

Abstract
Changes in the expression of α7 nicotinic acetylcholine receptors (α7 nAChRs) in the human brain are widely assumed to be associated with neurological and neurooncological processes. Investigation of these receptors in vivo depends on the availability of imaging agents such as radioactively labelled ligands applicable in positron emission tomography (PET). We report on a series of new ligands for α7 nAChRs designed by the combination of dibenzothiophene dioxide as a novel hydrogen bond acceptor functionality with diazabicyclononane as an established cationic center. To assess the structure-activity relationship (SAR) of this new basic structure, we further modified the cationic center systematically by introduction of three different piperazine-based scaffolds. Based on in vitro binding affinity and selectivity, assessed by radioligand displacement studies at different rat and human nAChR subtypes and at the structurally related human 5-HT3 receptor, we selected the compound 7-(1,4-diazabicyclo[3.2.2]nonan-4-yl)-2-fluorodibenzo-[b,d]thiophene 5,5-dioxide (10a) for radiolabeling and further evaluation in vivo. Radiosynthesis of [18F]10a was optimized and transferred to an automated module. Dynamic PET imaging studies with [18F]10a in piglets and a monkey demonstrated high uptake of radioactivity in the brain, followed by washout and target-region specific accumulation under baseline conditions. Kinetic analysis of [18F]10a in pig was performed using a two-tissue compartment model with arterial-derived input function. Our initial evaluation revealed that the dibenzothiophene-based PET radioligand [18F]10a ([18F]DBT-10) has high potential to provide clinically relevant information about the expression and availability of α7 nAChR in the brain.
Keywords: α7 nAChR, pharmacophore, positron emission tomography, neuroimaging, fluorine-18
1. Introduction
The long-standing interest in molecular imaging of nicotinic acetylcholine receptors (nAChRs) is driven by findings from preclinical and clinical studies, which have demonstrated that dysfunction of neuronal nAChRs is involved in the pathophysiology of many disorders [1]. The nAChRs are a superfamily of ligand-gated ion channels that consist of a pentamer of protein subunits. In the mammalian brain, different subunits assemble with much diversity; however, the α7 and α4β2 nAChR subtypes predominate [2]. The α7 subunit is highly expressed in the hippocampus and hypothalamus, as well as in cell types where receptor-mediated ion currents have not been reported. Therefore, α7 nAChR-mediated effects such as cognitive enhancement [3] may depend on the ionotropic signaling while other effects may be independent of ion-channel currents [4]. Altered functional availability of α7 nAChR has been implicated in a number of diseases of the human central nervous system (CNS), including Alzheimer’s and Parkinson’s disease, schizophrenia and autism, as well as in lung cancer and heart disease [5]. With highly selective ligands, α7 nAChRs are approachable targets not only for therapeutic interventions, but also for non-invasive imaging such as Positron Emission Tomography (PET), which can be used to investigate disease pathophysiology in vivo and support drug development.
While the cationic pharmacophore sufficient for full activation of all neuronal nAChRs is the tetramethylammonium cation, several α7 nAChR selective motifs have been identified based on the observation that an additional hydroxyl group present in choline or quinuclidinol activates largely the homomeric α7 but not the heteromeric nAChRs [6]. So far, at least nine structurally related families of high-affinity small-molecule ligands of the α7 nAChR have been characterized [6,7], including anabaseine-, pyrrolidine-, and diazabicyclononane-based PET radiotracers such as [11C]GTS-21 [8], [11C]A-844606 and [11C]A-582941 [9], [11C]CHIBA-1001 [10], [11C]NS14492 [11], [18F]NS10743 [12] and [18F]NS14490 [13]. Recent progress in α7 nAChR PET imaging comes from the discovery of the α7 nAChR selective binding of the antiviral interferon inducer tilorone [14], containing a tricyclic fluorenone nucleus. Further structural modification by replacing this fluorenone moiety with dibenzothiophene sulfone as alternative hydrogen bond acceptor (HBA) functionality and introduction of 1,4-diazabicyclo[3.2.2]nonane as cationic center resulted in a novel compound with markedly increased binding affinity for α7 nAChR (Ki = 56 nM for tilorone and 0.023 nM for compound 48, Figure 1, A1) [15]. This discovery by Schrimpf et al., in 2012 prompted us to develop a series of novel derivatives as references for 18F-labeled ligands of α7 nAChRs and to further investigate the SAR of this new pharmacophore. At the time of working on the radiolabeling of the most promising ligand for PET imaging studies [16], researchers from the Johns Hopkins University had successfully completed the radiosynthetic work on respective ligands [17]. While the evaluation of the resulting [18F]ASEM in baboons and humans was published [18,19,20], we felt encouraged by the inherent potential of the Abbott lead structure to continue our research on 18F-labelled diazabicyclononane-containing α7 nAChR ligands [16,21] by modifying both the HBA functionality and the cationic center.
Figure 1.

Structures of recent α7 nAChR ligands. Four examples of bicyclic amines coupled to dibenzothiophene sulfone (A) [15], a tropane derived compound (B) [24] and a propylene-bridged N-methylpiperazine (C) [25] are shown.
Besides the elaboration of a more effective approach towards compound [18F]10a ([18F]DBT-10), which we have selected for further development [16] and is identical to compound [18F]7c reported by Gao et al. [17], we have synthesized a series of derivatives of this lead structure to further analyze structural elements that determine the affinity of potential α7 nAChR ligands to other pentameric ligand-gated ion channels such as heteromeric nicotinic or 5-HT3 receptors. It has been shown previously that the affinity for α4β2 nAChR of dibenzothiophene-diazabicyclononanes (e.g., [18F]ASEM, Ki = 562 nM [17]) is notably higher than that of oxadiazolyl-substituted diazabicyclononanes (e.g., [18F]NS10743 Ki > 10 μM [12]). Therefore, we replaced the cationic center with alternative tertiary amine motifs. Insertion of three different diazabicycloalkanes resulted in N-methyl-substituted derivatives of the ethylene- and propylene-bridged piperazines diazabicyclo[3.2.1]octane and diazabicyclo[3.3.1]nonane, respectively [22,23].
Previous reports have also shown that structural changes on the amino side chain (cationic center) of a given aromatic unit may affect the binding profile with respect to α7/α4β2 nAChR affinity [26,27]. Furthermore, a small series of bicyclic amines coupled to dibenzothiophene sulfone, including chiral quinuclidines (Figure 1, A3 and A4) and a fused azetidine (3,6-diazabicyclo[3.2.0]heptane) (Figure 1, A2) have been investigated by Schrimpf et al. [15]. In addition to nitrogen bridgeheaded aza- or diazabicylic molecules derived from quinuclidine or diazabicyclo[3.2.2]nonane, several alkylene bridged piperidines (e.g., tropane derivatives such as tropisetron [24]) or piperazines (e.g., N-methyl-3-azagranatanine derivative such as NS12857 [25]) have been identified as potent α7 nAChR selective ligands (Figure 1B,C). Considering this, we were inspired to prepare a small series of fluorinated dibenzothiophene sulfones with a set of (three) differently arranged piperazine-based diazabicyclic scaffolds.
To test for the effect of the designed structural modifications and to evaluate the target selectivity of potential α7 nAChR imaging probes, the binding affinities of all fluoro-substituted derivatives for the human α7 nAChR, as well as the three most important subtypes of heteromeric nAChRs (human α4β2, human α3β4, and rat α6β2*) and the structurally similar 5-HT3 receptor, were determined. For the most suitable ligand 10a, a radiolabeling procedure was developed and successfully implemented, then subsequently optimized and transferred to a fully automated setup. Finally, the pharmacokinetics of [18F]10a was evaluated in both piglets and monkey by dynamic PET scans under baseline and blocking conditions.
2. Results and Discussion
2.1. Chemistry
2.1.1. Rationale
1,4-Diazabicyclo[3.2.2]nonane-substituted dibenzothiophene sulfone 10 was found to have exceptional high affinity for α7 nAChR and effective brain uptake [15,17]. This prompted us to develop novel derivatives as candidates for 18F-labeled ligands and to further investigate the SAR of this new pharmacophore (Table 1).
Table 1.
Structures of fluoro-substituted dibenzothiophene-based ligands for α7 nAChR.
