CCB02‐2.5‐Biotin
Under an argon atmosphere, 125 mg biotin (0.5 mmol, 1 eq.) was dissolved in 5 ml dry MeCN and the resulting reaction mixture was stirred to 0°C before 139 mg HOBt (1.0 mmol, 3 eq.) and 197 mg EDC·HCl (1 mmol, 3 eq.) was added. After 15 min, the free amine (90 mg, 0.34 mmol, 1 eq.) in 1 ml dry MeCN was added slowly. The resulting orange‐red reaction mixture was stirred for 20 h at room temperature, before 25 μl H2O was added. The reaction was directly purified by chromatography (CHCl3/MeOH = 1:0 → 1:1) to obtain the desired compound as an orange solid (121 mg, 0.25 mmol, 82%).
Author contributions
JG and AM conceived the concept and project. AM performed most of the experiments. KS and MS performed NMR experiments. KSc and KH performed drug screening. FZ, XZ, and HL performed in vitro microtubule and ITC biophysical experiments. MD, TK, AM, and H‐GS did chemical synthesis of CCB02. SM and HU did mass spectrometry. IM, MO, KG, RU, CY, and RA and RB performed mouse xenograft experiments. JS, AR, AW, and AAH involved in constructs and biochemical experiments. JG and AM wrote the manuscript. JG supervised the work.
Conflict of interest
The authors declare they have no conflict of interest. The University of Cologne has filed a European patent application related to the structures, syntheses, and uses of CCB02 and chemically related CCB02 inhibitors. Requests for CCB02 should be directed to JG (jay.gopalakrishnan@hhu.de).
Supporting information
Appendix
Expanded View Figures PDF
Table EV1
Table EV2
Movie EV1
Movie EV2
Movie EV3
Movie EV4
Movie EV5
Movie EV6
Review Process File
Acknowledgements
We thank Scarlett Dornauer and Ina Rothenaigner of the Assay Development and Screening Platform for helping us with the screening and data analysis. We thank Drs. Nigg and Tang for their kind gift of Cep152 and CDK5RAP2 antibodies. We are grateful to Dr. Susana Godinho for providing us inducible MCF10A cells overexpressing Plk4. We thank Dr. Anna Akhmanova for her kind gift of EB3‐EGFP plasmid. We thank Li Ming Gooi and Elke Gabriel for useful discussions, experiments, and their comments. We thank Marion Müller for technical assistance with the mouse experiment. K.S. is thankful for support by the IMPRS‐LS Graduate School. This work was supported by grants from the Human Frontier Science Program (HFSP) RGY0064/2015 to J.G. and H.L. Deutsche Forschungsgemeinschaft (DFG) GO 2301/2‐2 to J. G. and A.R.
The EMBO Journal (2019) 38: e99876
See also: Y Liu & L Pelletier (January 2019)
References
- Ahsan A (2016) Mechanisms of resistance to EGFR tyrosine kinase inhibitors and therapeutic approaches: an update. Adv Exp Med Biol 893: 137–153 [DOI] [PubMed] [Google Scholar]
- Avidor‐Reiss T, Gopalakrishnan J (2013) Building a centriole. Curr Opin Cell Biol 25: 72–77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basto R, Brunk K, Vinadogrova T, Peel N, Franz A, Khodjakov A, Raff JW (2008) Centrosome amplification can initiate tumorigenesis in flies. Cell 133: 1032–1042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bettencourt‐Dias M, Glover DM (2007) Centrosome biogenesis and function: centrosomics brings new understanding. Nat Rev Mol Cell Biol 8: 451–463 [DOI] [PubMed] [Google Scholar]
- Bieling P, Telley IA, Hentrich C, Piehler J, Surrey T (2010) Fluorescence microscopy assays on chemically functionalized surfaces for quantitative imaging of microtubule, motor, and +TIP dynamics. Methods Cell Biol 95: 555–580 [DOI] [PubMed] [Google Scholar]
- Bolanos‐Garcia VM, Blundell TL (2011) BUB1 and BUBR1: multifaceted kinases of the cell cycle. Trends Biochem Sci 36: 141–150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brugger W, Thomas M (2012) EGFR‐TKI resistant non‐small cell lung cancer (NSCLC): new developments and implications for future treatment. Lung Cancer 77: 2–8 [DOI] [PubMed] [Google Scholar]