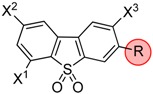
| Compound | R | X1 | X2 | X3 |
|---|---|---|---|---|
| 10 | 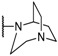 |
H | H | H |
| 10a | |
H | F | H |
| 10b | |
F | H | H |
| 10c | |
H | H | F |
| 12a | 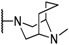 |
H | F | H |
| 12b | |
F | H | H |
| 13b | 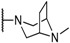 |
F | H | H |
| 14b | 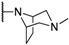 |
F | H | H |
Initially, the three fluoro-substituted 1,4-diazabicyclo[3.2.2]nonane-containing derivatives 10a–c were designed. While 10a and 10b are identical to compounds 7c and 7a, respectively, recently published by Gao et al. [17], the isomer 10c has not been described so far. To gain further insight into the SAR of dibenzothiophene-based scaffolds we embarked on the synthesis of a second series of compounds. Previous results on the systematic side chain rigidification have highlighted the importance of conformational restriction of the aminocycle for high α7 selectivity [15]. Since the tropane core has been identified as a structural motif that could provide this selectivity [4,6] it was of interest to include a set of three tropane-like diamines into the synthetic plan. With the carbon atom replaced at position 3 both in tropane and in N-methylgranatanine [28] by an nitrogen atom, the resulting secondary amino group of each N-methyl piperazine building block could serve as position of attachment to the tricyclic arene system.
Four novel fluorine-substituted derivatives were envisioned, in which the homopiperazine-based 1,4-diazabicyclo[3.2.2]nonane was replaced by conformationally restricted piperazine-based substituents, bridged by either ethylene (8-methyl-3,8-diazabicyclo[3.2.1]octane = azatropane (13b), 3-methyl-3,8-diazabicyclo[3.2.1]octane (14b)) or propylene units (9-methyl-3,9-diazabicyclo[3.3.1]nonane = N-methyl-3-azagranatanine (12a,b)) [22].
2.1.2. Synthesis of α7 nAChR Ligands
Dibenzo[b,d]thiophene (1a) and dibenzo[b,d]thiophene 4-boronic acid (1b) served as starting materials for the synthesis of dibenzo[b,d]thiophene-5,5-dioxides functionalized in the para- (Scheme 1) or ortho- position (Scheme 2) to the sulfone group. The final coupling to the N-atom of a diazabicyclic moiety was performed at position 7 or 3 of the respective tricyclic scaffold to afford the potential α7 nAChR ligands 10, 10a–c, 12a,b, 13b, and 14b, as shown in Table 1.
Scheme 1.

Synthesis of 3-bromo dibenzothiophene intermediates 3, 6a, 8a and 8c. Reagents and conditions: (a) H2O2 (50%), HOAc, 120 °C, 5 h, 97% for 2 and 96% for 5a (b) NBS, conc. H2SO4, r.t., 24 h, 44% for 3, 64% for 6a, 46% for 8c, and 18% for 8d; (c) HNO3 (65%), HOAc, 20–40 °C, 24 h, 26% for 4a; (d) TMAF·4 H2O, cyclohexane/DMSO, ↑↓, 6 h (azeotropic drying with separation of H2O), compound 5a or compound 6a, 95 °C, 5 h, 75% for 7a and 74% for 8a.
Scheme 2.

Synthesis of 3-bromo dibenzothiophene intermediates 6b and 8b. Reagents and conditions: (a) tert-butyl nitrite, MeCN, 55 °C, 24 h, mixture of 4/4b (~88:12); (b) H2O2 (50%), HOAc, 120 °C, 5 h, 72% (two steps); (c) NBS, conc. H2SO4, r.t., 24 h, 78%; (d) TMAF·4H2O, cyclohexane/DMSO, 6 h (azeotropic drying with separation of H2O), compound 6b, 95 °C, 5 h, 73%.
Synthesis of Bromo Dibenzothiophene Intermediates 3, 6a and 8a–8c
The brominated intermediates were obtained from dibenzo[b,d]thiophene (1a) by three different routes. The 3-bromo-derivative 3 was prepared via sulfide to sulfone oxidation of 1a with H2O2 in acetic acid followed by bromination with an excess of 2 in the NBS/H2SO4 system in 44% yield [29]. Isolation of compound 3 from the 3,7-dibrominated by-product (not shown) along with unreacted starting material was obtained by fractional crystallization of the dibromo compound, followed by a chromatographic separation of pure 3 from the filtrate. The 2-nitrodibenzothiophene sulfone 5a was prepared by nitration of 1a using HNO3 (65%) in acetic acid [30,31] affording the 2-nitro compound 4a in moderate yield (26%), followed by oxidation with H2O2 in acetic acid to give 5a [32]. The 2-nitro-7-bromo-dibenzothiophene sulfone 6a was obtained by bromination of 5a according to the corresponding conversion of 2 to 3. Because the one benzo ring in 5a is strongly deactivated by the NO2 and SO2R groups, no over-brominated by-products were detected and the 2-nitro-7-bromo-dibenzothiophene sulfone 6a was afforded in 64% yield. To obtain different mono-fluorinated building blocks in a short synthesis, the fluorine substitution either at positions para or ortho to the sulfone were intended to be carried out with a simple uniform synthetic pathway. In addition to the known three-step conversion of 6a into the 2-fluoro-7-bromo derivative 8a based on the Balz-Schiemann reaction with 45% overall yield [17], we deemed a one-step conversion from 6a to 8a via fluorodenitration as preferable [33,34]. Tetramethylammonium fluoride (TMAF) proved to be a good source for active fluoride after azeotropic drying of commercial TMAF tetrahydrate in refluxing cyclohexane/DMSO for 6 h. A smooth conversion of 6a occurred at 95 °C within 5 h to give 8a in 75%yield.Likewise, the nitro group in the 2-nitrodibenzothiophene 5,5-dioxide 5a was displaced by fluorine to give 7a in 74% yield. The vicinal 3-bromination of 7a was accomplished by applying NBS in H2SO4 (98%) at room temperature for 24 h to give compound 8b. In addition to 8c, the 3,7-dibromo derivative 8d was detected. Chromatographic separation gave 8d in 18% yield along with 2-fluoro-3-bromo compound 8c in 46% yield.
Synthesis of the 3-Bromo Dibenzothiophene Intermediates 6b and 8b
As result of the two-step synthesis of 5a from 1a (Scheme 1), the ortho-derivative 4b was concomitantly formed as a minor nitration product. However, because it was difficult to separate from the excess of 4a, this approach was considered inappropriate to gain an adequate amount of the 4-nitro dibenzothiophene sulfone 5b. The intermediate 5b was recently reported [17]. The described procedure for 4b is a nitration of commercially available arylboronic acid 1b with two equivalents of Bi(NO3)3 as described by Maiti et al. [35]. In contrast, we successfully applied a metal-free approach by performing an ipso nitrosation of 1b with tert-butyl nitrite in acetonitrile [36,37]. As shown in Scheme 2, the nitroso derivative 4 was formed as the main product along with a minor amount of 4b. Without further purification, the 4/4b mixture was readily oxidized in a one-pot reaction with H2O2/acetic acid to give 5b in 72% yield over two steps from 1b. After bromination, the resulting 6b was converted directly into 8b in 73% yield via our fluorodenitration protocol, in contrast to the earlier described independent four-step synthesis applying a Pschorr reaction [17].
Synthesis of 10, Fluorinated Reference Compounds 10a–c, and Nitro Precursor 11
The 3-bromodibenzo[b,d]thiophene sulfones 3, 6a and 8a–c were reacted with 1,4-diazabicyclo[3.2.2]nonane (9a) under Pd-catalyzed Buchwald-Hartwig conditions to provide the previously reported compounds 10a and 10b [17] and the novel fluorinated isomer 10c in 52%, 48% and 44% yields, respectively. The non-fluorinated compound 10 [15] and the nitro derivative 11a [17] were also obtained under Buchwald-Hartwig conditions in 73% and 53% yields, respectively, as depicted in Scheme 3.
Scheme 3.

Synthesis of 10, fluorinated reference compounds 10a–c, and nitro precursor 11. Reagents and conditions: (a) Pd2(dba)3, BINAP, Cs2CO3, toluene, 90 °C, 24–36 h, 52% for 10a, 48% for 10b, 44% for 10c, 73% for 10, and 53% for 11a.
Synthesis of the Fluorinated Reference Compounds 12a,b, 13b, and 14b
In order to expand the structural diversity of compounds derived from the novel pharmacophore dibenzothiophene for SAR characterization, in a second series of compounds the homopiperazine-based 1,4-diazabicyclo[3.2.2]nonane moiety was replaced by piperazine-based substituents, bridged by either ethylene or propylene units. The bromine substituent in the meta position to the sulfone group was used to incorporate these more rigid diamines. The Buchwald-Hartwig coupling of 3-bromodibenzo[b,d]thiophene sulfones 8a and 8b with 9-methyl-3,9-diazabicyclo[3.3.1]nonane (9b), 8-methyl-3,8-diazabicyclo[3.2.1]octane (9c) and 3-methyl-3,8-diazabicyclo[3.2.1]octane (9d) gave compounds 12a,b, 13b, and 14b in moderate yields (Scheme 4).
Scheme 4.

Synthesis of the fluorinated reference compounds 12a,b, 13b, and 14b. Reagents and conditions: (a) Pd2(dba)3, BINAP, Cs2CO3, toluene, 90 °C, 24–36 h, 38% for 12a, 42% for 12b, 47% for 13b, and 67% for 14b.
All final compounds (10–14) are crystalline solids and were fully characterized by NMR spectroscopy (1H, 13C, 19F, COSY, HSQC) and high resolution mass spectrometry.
2.2. In Vitro Affinity Assays
All compounds were evaluated in vitro to measure their affinity and selectivity for the target receptor α7 nAChR in relation to the heteromeric receptor subtypes α4β2, α6β2, and α3β4. The results of the respective binding assays are summarized in Table 2.
Table 2.
in vitro binding affinities towards human homomeric α7, heteromeric α4β2 and α3β4 nAChR, rat α6β2* nAChR subtypes, and human 5-HT3 receptor.
| Compound | Affinity (Ki in nM) | Selectivity (Ki ratio) | |||||
|---|---|---|---|---|---|---|---|
| nAChR Subtype | 5-HT3 d,e | ||||||
| hα7 a | hα4β2 b | hα3β4 b | rα6β2* c | α7/α4β2 | α7/α3β4 | ||
| 10 | 0.51 ± 0.32 | 318 ± 43.3 | 49.6 ± 14.7 | 517 ± 186 | (35%) | 623 | 97 |
| 10a | 0.60 ± 0.44 | 517 ± 375 | 119 ± 29.0 | 589 ± 217 | 440 (2%) | 862 | 198 |
| 10b | 0.84 ± 0.16 | 211 ± 108 | 42.3 ± 4.73 | 435 ± 152 | (42%) | 251 | 50 |
| 10c | 8.53 ± 1.74 | 507 ± 212 | 279 ± 24.4 | 1390 ± 340 | (26%) | 59 | 33 |
| 12a | 30.9 ± 8.72 | 141 ± 11.7 | 96.0 ± 1.48 | 1180 ± 360 | (10%) | 5 | 3 |
| 12b | 105 ± 23.9 | 301 ± 148 | 94.8 ± 6.48 | 1190 ± 230 | (11%) | 3 | 1 |
| 13b | 40.9 ± 7.77 | 426 ± 197 | 224 ± 51.7 | 2260 ± 540 | (10%) | 10 | 5 |
| 14b | 9.26 ± 2.23 | >4000 | >5000 | 1450 ± 370 | (4%) | >400 | >500 |
a Human α7 nAChR in stably transfected SH-SY5Y cells, with radiotracer [3H]methyllycaconitine (0.5–1 nM), KD = 2.0 nM. b Human α4β2 and α3β4 nAChR in stably transfected HEK-293 cells, with radiotracer [3H]epibatidine (0.5–1 nM), KD = 0.025 nM for hα4β2 nAChR, KD = 0.117 nM for hα3β4 nAChR. c Rat α6β2* obtained from rat striatum by immunoimmobilization using anti-rα6 nAChR antibody, with radiotracer [3H]epibatidine (0.1 nM), KD = 0.025 nM. d Ki value in nM; human 5-HT3 receptor recombinant-HEK293 cells, with radiotracer [3H]GR65630 (working concentration n = 0.69 nM; KD = 0.2 nM). e percentage of inhibition at 0.1 μM concentration of test compound.
2.2.1. Affinity for α7 nAChR
The affinity for α7 nAChR was assessed for the human receptor protein expressed in a stably transfected cell line and labeled with the selective α7 nAChR antagonist [3H]methyllycaconitine. The affinity of the lead of the series (compound 10), Ki = 0.51 nM, is almost identical to the value reported by Gao et al., obtained on [125I]α-bungarotoxin labelled rat cortical membranes. The binding affinities of the fluoro-substituted compounds 10a (Ki = 0.6 nM vs. 1.4 nM of compound 7c [17]) and 10b (Ki = 0.84 nM vs. 0.4 nM of compound 7a [17]) are also comparable with previously published results. All novel fluoro-containing compounds 10c, 12a,b, 13b, and 14b bind with remarkably lower affinity to α7 nAChR. Interestingly, the vicinal substitution of the dibenzothiophene with both the fluorine and the diazabicycle is inappropriate in terms of binding as reflected by the 2-fluoro-3-amino derivative 10c, which possesses an about 10-fold lower affinity at the α7 subtype relative to the 2-fluoro-7-amino derivative 10a and the 6-fluoro-3-amino derivative 10b.
In particular, we assume that steric effects impair the interactions between the cationic center of compound 10c and the binding site of α7 nAChR [38]. Furthermore, as noticed for the new series of dibenzothiophene derivatives (12a,b, 13b and 14b) an increase in the flexibility of the tertiary amine in the cationic center is also not tolerated. Replacement of the NC-bridged homopiperazine moiety, containing a bicyclic tertiary amine as a basic structural element of the reference compound 10 by three different CC-bridged piperazine ring systems and carrying a methyl-substituted tertiary amino group, has negative effects. The 9-methyl-3,9-diazabicyclo[3.3.1]nonane substituted compounds 12a and 12b have significantly diminished affinity for α7 nAChR relative to the matched pairs of 1,4-diazabicyclo[3.2.2]nonane-substituted 10a and 10b. A similar effect is observed by substitution with an azatropane in compound 13b (8-methyl-3,8-diazabicyclo[3.2.1]octane) and its isomer 14b (3-methyl-3,8-diazabicyclo[3.2.1]octane).
Thus, in terms of affinity for the α7 nAChR we considered compound 10a as the most suitable compound reported herein. The structural modifications within the cationic center of the molecules, which we performed to increase selectivity by reducing off-target binding, unexpectedly impaired the binding to α7 nAChR to such an extent that the resulting compounds were not included in the development of radioligands within this study.
2.2.2. Affinities for α4β2, α3β4, and α6β2 nAChR
To evaluate off-target binding of the compounds towards heteromeric nAChR subtypes, respective binding assays were performed with [3H]epibatidine-labeled human α4β2 or human α3β4 nAChR, both stably expressed on human HEK cells, as well as native rat α6β2* nAChR immobilized by a subunit-specific antibody.
Besides the α4β2* nAChR, the α6β2* nAChR belongs to the high-affinity family of β2-containing nAChRs [39]. While functionally similar to α4β2* nAChR, the expression of the α6β2* subtype is rather selective, primarily in dopamine neurons in the brain [40,41,42,43]. Overlapping expression of α7 nAChR with α4β2* nAChR and also α6β2* nAChR within the mesolimbic axis [44,45] requires careful analysis of subtype affinity of radioligands for nAChR. In heteromeric nAChR subtypes, the orthosteric ligand binding site is formed at the interface of an α subunit (principal component) and an adjacent non-α subunit (complementary component), equivalent to the (+) surface and the (−) surface of an α subunit in homomeric nAChRs [46,47].
Consistent with the observation that in heteromeric neuronal nAChRs the non-α subunit is of particular importance [38], as it makes the affinity for nicotine dependent on the presence of a β2 subunit regardless of the α subunit [47], the herein investigated compounds bind with generally moderate (Ki values > 100 nM) and almost equal affinities towards both human α4β2 and rat α6β2* nAChRs. It is worth noting that the affinities for the α4β2 subtype of the α7 nAChR ligands investigated herein, with the exception of 14b, are significantly higher than those of the compounds in the NeuroSearch (NS) series [12]. Because these compounds contain identical cationic centers, differences in the receptor subtype binding affinities are probably related to differences in the HBA and hydrophobic functionalities. In the NeuroSearch compound series, an oxadiazole moiety appears as HBA which is spatially more separated from and not forced in plane with the hydrophobic fluorophenyl moiety, while compounds 10a–10c possess fused functionalities with both the HBA and the hydrophobic moiety represented by the fluorine-substituted dibenzothiophene dioxide ring system. We hypothesize that in particular the sulfonyl moiety of these novel compounds promotes binding to the α4β2 subtype, comparable to the carbonyl moiety acting as a hydrogen acceptor along with a cationic pharmacophore element in the off-target binding of α7 nAChR ligand CHIBA-1001 [48,49]. However, for the most selective fluorine-containing ligand of the current study, compound 10a (selectivity > 800), interfering effects on α7 nAChR PET imaging due to its binding to β2-containing receptor subtypes are highly unlikely. The conformational changes due to the shift of the bridgehead carbons in the diazabicyclo[3.2.1]octane that we assume to contribute to the significantly improved selectivity of compound 14b in comparison to 13b will be analyzed in future studies. It appears that the basic N-methyl group is sterically more deshielded in contrast to the azatropane 13b, resulting in a detrimental effect both on α4β2 and α3β4 nAChR binding with only weakly attenuated affinity to the α7 subtype (Table 2).