- Chavali PL, Putz M, Gergely F (2014) Small organelle, big responsibility: the role of centrosomes in development and disease. Philos Trans R Soc Lond B Biol Sci 369: 20130468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chavali PL, Chandrasekaran G, Barr AR, Tatrai P, Taylor C, Papachristou EK, Woods CG, Chavali S, Gergely F (2016) A CEP215‐HSET complex links centrosomes with spindle poles and drives centrosome clustering in cancer. Nat Commun 7: 11005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi YK, Liu P, Sze SK, Dai C, Qi RZ (2010) CDK5RAP2 stimulates microtubule nucleation by the gamma‐tubulin ring complex. J Cell Biol 191: 1089–1095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou EJ, Hung LY, Tang CJ, Hsu WB, Wu HY, Liao PC, Tang TK (2016) Phosphorylation of CPAP by aurora‐A maintains spindle pole integrity during mitosis. Cell Rep 14: 2975–2987 [DOI] [PubMed] [Google Scholar]
- Conduit PT, Brunk K, Dobbelaere J, Dix CI, Lucas EP, Raff JW (2010) Centrioles regulate centrosome size by controlling the rate of Cnn incorporation into the PCM. Curr Biol 20: 2178–2186 [DOI] [PubMed] [Google Scholar]
- Conduit PT, Raff JW (2010) Cnn dynamics drive centrosome size asymmetry to ensure daughter centriole retention in Drosophila neuroblasts. Curr Biol 20: 2187–2192 [DOI] [PubMed] [Google Scholar]
- Conduit PT, Feng Z, Richens JH, Baumbach J, Wainman A, Bakshi SD, Dobbelaere J, Johnson S, Lea SM, Raff JW (2014) The centrosome‐specific phosphorylation of Cnn by Polo/Plk1 drives Cnn scaffold assembly and centrosome maturation. Dev Cell 28: 659–669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conduit PT, Wainman A, Novak ZA, Weil TT, Raff JW (2015) Re‐examining the role of Drosophila Sas‐4 in centrosome assembly using two‐colour‐3D‐SIM FRAP. Elife 4: e08483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cormier A, Clement MJ, Knossow M, Lachkar S, Savarin P, Toma F, Sobel A, Gigant B, Curmi PA (2009) The PN2‐3 domain of centrosomal P4.1‐associated protein implements a novel mechanism for tubulin sequestration. J Biol Chem 284: 6909–6917 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox J, Mann M (2008) MaxQuant enables high peptide identification rates, individualized p.p.b.‐range mass accuracies and proteome‐wide protein quantification. Nat Biotechnol 26: 1367–1372 [DOI] [PubMed] [Google Scholar]
- Cox J, Neuhauser N, Michalski A, Scheltema RA, Olsen JV, Mann M (2011) Andromeda: a peptide search engine integrated into the MaxQuant environment. J Proteome Res 10: 1794–1805 [DOI] [PubMed] [Google Scholar]
- Delaglio F, Grzesiek S, Vuister GW, Zhu G, Pfeifer J, Bax A (1995) NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J Biomol NMR 6: 277–293 [DOI] [PubMed] [Google Scholar]
- Engelman JA, Zejnullahu K, Mitsudomi T, Song Y, Hyland C, Park JO, Lindeman N, Gale CM, Zhao X, Christensen J, Kosaka T, Holmes AJ, Rogers AM, Cappuzzo F, Mok T, Lee C, Johnson BE, Cantley LC, Janne PA (2007) MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science 316: 1039–1043 [DOI] [PubMed] [Google Scholar]
- Fabian MA, Biggs WH III, Treiber DK, Atteridge CE, Azimioara MD, Benedetti MG, Carter TA, Ciceri P, Edeen PT, Floyd M, Ford JM, Galvin M, Gerlach JL, Grotzfeld RM, Herrgard S, Insko DE, Insko MA, Lai AG, Lelias JM, Mehta SA et al (2005) A small molecule‐kinase interaction map for clinical kinase inhibitors. Nat Biotechnol 23: 329–336 [DOI] [PubMed] [Google Scholar]
- Fielding AB, Lim S, Montgomery K, Dobreva I, Dedhar S (2011) A critical role of integrin‐linked kinase, ch‐TOG and TACC3 in centrosome clustering in cancer cells. Oncogene 30: 521–534 [DOI] [PubMed] [Google Scholar]