An overlap in the receptor expression between α7 and α3β4* nAChRs, in particular within autonomic neurons [50,51], necessitates the investigation of the selectivity of potential ligands. By comparing data obtained for compounds 10a and 10b (Ki: 119 nM and 43 nM) with the affinity values of the corresponding compounds 7c and 7a reported by Gao et al., (Ki: 5000 nM and 709 nM) [17], species-specific differences become obvious. Although the most selective compound 10a (selectivity ~200) binds with sufficiently low affinity to human α3β4 nAChRs to afford imaging of the α7 subtype in human brain, more than 10-fold higher affinities at the human than the rat receptor subtype illustrate the significance of species-specific targeting at the early stage of the development of PET ligands.
The in vitro binding affinity of all compounds from this series at the 5-HT3 receptor, as determined by percentage of inhibition of the binding of [3H]GR65630 at 10 μM, indicates high α7 nAChR over 5-HT3 selectivity (Table 2). From this direct comparison a much higher affinity of ASEM (10b) than that of 10a for 5-HT3 may be expected considering the inhibition data at 100 nM (2% for 10a, 42% for ASEM). Based on this value, compound 10a is assumed to possess an at least fivefold higher selectivity than all other compounds, except 14b, reported herein. The Ki of 10a was estimated to be 440 nM for 5-HT3.
In addition, investigation of interaction of 10a at 1 μM with further off-targets (α1 nAChR, SERT, DAT, NET, VMAT, and choline transporter) by radioligand binding assays revealed no significant binding. Altogether, the target affinity and nAChR subtype specificity of 10a, the most suitable fluorine-substituted α7 nAChR ligand reported herein, is certainly sufficient to ensure that assessment of α7 nAChR in humans will not be confounded by binding of the respective PET radiotracer to relevant off-target sites. Therefore, compound 10a was selected for radiolabeling and further evaluation in vivo.
2.3. Optimization of Manual Radiosynthesis of [18F]10a for Translation to an Automated Module
Manual radiosynthesis of [18F]10a was performed starting from the nitro precursor 11a and systematically optimized by varying the base, solvent, reaction time and heating system (Scheme 5). The amount of nitro precursor 11a was kept constant at 1 mg according to our previous practice [52]. A nitro-to-fluoro substitution of activated aryl moieties in general requires moderately high temperatures (e.g., 130–160 °C) [53], reflected by low labeling efficiencies of <1% at 90 °C using MeCN. With increasing temperature (150 °C) and the use of DMSO, labeling efficiency of up to 30% were achieved.
Scheme 5.

Radiosynthesis of [18F]10a. Solvents tested: MeCN, DMSO, and DMF at different temperatures with microwave irradiation or conventional heating.
In contrast to decomposition of the corresponding [18F]7c reported by Gao et al. [17], we did not observe decomposition of [18F]10a in the presence of the strong base potassium carbonate in DMSO at this temperature. In an attempt to improve labelling efficiencies prior to high-scale production of [18F]10a in a commercially available automated device, we investigated the aromatic radiofluorination of 11a using DMF as solvent based on our recent experience [52] under different conditions (thermal vs. microwave heating). Influence of the basicity of the metal salt on the labelling yield (potassium carbonate vs. potassium oxalate) was also investigated.
Under thermal heating and the use of K2CO3, we determined a time- and temperature-dependent increase in the nitro-to-fluoro conversion with maximum labelling efficiencies of about 90% after 10 min of reaction time at 120 °C. Under these conditions, K2CO3 is preferred, since we observed a significantly lower labelling efficiency of ~30% using the weaker base K2C2O4. Our attempt to combine microwave dielectric heating with DMF as solvent with a mean-to-high dielectric constant rendered comparably high labeling efficiencies (≈94%) under microwave pulse mode (power cycling mode, 150 °C, 75 W, 5 min).
A semi-automated radiosynthesis coupled with microwave was recently reported for [18F]10b ([18F]ASEM) using the DMSO/K2CO3 system at 160 °C, resulting in a labeling efficiency of 45%–50% [54]. In the attempt to gain insight into the influence of the solvent on labeling efficiencies, we radiolabeled the nitro precursor 11b to obtain [18F]10b using the DMF/K2CO3 system under conventional and microwave-assisted heating at 120 °C. With only 1 mg of the nitro precursor 11b, labeling efficiencies of 60%–70% were achieved with DMF as solvent in both heating modes. These findings reinforce the superiority of DMF as a solvent of choice for this compound class, and allows the fully automated radiosynthesis of [18F]10a and [18F]10b for transfer to clinical radiopharmacies.
Overall, the radiolabelling reactions proceeded cleanly. No significant amount of 18F-labeled by-products was detected according to radio-TLC analyses (data not shown). The crude reaction mixture was applied directly onto a semi-preparative HPLC column (Figure 2a) and [18F]10a was successfully isolated in high radiochemical purities (≥98%) with a retention time of about 17 min. Analytical radio-HPLC analysis of the final product spiked with the reference compound confirmed the identity of [18F]10a (Figure 2b). [18F]10a proved to be stable in physiological solutions and organic solvents at 40 °C for up to 90 min.
Figure 2.


(a) Representative radio- and UV-chromatograms obtained for isolation of [18F]10a from crude reaction mixture by semi-preparative HPLC (Reprosil-Pur C18-AQ column, 35% MeCN/H2O/0.05% TFA, Flow rate: 10 mL∙min−1); (b) Analytical radio-chromatogram (top) and UV-chromatogram (bottom) of purified [18F]10a spiked with the reference compound 10a.
We determined the distribution coefficient of [18F]10a in the n-octanol-PBS system experimentally by the shake-flask method with a log D7.2 value of 1.3 ± 0.1 (n = 3), making sufficient blood-brain barrier permeability of [18F]10a likely.
The translation to remotely controlled radiosynthesis of [18F]10a was performed using the TRACERLAB™ FXFN module. The reaction was performed using the Kryptofix®222/K2CO3 system at 120 °C in 10 min with 1 mg of precursor 11a in DMF. Also in the automated process [18F]10a was obtained with extremely high labeling efficiencies of 86% ± 3%. After isolation via semi-preparative HPLC, [18F]10a was trapped on a pre-conditioned Sep-Pak® (Waters, Milford, MA, USA) C18 light cartridge, and formulated in isotonic saline containing 10% of EtOH (v/v). The average decay-corrected radiochemical yield was 35% ± 9% (n = 6) calculated at the end of the synthesis (EOS). Radiochemical purity of >99% and high specific activities of 855 ± 302 GBq·μmol−1 (n = 6) were obtained in a total synthesis time of 70 min. This rapid and versatile automated radiosynthesis will enhance the accessibility of [18F]10a for widespread production in future clinical studies.
2.4. Dynamic PET Studies of [18F]10a in Piglets
2.4.1. Baseline Studies
After intravenous injection of 425 ± 78 MBq [18F]10a (n = 3), we observed high uptake of radioactivity in the brain with peak concentrations at 8–10 min p.i., followed by washout. The highest accumulation of radioactivity (SUVmax > 2.2) occurred in the thalamus, colliculi, and midbrain. Somewhat lower accumulation (SUVmax ~1.9–2.1) was observed in other brain regions (Figure 3). The summed PET image (inset of Figure 3) shows the corresponding distribution pattern in the brain of a female piglet from 0 to 20 min p.i.
Figure 3.

Time-activity curves in different brain regions obtained by dynamic PET scans of [18F]10a in piglets under baseline and blocking conditions (mean values; n = 3). Standard deviations are shown for two examples (hippocampus baseline and cerebellum block). *** p < 0.001 vs. baseline. Inset: Brain image in sagittal plane acquired from 0 to 20 min p.i. after injection of [18F]10a in a female piglet. Data are expressed as standardized uptake value (SUV). Regions of interest (ROI) were drawn based on an overlay with T1-weighted MR images of a pig brain. Abbreviations: CB = Cerebellum, CC = Corpus callosum, St = Striatum, Th = Thalamus.