- Friedrich J, Seidel C, Ebner R, Kunz‐Schughart LA (2009) Spheroid‐based drug screen: considerations and practical approach. Nat Protoc 4: 309–324 [DOI] [PubMed] [Google Scholar]
- Gabriel E, Wason A, Ramani A, Gooi LM, Keller P, Pozniakovsky A, Poser I, Noack F, Telugu NS, Calegari F, Saric T, Hescheler J, Hyman AA, Gottardo M, Callaini G, Alkuraya FS, Gopalakrishnan J (2016) CPAP promotes timely cilium disassembly to maintain neural progenitor pool. EMBO J 35: 803–819 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganem NJ, Godinho SA, Pellman D (2009) A mechanism linking extra centrosomes to chromosomal instability. Nature 460: 278–282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganier O, Schnerch D, Oertle P, Lim RY, Plodinec M, Nigg EA (2018) Structural centrosome aberrations promote non‐cell‐autonomous invasiveness. EMBO J 37: e98576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gergely F, Basto R (2008) Multiple centrosomes: together they stand, divided they fall. Genes Dev 22: 2291–2296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gigant B, Wang C, Ravelli RB, Roussi F, Steinmetz MO, Curmi PA, Sobel A, Knossow M (2005) Structural basis for the regulation of tubulin by vinblastine. Nature 435: 519–522 [DOI] [PubMed] [Google Scholar]
- Godinho SA, Pellman D (2014) Causes and consequences of centrosome abnormalities in cancer. Philos Trans R Soc Lond B Biol Sci 369: 20130467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Godinho SA, Picone R, Burute M, Dagher R, Su Y, Leung CT, Polyak K, Brugge JS, Thery M, Pellman D (2014) Oncogene‐like induction of cellular invasion from centrosome amplification. Nature 510: 167–171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gopalakrishnan J, Mennella V, Blachon S, Zhai B, Smith AH, Megraw TL, Nicastro D, Gygi SP, Agard DA, Avidor‐Reiss T (2011) Sas‐4 provides a scaffold for cytoplasmic complexes and tethers them in a centrosome. Nat Commun 2: 359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gopalakrishnan J, Frederick Chim YC, Ha A, Basiri ML, Lerit DA, Rusan NM, Avidor‐Reiss T (2012) Tubulin nucleotide status controls Sas‐4‐dependent pericentriolar material recruitment. Nat Cell Biol 14: 865–873 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo A, Villen J, Kornhauser J, Lee KA, Stokes MP, Rikova K, Possemato A, Nardone J, Innocenti G, Wetzel R, Wang Y, MacNeill J, Mitchell J, Gygi SP, Rush J, Polakiewicz RD, Comb MJ (2008) Signaling networks assembled by oncogenic EGFR and c‐Met. Proc Natl Acad Sci USA 105: 692–697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harper JV (2005) Synchronization of cell populations in G1/S and G2/M phases of the cell cycle. Methods Mol Biol 296: 157–166 [DOI] [PubMed] [Google Scholar]
- Hochegger H, Takeda S, Hunt T (2008) Cyclin‐dependent kinases and cell‐cycle transitions: does one fit all? Nat Rev Mol Cell Biol 9: 910–916 [DOI] [PubMed] [Google Scholar]
- Hsu WB, Hung LY, Tang CJ, Su CL, Chang Y, Tang TK (2008) Functional characterization of the microtubule‐binding and ‐destabilizing domains of CPAP and d‐SAS‐4. Exp Cell Res 314: 2591–2602 [DOI] [PubMed] [Google Scholar]
- Hubner NC, Bird AW, Cox J, Splettstoesser B, Bandilla P, Poser I, Hyman A, Mann M (2010) Quantitative proteomics combined with BAC TransgeneOmics reveals in vivo protein interactions. J Cell Biol 189: 739–754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivanov AS, Tugusheva NZ, Soloveva NP, Granik VG (2002) Study of the reactions of 3‐chloro‐4‐cyanobenzo[b][1,6]naphthyridine with nucleophilic reagents. Russ Chem Bull, Int Ed 51: 2121–2128 [Google Scholar]
- Ivanov AS, Tugusheva NZ, Alekseeva LM, Granik VG (2004) Synthesis and hydrolytic cleavage of 1‐aryl‐5‐cyano‐6‐(2‐dimethylaminovinyl)‐4‐oxo(thioxo)‐1,4‐dihydropyrimidines. Russ Chem Bull, Int Ed 53: 873–881 [Google Scholar]
- Ivascu A, Kubbies M (2007) Diversity of cell‐mediated adhesions in breast cancer spheroids. Int J Oncol 31: 1403–1413 [PubMed] [Google Scholar]
- Johnson VL, Scott MI, Holt SV, Hussein D, Taylor SS (2004) Bub1 is required for kinetochore localization of BubR1, Cenp‐E, Cenp‐F and Mad2, and chromosome congression. J Cell Sci 117: 1577–1589 [DOI] [PubMed] [Google Scholar]
- Jonkman JE, Cathcart JA, Xu F, Bartolini ME, Amon JE, Stevens KM, Colarusso P (2014) An introduction to the wound healing assay using live‐cell microscopy. Cell Adh Migr 8: 440–451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karki M, Keyhaninejad N, Shuster CB (2017) Precocious centriole disengagement and centrosome fragmentation induced by mitotic delay. Nat Commun 8: 15803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kavallaris M (2010) Microtubules and resistance to tubulin‐binding agents. Nat Rev Cancer 10: 194–204 [DOI] [PubMed] [Google Scholar]
- Kawamura E, Fielding AB, Kannan N, Balgi A, Eaves CJ, Roberge M, Dedhar S (2013) Identification of novel small molecule inhibitors of centrosome clustering in cancer cells. Oncotarget 4: 1763–1776 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim T, Moyle MW, Lara‐Gonzalez P, De Groot C, Oegema K, Desai A (2015) Kinetochore‐localized BUB‐1/BUB‐3 complex promotes anaphase onset in C. elegans. J Cell Biol 209: 507–517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohlmaier G, Loncarek J, Meng X, McEwen BF, Mogensen MM, Spektor A, Dynlacht BD, Khodjakov A, Gonczy P (2009) Overly long centrioles and defective cell division upon excess of the SAS‐4‐related protein CPAP. Curr Biol 19: 1012–1018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kramer A, Maier B, Bartek J (2011) Centrosome clustering and chromosomal (in)stability: a matter of life and death. Mol Oncol 5: 324–335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon M, Godinho SA, Chandhok NS, Ganem NJ, Azioune A, Thery M, Pellman D (2008) Mechanisms to suppress multipolar divisions in cancer cells with extra centrosomes. Genes Dev 22: 2189–2203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawo S, Hasegan M, Gupta GD, Pelletier L (2012) Subdiffraction imaging of centrosomes reveals higher‐order organizational features of pericentriolar material. Nat Cell Biol 14: 1148–1158 [DOI] [PubMed] [Google Scholar]
- Leber B, Maier B, Fuchs F, Chi J, Riffel P, Anderhub S, Wagner L, Ho AD, Salisbury JL, Boutros M, Kramer A (2010) Proteins required for centrosome clustering in cancer cells. Sci Transl Med 2: 33ra38 [DOI] [PubMed] [Google Scholar]
- Lee K, Rhee K (2011) PLK1 phosphorylation of pericentrin initiates centrosome maturation at the onset of mitosis. J Cell Biol 195: 1093–1101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Y, Chen J, Xiao M, Li W, Miller DD (2012) An overview of tubulin inhibitors that interact with the colchicine binding site. Pharm Res 29: 2943–2971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marteil G, Guerrero A, Vieira AF, de Almeida BP, Machado P, Mendonca S, Mesquita M, Villarreal B, Fonseca I, Francia ME, Dores K, Martins NP, Jana SC, Tranfield EM, Barbosa‐Morais NL, Paredes J, Pellman D, Godinho SA, Bettencourt‐Dias M (2018) Over‐elongation of centrioles in cancer promotes centriole amplification and chromosome missegregation. Nat Commun 9: 1258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marthiens V, Rujano MA, Pennetier C, Tessier S, Paul‐Gilloteaux P, Basto R (2013) Centrosome amplification causes microcephaly. Nat Cell Biol 15: 731–740 [DOI] [PubMed] [Google Scholar]