2.4.2. Metabolite Analysis
Radioactive metabolites of [18F]10a were assessed in plasma of pigs for up to 120 min p.i. (Figure 4a). For preparation of RP-HPLC samples, the proteins were precipitated and extracted two times with MeCN with a reproducible recovery of >90% of the starting radioactivity in the supernatant. The patterns of radioactive metabolites obtained by radio-TLC and radio-HPLC correlated well with each other. Parent fraction accounted for 88% ± 6%, 24% ± 4%, and 19% ± 4% of total radioactivity at 2, 60, and 120 min p.i., respectively.
Figure 4.

(a) Tri-exponential function fitting of the percentage of [18F]10a under baseline conditions (n = 3) which was used to obtain metabolite-corrected arterial plasma input functions for modelling; (b) Representative radio-chromatogram of a pig plasma sample obtained at 30 min after intravenous injection of [18F]10a under baseline conditions.
One major radioactive metabolite was detected, which was more hydrophilic than [18F]10a and is therefore assumed not to pass the blood-brain barrier (Figure 4b). Based on previous experience from our group with the diazabicyclononane-containing α7 nAChR ligand [18F]NS10743 [21], we believe that this metabolite is formed by enzymatic oxidation of the nitrogen at position 1 in the identical motif of 10a.
2.4.3. Blocking Studies
To evaluate the binding specificity of [18F]10a in vivo, we performed blocking studies (n = 3). Animals were pretreated with 3 mg∙kg−1 of the α7 nAChR ligand NS6740 [3] at 10 min prior to injection of 429 ± 45 MBq [18F]10a, followed by a continuous infusion of NS6740 at 1 mg∙kg−1∙h−1 during the course of the PET scan [21].
Despite a significant increase of about 40%–50%, in the brain uptake, radioactivity uptake under blocking conditions was about 15%–20% lower than that under baseline conditions in all brain regions evaluated from about 90 min p.i. until the end of the study (Figure 3). The early increase of brain uptake is accompanied by a significant increase of the influx rate constant K1 (Table 4). The most likely explanation is a blood flow driven effect. There is strong evidence that α7 nAChRs are expressed on vascular endothelial and smooth muscle cells [55,56,57], which we assume were activated by NS6740 despite its classification as a “weak agonist” [3].
Table 4.
Kinetic rate constants, non-displaceable volume of distribution (VND), and non-displaceable binding potential (BPND) of [18F]10a in different regions of piglet brain using 240 min of scan data under blocking conditions (n = 3).
| Brain Region | Rate Constant | VND | BPND | |||
|---|---|---|---|---|---|---|
| K1 (mL·cm−3·min−1) | k2 (min−1) | k3 (min−1) | k4 (min−1) | |||
| Whole | 0.598 ± 0.108 * | 0.062 ± 0.015 | 0.018 ± 0.022 | 0.011 ± 0.018 | 10.27 ± 3.94 * | 0.84 ± 1.07 * |
| Frontal Cortex | 0.647 ± 0.117 * | 0.055 ± 0.011 | 0.014 ± 0.019 | 0.010 ± 0.020 | 12.34 ± 4.27 * | 0.86 ± 0.92 |
| Parietal Cortex | 0.659 ± 0.120 * | 0.055 ± 0.013 | 0.015 ± 0.020 | 0.011 ± 0.021 | 12.58 ± 4.52 * | 0.82 ± 0.88 * |
| Occipital Cortex | 0.668 ± 0.103 * | 0.064 ± 0.019 | 0.022 ± 0.032 | 0.013 ± 0.023 | 11.36 ± 4.41 * | 0.95 ± 1.03 * |
| Hippocampus | 0.603 ± 0.118 * | 0.063 ± 0.023 * | 0.024 ± 0.030 | 0.011 ± 0.015 | 10.81 ± 5.02 * | 0.84 ± 1.70 * |
| Striatum | 0.624 ± 0.143 * | 0.057 ± 0.015 | 0.020 ± 0.025 | 0.011 ± 0.017 | 11.78 ± 4.91 * | 0.75 ± 1.29 * |
| Thalamus | 0.762 ± 0.129 * | 0.070 ± 0.025 | 0.023 ± 0.031 | 0.012 ± 0.021 | 12.09 ± 5.31 * | 0.88 ± 1.00 * |
| Colliculi | 0.735 ± 0.182 * | 0.066 ± 0.007 | 0.011 ± 0.013 | 0.007 ± 0.015 | 11.41 ± 3.90 * | 0.73 ± 0.74 * |
| Midbrain | 0.685 ± 0.131 * | 0.063 ± 0.010 | 0.013 ± 0.014 | 0.012 ± 0.018 | 11.17 ± 3.64 * | 0.61 ± 0.77 * |
| Pons | 0.670 ± 0.092 * | 0.084 ± 0.030 | 0.019 ± 0.023 | 0.012 ± 0.016 | 8.93 ± 3.96 * | 0.76 ± 1.16 * |
| Cerebellum | 0.711 ± 0.148 * | 0.076 ± 0.014 | 0.016 ± 0.019 | 0.011 ± 0.016 | 9.75 ± 3.57 * | 0.71 ± 1.10 * |
* p < 0.05 vs. baseline (Table 3).
2.4.4. Modeling
One-tissue and two-tissue compartment models (1TCM and 2TCM) were evaluated for mathematical description of [18F]10a time-activity curves (TACs) in pig brain regions (Figure 3) using metabolite-corrected arterial plasma input functions. The 2TCM was identified as the more appropriate model based on its smaller Akaike information criterion (AIC) value. The four rate constants K1, k2, k3, and k4 were fitted and the non-displaceable volume of distribution (VND = K1/k2) and the binding potential (BPND = k3/k4) as defined by Innis et al. [58] were calculated for each region of interest (Table 3 and Table 4).
Table 3.
Kinetic rate constants, non-displaceable volume of distribution (VND), and non-displaceable binding potential (BPND) of [18F]10a in different regions of piglet brain using 240 min of scan data under baseline conditions (n = 3).
| Brain Region | Rate Constant | VND | BPND | |||
|---|---|---|---|---|---|---|
| K1 (mL·cm−3·min−1) | k2 (min−1) | k3 (min−1) | k4 (min−1) | |||
| Whole | 0.362 ± 0.038 | 0.131 ± 0.062 | 0.093 ± 0.074 | 0.027 ± 0.018 | 3.32 ± 1.74 | 3.31 ± 1.02 |
| Frontal Cortex | 0.387 ± 0.059 | 0.182 ± 0.153 | 0.142 ± 0.165 | 0.027 ± 0.015 | 3.32 ± 2.06 | 4.22 ± 2.93 |
| Parietal Cortex | 0.399 ± 0.038 | 0.140 ± 0.089 | 0.126 ± 0.135 | 0.031 ± 0.021 | 3.88 ± 2.49 | 3.47 ± 1.65 |
| Occipital Cortex | 0.395 ± 0.053 | 0.123 ± 0.063 | 0.099 ± 0.087 | 0.030 ± 0.023 | 4.07 ± 2.23 | 3.16 ± 1.11 |
| Hippocampus | 0.400 ± 0.023 | 0.188 ± 0.083 | 0.145 ± 0.101 | 0.025 ± 0.013 | 2.43 ± 0.96 | 5.50 ± 1.01 |
| Striatum | 0.382 ± 0.028 | 0.144 ± 0.079 | 0.114 ± 0.093 | 0.027 ± 0.014 | 3.35 ± 1.81 | 3.98 ± 1.40 |
| Thalamus | 0.448 ± 0.027 | 0.150 ± 0.071 | 0.111 ± 0.085 | 0.030 ± 0.018 | 3.67 ± 2.10 | 3.51 ± 1.13 |
| Colliculi | 0.450 ± 0.072 | 0.134 ± 0.074 | 0.070 ± 0.042 | 0.027 ± 0.018 | 4.19 ± 2.37 | 2.68 ± 1.42 |
| Midbrain | 0.436 ± 0.045 | 0.162 ± 0.117 | 0.121 ± 0.132 | 0.029 ± 0.022 | 4.04 ± 3.06 | 3.40 ± 1.73 |
| Pons | 0.440 ± 0.045 | 0.149 ± 0.059 | 0.068 ± 0.042 | 0.026 ± 0.019 | 3.32 ± 1.40 | 2.76 ± 0.81 |
| Cerebellum | 0.421 ± 0.048 | 0.131 ± 0.052 | 0.067 ± 0.051 | 0.026 ± 0.020 | 3.62 ± 1.47 | 2.53 ± 0.57 |
The values of the influx rate constant K1 and the clearance rate constant k2 were comparable between the different brain regions with a mean of 0.362 mL·cm−3·min−1 and 0.131 mL·cm−3·min−1, respectively, in the whole brain. The total volume of distribution VT calculated from K1/k2(1 + k3/k4) [58] was 17 mL∙cm−3 in thalamus and 13 mL∙cm−3 in cerebellum, i.e., slightly lower than the comparable values for [18F]ASEM in baboons [18].