- Mason JM, Lin DC, Wei X, Che Y, Yao Y, Kiarash R, Cescon DW, Fletcher GC, Awrey DE, Bray MR, Pan G, Mak TW (2014) Functional characterization of CFI‐400945, a Polo‐like kinase 4 inhibitor, as a potential anticancer agent. Cancer Cell 26: 163–176 [DOI] [PubMed] [Google Scholar]
- Moritz M, Braunfeld MB, Fung JC, Sedat JW, Alberts BM, Agard DA (1995) Three‐dimensional structural characterization of centrosomes from early Drosophila embryos. J Cell Biol 130: 1149–1159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Musacchio A (2015) The molecular biology of spindle assembly checkpoint signaling dynamics. Curr Biol 25: R1002–R1018 [DOI] [PubMed] [Google Scholar]
- Nigg EA (2002) Centrosome aberrations: cause or consequence of cancer progression? Nat Rev Cancer 2: 815–825 [DOI] [PubMed] [Google Scholar]
- Nigg EA (2004) Centrosomes in development and disease. Weinheim: Wiley‐VCH; [Google Scholar]
- Nigg EA, Raff JW (2009) Centrioles, centrosomes, and cilia in health and disease. Cell 139: 663–678 [DOI] [PubMed] [Google Scholar]
- Nigg EA, Stearns T (2011) The centrosome cycle: centriole biogenesis, duplication and inherent asymmetries. Nat Cell Biol 13: 1154–1160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oegema K, Wiese C, Martin OC, Milligan RA, Iwamatsu A, Mitchison TJ, Zheng Y (1999) Characterization of two related Drosophila gamma‐tubulin complexes that differ in their ability to nucleate microtubules. J Cell Biol 144: 721–733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogden A, Rida PC, Aneja R (2012) Let's huddle to prevent a muddle: centrosome declustering as an attractive anticancer strategy. Cell Death Differ 19: 1255–1267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olsen JV, de Godoy LM, Li G, Macek B, Mortensen P, Pesch R, Makarov A, Lange O, Horning S, Mann M (2005) Parts per million mass accuracy on an Orbitrap mass spectrometer via lock mass injection into a C‐trap. Mol Cell Proteomics 4: 2010–2021 [DOI] [PubMed] [Google Scholar]
- Orts J, Griesinger C, Carlomagno T (2009) The INPHARMA technique for pharmacophore mapping: a theoretical guide to the method. J Magn Reson 200: 64–73 [DOI] [PubMed] [Google Scholar]
- Pagliarini R, Shao W, Sellers WR (2015) Oncogene addiction: pathways of therapeutic response, resistance, and road maps toward a cure. EMBO Rep 16: 280–296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pannu V, Rida PC, Celik B, Turaga RC, Ogden A, Cantuaria G, Gopalakrishnan J, Aneja R (2014) Centrosome‐declustering drugs mediate a two‐pronged attack on interphase and mitosis in supercentrosomal cancer cells. Cell Death Dis 5: e1538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pannu V, Rida PC, Ogden A, Turaga RC, Donthamsetty S, Bowen NJ, Rudd K, Gupta MV, Reid MD, Cantuaria G, Walczak CE, Aneja R (2015) HSET overexpression fuels tumor progression via centrosome clustering‐independent mechanisms in breast cancer patients. Oncotarget 6: 6076–6091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE (2004) UCSF Chimera–a visualization system for exploratory research and analysis. J Comput Chem 25: 1605–1612 [DOI] [PubMed] [Google Scholar]
- Piehl M, Tulu US, Wadsworth P, Cassimeris L (2004) Centrosome maturation: measurement of microtubule nucleation throughout the cell cycle by using GFP‐tagged EB1. Proc Natl Acad Sci USA 101: 1584–1588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prota AE, Bargsten K, Zurwerra D, Field JJ, Diaz JF, Altmann KH, Steinmetz MO (2013) Molecular mechanism of action of microtubule‐stabilizing anticancer agents. Science 339: 587–590 [DOI] [PubMed] [Google Scholar]
- Pryor DE, O'Brate A, Bilcer G, Diaz JF, Wang Y, Wang Y, Kabaki M, Jung MK, Andreu JM, Ghosh AK, Giannakakou P, Hamel E (2002) The microtubule stabilizing agent laulimalide does not bind in the taxoid site, kills cells resistant to paclitaxel and epothilones, and may not require its epoxide moiety for activity. Biochemistry 41: 9109–9115 [DOI] [PubMed] [Google Scholar]
- Raab MS, Breitkreutz I, Anderhub S, Ronnest MH, Leber B, Larsen TO, Weiz L, Konotop G, Hayden PJ, Podar K, Fruehauf J, Nissen F, Mier W, Haberkorn U, Ho AD, Goldschmidt H, Anderson KC, Clausen MH, Kramer A (2012) GF‐15, a novel inhibitor of centrosomal clustering, suppresses tumor cell growth in vitro and in vivo . Can Res 72: 5374–5385 [DOI] [PubMed] [Google Scholar]
- Ravelli RB, Gigant B, Curmi PA, Jourdain I, Lachkar S, Sobel A, Knossow M (2004) Insight into tubulin regulation from a complex with colchicine and a stathmin‐like domain. Nature 428: 198–202 [DOI] [PubMed] [Google Scholar]
- Rebacz B, Larsen TO, Clausen MH, Ronnest MH, Loffler H, Ho AD, Kramer A (2007) Identification of griseofulvin as an inhibitor of centrosomal clustering in a phenotype‐based screen. Can Res 67: 6342–6350 [DOI] [PubMed] [Google Scholar]
- Ronnest MH, Raab MS, Anderhub S, Boesen S, Kramer A, Larsen TO, Clausen MH (2012) Disparate SAR data of griseofulvin analogues for the dermatophytes Trichophyton mentagrophytes, T. rubrum, and MDA‐MB‐231 cancer cells. J Med Chem 55: 652–660 [DOI] [PubMed] [Google Scholar]
- Roostalu J, Surrey T (2017) Microtubule nucleation: beyond the template. Nat Rev Mol Cell Biol 18: 702–710 [DOI] [PubMed] [Google Scholar]
- Rusan N, Peifer M (2007) A role for a novel centrosome cycle in asymmetric cell division. J Cell Biol 177: 13–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabino D, Gogendeau D, Gambarotto D, Nano M, Pennetier C, Dingli F, Arras G, Loew D, Basto R (2015) Moesin is a major regulator of centrosome behavior in epithelial cells with extra centrosomes. Curr Biol 25: 879–889 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez‐Pedregal VM, Reese M, Meiler J, Blommers MJ, Griesinger C, Carlomagno T (2005) The INPHARMA method: protein‐mediated interligand NOEs for pharmacophore mapping. Angew Chem 44: 4172–4175 [DOI] [PubMed] [Google Scholar]
- Sankaran S, Starita LM, Groen AC, Ko MJ, Parvin JD (2005) Centrosomal microtubule nucleation activity is inhibited by BRCA1‐dependent ubiquitination. Mol Cell Biol 25: 8656–8668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schindelin J, Arganda‐Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, Tinevez JY, White DJ, Hartenstein V, Eliceiri K, Tomancak P, Cardona A (2012) Fiji: an open‐source platform for biological‐image analysis. Nat Methods 9: 676–682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schorpp K, Rothenaigner I, Salmina E, Reinshagen J, Low T, Brenke JK, Gopalakrishnan J, Tetko IV, Gul S, Hadian K (2013) Identification of small‐molecule frequent hitters from alphascreen high‐throughput screens. J Biomol Screen 19: 715–726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah JV, Cleveland DW (2000) Waiting for anaphase: Mad2 and the spindle assembly checkpoint. Cell 103: 997–1000 [DOI] [PubMed] [Google Scholar]
- Sharma A, Aher A, Dynes NJ, Frey D, Katrukha EA, Jaussi R, Grigoriev I, Croisier M, Kammerer RA, Akhmanova A, Gonczy P, Steinmetz MO (2016) Centriolar CPAP/SAS‐4 imparts slow processive microtubule growth. Dev Cell 37: 362–376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang CJ, Fu RH, Wu KS, Hsu WB, Tang TK (2009) CPAP is a cell‐cycle regulated protein that controls centriole length. Nat Cell Biol 11: 825–831 [DOI] [PubMed] [Google Scholar]