Under blocking conditions K1 was significantly increased by 50% to 70% in all brain regions while k2 was decreased between 42% and 70% resulting in a significant (up to 4-fold) increase of VND (Table 4) and preventing the use of the occupancy plot [59] for calculation of BPND. Therefore BPND, as the ratio (at equilibrium) of specifically bound radioligand to that of nondisplaceable radioligand in tissue [58], was directly calculated form the rate constants k3, which is proportional to the association rate constant, and k4, which is proportional to the dissociation rate constant from the specific compartment. The baseline study revealed that the rate constant k3 was highest in the hippocampus and lowest in the cerebellum and the values of k4 were low, rather uniform and ranged from 0.025 to 0.031 min−1. BPND values between 5.5 (hippocampus) and 2.5 (cerebellum) were reliably calculated from these data (SDwhole brain ~30%). A similarly high BPND has recently been reported in baboons for the structurally related [18F]ASEM [18] but not for any previous α7 nAChR PET radioligands [60]. Under blocking conditions k3 was decreased by 70% to 90% in various brain regions while k4 was decreased between 60% and 70%, resulting in a significant decrease of BPND by 72% to 85% in all regions but the frontal cortex (Table 4), suggesting specific binding of [18F]10a to α7 nAChR in a similar range as the recently reported [18F]ASEM [18].
2.5. Comparative PET Study of [18F]10a ([18F]DBT-10) and [18F]10b ([18F]ASEM) in a Rhesus Monkey
A PET study in a single rhesus monkey was performed for direct comparison of [18F]DBT-10 (injected dose: 167 MBq) and [18F]ASEM (injected dose: 185 MBq). Regional time-activity curves for both radioligands in the monkey brain are presented in Figure 5. Initial uptake levels were lower, and kinetics slower for [18F]DBT-10 than [18F]ASEM. For both radioligands the uptake levels follow the order of thalamus > frontal cortex = putamen > caudate > hippocampus > occipital cortex > cerebellum.
Figure 5.

Time-activity curves of [18F]DBT-10 and [18F]ASEM in selected brain regions of a rhesus monkey.
The total volume of distribution VT for both radioligands was similar with the highest values in the thalamus ([18F]DBT-10: 48.1 mL∙cm−3; [18F]ASEM: 46.7 mL∙cm−3) and the lowest in the cerebellum ([18F]DBT-10: 28.5 mL∙cm−3; [18F]ASEM: 26.8 mL∙cm−3). These values are almost two-fold higher than those reported for [18F]ASEM in baboons [18], supporting the species differences in α7 nAChR binding and distribution.
Altogether these preliminary data support the equal potency of both [18F]DBT-10 and [18F]ASEM for PET imaging of α7 nAChRs, with [18F]ASEM having slightly faster kinetics.
2.6. Toxicity Studies in Rats
To prepare [18F]10a for use in humans, toxicity studies were performed in rats according to EU cGLP. Compound 10a was administered by a single intravenous injection to rats followed by an observation period of 2 or 15 day. The study was performed with 4 test groups, including 1 control and 3 dose groups (6.2, 62 and 620 μg∙kg−1), with 60 male and 60 female Wistar rats divided into two experiments: Day 2 (40 males and 40 females) and Day 15 (20 males and 20 females). During clinical observation the animals displayed no notable clinical effects. No statistically significant differences in body weights between control and treated groups in either gender were detected. Food consumption corresponded with body weight development.
All haematology parameters on Day 2 and Day 15 were within physiological range for this species. Individual divergences of some haematology parameters were small and not correlated with treatment. No test item effect on haematology parameters was observed.
There were no findings in clinical chemistry parameters which could be definitively considered as adverse. The average values of all test groups were within the historical control ranges. Occasional changes had no dose relationship, and they were therefore considered as a result of intra- and inter-individual variability for this species. The results of pathology examination indicated that 10a after single intravenous administration did not result in changes in pathological and histopathological parameters on either Day 2 or Day 15. The no observed effect level (NOEL) of 10a was determined to be 620 μg∙kg−1.
3. Experimental Section
3.1. General Information
Analytical thin-layer chromatography (TLC) was performed with Macherey-Nagel precoated plastic sheets with fluorescent indicator UV254 (Polygram® SIL G/UV254, Düren, Germany). Visualization of the spots was effected by illumination with an UV lamp (254 nm and 366 nm). Dry-column flash chromatography (DCFC) [61] was performed with vacuum on silica gel 60 (particle size 15–40 μm, Ref. 815650) from Macherey-Nagel (Macherey-Nagel GmbH & Co. KG, Düren, Germany). NMR spectra (1H, 13C, 13C-APT, 19F, COSY, HSQC, HMBC) were recorded with Varian spectrometer (Varian Mercury-300BB and Mercury-400BB; Agilent Technologies, Santa Clara, CA, USA). Chemical shifts are reported as δ (δH, δC, δF) values. Coupling constants are reported in Hz. Multiplicity is defined by s (singlet), d (doublet), t (triplet), and combinations thereof; br (broad) and m (multiplet). ESI/Ion trap mass spectra (LRMS) were recorded with a Bruker Esquire 3000 plus instrument (Billerica, MA, USA). High resolution mass spectra were recorded on an FT-ICR APEX II spectrometer (Bruker Daltonics; Bruker Corporation, Billerica, MA, USA) using electrospray ionization (ESI) in positive ion mode. Melting points were determined on a Linström capillary apparatus (Wagner & Munz GmbH, Vienna, Austria) in open capillary tubes and are uncorrected. Reagents and solvents were purchased from commercial sources and used without further purification unless otherwise noted. Compounds 2, 4a, and 5a were prepared starting from 1a (dibenzo[b,d]thiophene; Acros Organics, Geel, Belgium) according to literature methods [30,31,32]. The preparation of intermediates 3, 5b, 8a, 8b, and the final compound 10 have been described previously, but with different synthetic routes or from starting materials not used in our protocols [17,31,62]. Intermediate 6a (two steps from 4a), 6b (one step from 5b) and final compounds 10a, 10b, 11a and 11b (one step each, from amine 9a), which have been prepared according to the literature [62], were also reported by Gao et al. [17]. To the best of our knowledge, intermediates 7a, 8c, 8d and final compounds 10c, 12a,b, 13b, and 14b have not been reported so far.
3.2. Chemistry
4-Nitrosodibenzo[b,d]thiophene (4). tert-Butyl nitrite (2.14 mL, 1.86 g, 18 mmol) was added in one portion to a stirred suspension of dibenzo[b,d]thiophen-4-yl boronic acid (1b, 1.37 g, 6 mmol; Frontier Scientific, Logan, UT, USA) in MeCN (24 mL) under argon at 22 °C. The mixture was stirred at 50–55 °C while its colour turned from yellow to dark brown in the course of 24 h. The solvent was evaporated and the residual solid was dissolved in CH2Cl2 (80 mL), washed with H2O (25 mL), dried (MgSO4) and evaporated to leave a dark greenish brown residue (1.38 g), which was used for the oxidation step. A sample (40 mg) was dissolved in cyclohexane/CH2Cl2 (2:3, v/v, 3 mL) and filtered through a short plug of silica gel (15–40 μm, 2 g). Subsequent elution with cyclohexane/CH2Cl2 (2:3, v/v, 50 mL) gave a green eluate which was evaporated to yield 30 mg of the title compound 4 (Rf = 0.56, heptane/EtOAc, 3:1) as a green powder, m.p. 113–115 °C (lit. m.p. 115–117 °C [36]). 1H-NMR (300 MHz, CDCl3): δ 7.48–7.63 (m, 2H, 7-H, 8-H), 7.88–7.94 (m, 1H, 6-H), 7.94 (t, J = 7.7 Hz, 1H, 2-H), 8.13–8.22 (m, 1H, 9-H), 8.49 (dd, J = 7.7, 1.1 Hz, 1H, 3-H), 9.58 (dd, J = 7.6, 1.1 Hz, 1H, 1-H). 13C-NMR (75 MHz, CDCl3): δ 120.10 (C), 121.68 (CH), 123.67 (CH), 125.44 (CH), 125.84 (CH), 127.82 (CH), 128.21 (CH), 132.38 (C), 137.94 (C), 138.11 (C), 142.04 (C), 161.91 (C), 165.95 (C). The product is 88% pure, containing 12% of 4-nitrodibenzo[b,d]thiophene (4b), as determined by comparison with 1H-NMR of a pure sample.