- Trott O, Olson AJ (2010) AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem 31: 455–461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyanova S, Temu T, Sinitcyn P, Carlson A, Hein MY, Geiger T, Mann M, Cox J (2016) The Perseus computational platform for comprehensive analysis of (prote)omics data. Nat Methods 13: 731–740 [DOI] [PubMed] [Google Scholar]
- Vranken WF, Boucher W, Stevens TJ, Fogh RH, Pajon A, Llinas M, Ulrich EL, Markley JL, Ionides J, Laue ED (2005) The CCPN data model for NMR spectroscopy: development of a software pipeline. Proteins 59: 687–696 [DOI] [PubMed] [Google Scholar]
- Wang W, Zhang X, Qin JJ, Voruganti S, Nag SA, Wang MH, Wang H, Zhang R (2012) Natural product ginsenoside 25‐OCH3‐PPD inhibits breast cancer growth and metastasis through down‐regulating MDM2. PLoS One 7: e41586 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watts CA, Richards FM, Bender A, Bond PJ, Korb O, Kern O, Riddick M, Owen P, Myers RM, Raff J, Gergely F, Jodrell DI, Ley SV (2013) Design, synthesis, and biological evaluation of an allosteric inhibitor of HSET that targets cancer cells with supernumerary centrosomes. Chem Biol 20: 1399–1410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiese C, Zheng Y (2006) Microtubule nucleation: gamma‐tubulin and beyond. J Cell Sci 119: 4143–4153 [DOI] [PubMed] [Google Scholar]
- Wong YL, Anzola JV, Davis RL, Yoon M, Motamedi A, Kroll A, Seo CP, Hsia JE, Kim SK, Mitchell JW, Mitchell BJ, Desai A, Gahman TC, Shiau AK, Oegema K (2015) Cell biology. Reversible centriole depletion with an inhibitor of Polo‐like kinase 4. Science 348: 1155–1160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yalysheva NZ, Soloveva NP, Chistyakov VV, Sheinker YN, Granik VG (1986) Acetals of lactams and acid amides. 47. Investigation of the behavior of substituted 6‐(B‐dimethylamino) vinyl‐4‐pyrimidinones in acidic media. Synthesis of 3‐cyano‐4‐anilino‐5‐formyl‐2‐pyridone and 3‐chloro‐4‐cyanobenzo[b] [1,6]‐naphthyridine. Chem Heterocycl Compd 22: 909–914 [Google Scholar]
- Zhao L, Jin C, Chu Y, Varghese C, Hua S, Yan F, Miao Y, Liu J, Mann D, Ding X, Zhang J, Wang Z, Dou Z, Yao X (2010) Dimerization of CPAP orchestrates centrosome cohesion plasticity. J Biol Chem 285: 2488–2497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng X, Ramani A, Soni K, Gottardo M, Zheng S, Ming Gooi L, Li W, Feng S, Mariappan A, Wason A, Widlund P, Pozniakovsky A, Poser I, Deng H, Ou G, Riparbelli M, Giuliano C, Hyman AA, Sattler M, Gopalakrishnan J, et al (2016) Molecular basis for CPAP–tubulin interaction in controlling centriolar and ciliary length. Nature Commun 7: 11874 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng Y, Wong ML, Alberts B, Mitchison T (1995) Nucleation of microtubule assembly by a gamma‐tubulin‐containing ring complex. Nature 378: 578–583 [DOI] [PubMed] [Google Scholar]
- Zheng X, Gooi LM, Wason A, Gabriel E, Mehrjardi NZ, Yang Q, Zhang X, Debec A, Basiri ML, Avidor‐Reiss T, Pozniakovsky A, Poser I, Saric T, Hyman AA, Li H, Gopalakrishnan J (2014) Conserved TCP domain of Sas‐4/CPAP is essential for pericentriolar material tethering during centrosome biogenesis. Proc Natl Acad Sci USA 111: E354–E363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng X, Ramani A, Soni K, Gottardo M, Zheng S, Ming Gooi L, Li W, Feng S, Mariappan A, Wason A, Widlund P, Pozniakovsky A, Poser I, Deng H, Ou G, Riparbelli M, Giuliano C, Hyman AA, Sattler M, Gopalakrishnan J et al (2016) Molecular basis for CPAP‐tubulin interaction in controlling centriolar and ciliary length. Nat Commun 7: 11874 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Expanded View Figures PDF
Table EV1
Table EV2
Movie EV1
Movie EV2
Movie EV3
Movie EV4
Movie EV5
Movie EV6
Review Process File