2-Nitrodibenzo[b,d]thiophene 5,5-dioxide (5a) [32]. A mixture of 2-nitrodibenzo[b,d]thiophene (4a, 2.3 g, 10 mmol) in acetic acid (64 mL) was stirred at 80 °C while H2O2 (50%, 6.8 mL, 0.12 mol) was added. The temperature was raised to 120 °C and after 1 h a second portion H2O2 (50%, 4.5 mL, 0.08 mol) was added. Stirring was continued at 120 °C for 1 h and at 80 °C for 3 h. After cooling, the mixture was poured into water (180 mL) to form a precipitate which was filtered, washed and dried to yield the pure title compound 5a (2.52 g, 9.64 mmol, 96% yield, Rf = 0.22, cyclohexane/CHCl3, 1:3) as a pale yellow powder, m.p. 256.5–257.5 °C (lit. m.p. 258 °C [32]); 1H-NMR (400 MHz, DMSO-d6) δH 7.74 (td, J = 7.6, 0.9 Hz, 1HAr, 7-H), 7.87 (td, J = 7.6, 1.0 Hz, 1HAr, 8-H), 8.07 (d, J = 7.7 Hz, 1HAr, 6-H), 8.29 (m, 1HAr, 9-H), 8.42 (dd, J = 8.4, 2.0 Hz, 1HAr, 3-H), 8.48 (d, J = 7.8 Hz, 1HAr, 4-H), 9.04 (d, J = 2.0 Hz, 1HAr, 1-H). 13C-NMR (100 MHz, DMSO-d6): δC 118.26 (CH), 122.28 (CH), 123.60 (CH), 123.95 (CH), 126.10 (CH), 129.10 (C), 132.09 (CH), 132.98 (C), 135.04 (CH), 137.08 (C), 141.44 (C), 151.72 (C). LRMS: m/z (ESI) = 284.0 (M + Na)+.
4-Nitrodibenzo[b,d]thiophene 5,5-dioxide (5b). In a procedure similar to the preparation of 5a, compound 4 (1.33 g, 5.8 mmol) was reacted with H2O2 (6.6 mL, 0.11 g, 116 mmol) in acetic acid to give 1.33 g of a dark yellow solid. The product was further purified by column chromatogaphy (silica gel 15–40 μm, 24 g) with cyclohexane/CH2Cl2 (1:1→1:2) to yield the pure title compound 5b (1.1 g, 4.2 mmol, 72% yield, Rf = 0.2, CHCl3) as pale yellow crystals, m.p. 245.5–251 °C. 1H-NMR (400 MHz, DMSO-d6): δH 7.75 (td, J = 7.6, 0.8 Hz, 1HAr, 7-H), 7.86 (td, J = 7.6, 1.0 Hz, 1HAr, 8-H), 8.04 (d, J = 7.7 Hz, 1HAr), 8.08 (t, J = 8.0 Hz, 1HAr), 8.29 (d, J = 7.7 Hz, 1HAr), 8.39 (dd, J = 8.2, 0.7 Hz, 1HAr), 8.65 (dd, J = 7.7, 0.7 Hz, 1HAr). 13C-NMR (100 MHz, DMSO-d6): δC 122.19 (CH), 123.21 (CH), 125.91 (CH), 128.29 (C), 129.16 (CH), 130.58 (C), 132.18 (CH), 134.45 (C), 134.71 (CH), 136.21 (CH), 137.26 (C), 143.39 (C, 4-C). LRMS: m/z (ESI) = 284.0 (M + Na)+.
7-Bromo-2-nitrodibenzo[b,d]thiophene 5,5-dioxide (6a). Compound 5a (1.75 g, 6.7 mmol) was dissolved in H2SO4 (96%, 30 mL). N-Bromosuccinimide (1.28 g, 7.2 mmol) was added to this solution in several portions and the mixture was stirred at room temperature for 24 h. The suspension was poured into ice-water (90 mL) and stirred for 10 min. The precipitate was filtered off, washed with H2O until neutral, followed by MeOH, and dried at 60 °C to obtain a yellowish solid (2.29 g). The raw material was recrystallized twice from MeCN to afford the pure title compound 6a (1.45 g, 4.27 mmol, 63.8% yield, Rf = 0.30, cyclohexane/CHCl3, 1:4) as a pale yellow powder, m.p. 281.0–282.5 °C. 1H-NMR (400 MHz, DMSO-d6): δH 8.08 (dd, J = 8.4, 1.8 Hz, 1HAr, 8-H), 8.31 (d, J = 8.4 Hz, 1HAr, 4-H), 8.41–8.47 (m, 3HAr, 9-H, 3-H, 6-H), 9.07 (d, J = 1.8 Hz, 1HAr, 1-H).13C-NMR (100 MHz, DMSO-d6): δC 118.52 (CH), 123.72 (CH), 125.11 (C), 125.36 (CH), 125.85 (CH), 126.35 (CH), 128.41 (C), 132.19 (C), 137.90 (CH), 138.66 (C), 141.11 (C), 151.78 (C). LRMS: m/z (ESI) = 364.0, 362.0 (M + Na)+.
7-Bromo-2-fluorodibenzo[b,d]thiophene 5,5-dioxide (8a). TMAF tetrahydrate (0.22 g, 1.3 mmol, Acros) was dried under an atmosphere of argon by azeotropic distillation with a mixture of DMSO (6 mL) and cyclohexane (12 mL) using a water separator for 6 h (bath temperature: 115–120 °C). The bath temperature was allowed to cool to 80 °C and the nitro derivative 6a (0.34 g, 1 mmol) was added under stirring to the suspension of the dried TMAF in one portion. The mixture immediately turned dark brown and was stirred for additional 5 h at 95 °C. Reaction progress was monitored by TLC (cyclohexane/CHCl3, 1:4). The portion of cyclohexane was evaporated. The residual oil was poured into water (60 mL) and extracted with CH2Cl2 (4 × 15mL).
The extracts were combined and washed with water (10 mL), dried (MgSO4) and evaporated. The residual yellow solid (0.283 g) was purified by column chromatography (silica gel 15–40 μm, 6 g) with CH2Cl2 to afford the pure title compound 8a, (0.23 g, 0.74 mmol, 73.7% yield, Rf = 0.39, cyclohexane/CHCl3, 1:4) as a colorless powder, m.p. 266.5–268.5 °C. 1H-NMR (300 MHz, DMSO-d6): δH 7.52 (dt-like, J = 8.8, 8.7, 2.4 Hz, 1HAr, 3-H), 8.05 (dd, J = 8.3, 1.8 Hz, 1HAr, 8-H), 8.11 (dd, J = 8.6, 4.9 Hz, 1HAr, 4-H), 8.16 (d, J = 8.3 Hz, 1HAr, 9-H), 8.18 (dd, J = 9.2, 2.3 Hz, 1HAr, 1-H), 8.35 (d, J = 1.6 Hz, 1HAr, 6-H); 13C-NMR (75 MHz, DMSO-d6): δC 110.74 (d, 2JCF = 25.6 Hz, CH), 118.22 (d, 2JCF = 24.1 Hz, CH), 124.53 (s, C), 124.89 (d, 3JCF = 10.2 Hz, CH), 125.09 (s, CH), 125.16 (s, CH), 128.98 (d, 4JCF = 2.4 Hz, C), 132.82 (d, 4JCF = 3.0 Hz, C), 133.45 (d, 3JCF = 10.7 Hz, C), 137.49 (s, CH), 139.31 (s, C), 165.85 (d, 1JCF = 252.2 Hz, C); 19F-NMR (282.36 MHz, DMSO-d6): δF −103.8 (m, FAr, 2-F). LRMS: m/z (ESI) = 334.9, 336.9 (M + Na)+.
2-Fluorodibenzo[b,d]thiophene 5,5-dioxide (7a). Similar to the preparation of 8a, TMAF tetrahydrate (1.35 g, 8.18 mmol) was azeotropically dried with cyclohexane/DMSO and subsequently reacted with 5a (1.525 g, 5.84 mmol). After work-up, the yellow solid (1.25 g) was purified by column chromatography (silica gel 15–40 μm, 18 g) with cyclohexane/CH2Cl2 (1:1→1:3) as eluent to yield the pure title compound 7a (1.03 g, 4.4 mmol, 75.2% yield, Rf = 0.4, cyclohexane/CHCl3, 1:4) as a colorless powder, m.p. 227–228 °C. 1H-NMR (400 MHz, CDCl3): δH 7.21 (dt, J = 8.45, 8.45, 2.27 Hz, 1HAr, H-3), 7.45 (dd, J = 8.37, 2.26 Hz, 1HAr, H-1), 7.57 (dt, J = 7.56, 7.53, 1.09 Hz, 1HAr, H-7), 7.66 (dt, J = 7.65, 7.58, 1.17 Hz, 1HAr, H-8), 7.75 (ddd, J = 7.70, 1.01, 0.57 Hz, 1HAr, 9-H), 7.82 (dd, J = 8.42, 4.92 Hz, 1HAr, H-4), 7.83 (ddd, J = 7.60, 1.09, 0.69 Hz, 1HAr, 6-H); 13C-NMR (100 MHz, CDCl3): δC 109.34 (d, 2JCF = 24.6 Hz, CH), 117.61 (d, 2JCF = 23.9 Hz, CH), 121.98 (s, CH), 122.40 (s, CH), 124.58 (d, 3JCF = 9.9 Hz, CH), 130.47 (d, 4JCF = 2.5 Hz, C), 131.23 (s, CH), 133.72 (d, 4JCF = 3.2 Hz, C), 134.13 (s, CH), 134.92 (d, 3JCF = 9.7 Hz, C), 138.68 (s, C), 166.37 (d, 1JCF = 255.0 Hz, C); 19F-NMR (282.36 MHz, DMSO-d6): δF ppm −103.63 (m. 1F, 2-F); LRMS: m/z (ESI) = 257.0, 258.0 (M + Na)+.
3-Bromo-2-fluorodibenzo[b,d]thiophene 5,5-dioxide (8c) and 3,7-dibromo-2-fluoro-dibenzo[b,d]-thiophene 5,5-dioxide (8d). Similar to the preparation of compound 6a, compound 7a (0.56 g, 2.4 mmol) was reacted with NBS (0.43 g, 2.4 mmol) in H2SO4. The solid residue obtained upon work-up (0.75 g) consisted of a mixture of starting material (Rf = 0.32), mono- (Rf = 0.43) and dibrominated derivative (Rf = 0.56) as shown by TLC analysis (cyclohexane/CHCl3, 1:4). The mixture was subjected to chromatographic purification (silica gel 15–40 μm, 28 g) with cyclohexane/CHCl3 (1:3) to yield the 2-fluoro-3,7-dibromo derivative 8d (0.17 g, 0.43 mmol, 18%) from the first fraction and the 2-fluoro-3-bromo derivative 8c (0.35 g, 1.12 mmol, 46.5%) from the second fraction. Compound 8c: m.p. 269–274.5 °C. 1H-NMR (400 MHz, DMSO-d6): δH 7.72 (t, J = 7.6 Hz, 1HAr, 7-H), 7.84 (t, J = 7.6 Hz, 1HAr, 8-H), 8.01 (d, J = 7.7 Hz, 1HAr, 6-H), 8.22 (d, J = 7.7 Hz, 1HAr, 9-H), 8.32 (d, J = 8.9 Hz, 1HAr, 1-H), 8.55 (d, J = 6.2 Hz, 1HAr, 4-H). 13C-NMR (100 MHz, DMSO-d6): δC 110.78 (d, 2JCF = 23.4 Hz, C), 111.57 (d, 2JCF = 26.5 Hz, CH), 122.07 (s, CH), 123.33 (s, CH), 127.71 (d, 3JCF = 1.5 Hz, CH), 129.16 (d, 4JCF = 2.1 Hz, C), 131.80 (s, CH), 133.30 (d, 3JCF = 9.9 Hz, C), 134.06 (d, 4JCF = 3.5 Hz, C), 134.77 (s, CH), 137.28 (s, C), 162.00 (d, 1JCF = 251.8 Hz, C). 19F-NMR (376.4 MHz, DMSO-d6): δF −98.68 (m, 1 FAr, 2-F). LRMS: m/z (ESI) = 336.9, 335.0 (M + Na)+. Compound 8d: m.p. 282.5–285 °C. 1H-NMR (300 MHz, CDCl3): δH 7.49 (d, J = 7.8 Hz, 1HAr, 1-H), 7.60 (d, J = 8.2 Hz, 1HAr, 9-H), 7.79 (dd, J = 8.2, 1.8 Hz, 1HAr, 8-H), 7.94 (d, J = 1.7 Hz, 1HAr, 6-H), 8.01 (d, J = 6.0 Hz, 1HAr, 4-H). 13C-NMR (75 MHz, CDCl3): δC 110.07 (d, 2JCF = 25.8 Hz, CH), 111.88 (d, 2JCF = 12.2 Hz, C), 123.28 (s, CH), 125.53 (s, C), 125.84 (s, CH), 128.11 (d, 3JCF = 1.9 Hz, CH), 128.75 (s, C), 132.68 (d, 3JCF = 8.8 Hz, C), 134.32 (d, 4JCF = 3.8 Hz, C), 137.41 (s, CH), 139.67 (s, C), 162.85 (d, J = 256.4 Hz, C). 19F-NMR (282.4 MHz, CDCl3) δF −98.68 (m, 1 FAr, 2-F). LRMS: m/z (ESI) = 414.9, 416.9, 413.0 (M + Na)+.
7-(1,4-Diazabicyclo[3.2.2]nonan-4-yl)-2-fluorodibenzo[b,d]thiophene 5,5-dioxide (10a). A mixture of Pd2(dba)3 (11 mg, 0.012 mmol) and BINAP (15 mg, 0.024 mmol) in toluene (1.5 mL) was stirred for 30 min at 90 °C. The red-orange colored solution of the catalyst was allowed to cool (22 °C) and added to a mixture of 1,4-diazabicyclo[3.2.2]nonane (9a, 53 mg, 0.42 mmol) and 8a (0.126 g, 0.40 mmol) in toluene (2 mL). Cs2CO3 (Alfa Aesar, Karlsruhe, Germany; previously dried for 2 h at 120 °C, 4 mbar; 0.39 g, 1.2 mmol) was then added, and the reaction mixture was stirred under an atmosphere of argon for 24 h at 90° C. After cooling to room temperature, the solid was filtered off and washed with CH2Cl2 (2 × 4 mL). The filtrate was evaporated and chromatographically purified (silica gel 15–40 μm, 8 g) with a gradient from CHCl3 (100%) to CHCl3/MeOH/NH3 (aq) (100:8:0.8). The fractions containing the product were combined, evaporated and the solid residue was recrystallized from EtOH to afford the title compound 10a (0.075 g, 0.21 mmol, 52% yield, Rf = 0.18, CHCl3/MeOH/NH3 (aq.), 100:10:1) as yellow crystals, m.p. 317–319 °C (dec.). 1H-NMR (400 MHz, CDCl3): δH 1.78 (qd-like, J = 9.7, 4.6 Hz, 2H, 6′-Ha, 9′-Ha), 2.11 (m, 2H, 6′-Hb, 9′-Hb), 2.99 (m, 2H, 7′-Ha, 8′-Ha), 3.09 (A-part of AA′BB′, 2H, 2′-H2), 3.14 (m, 2H, 7′-Hb, 8′-Hba), 3.61 (B-part of AA′BB′, 2H, 3′-H2), 4.08 (m, not resolved, 1H, 5′-H),6.89 (dd, J = 8.8, 2.5 Hz, 1HAr, 8-H), 7.00 (td-like, J = 8.5, 8.5, 2.2 Hz, 1HAr, 3-H), 7.10 (d, J = 2.5 Hz,1HAr,6-H), 7.24 (dd, J = 8.8, 2.2 Hz, 1HAr, 1-H), 7.49 (d, J = 8.7 Hz, 1HAr, 9-H), 7.70 (dd, J = 8.4, 4.9 Hz, 1HAr, 4-H). 13C-NMR (100 MHz, CDCl3): δC 26.77 (s, 2 CH2), 44.69 (s, CH2), 46.51 (s, 2 CH2), 51.74 (s, CH), 57.05 (s, CH2), 105.19 (s, CH), 107.66 (d, 2JCF = 25.1 Hz, CH), 114.70 (d, 2JCF = 24.3 Hz, CH), 117.03 (s, CH), 117.28 (d, 4JCF = 2.2 Hz, C), 123.15 (s, CH), 124.27 (d, 3JCF = 10.3 Hz, CH), 132.88 (d, 4JCF = 3.0 Hz, C), 136.19 (d, 3JCF = 9.6 Hz, C), 140.61 (s, C), 151.15 (s, C), 166.58 (d, 1JCF = 253.6 Hz, C). 19F-NMR (282.4 MHz, CDCl3): δF −104.5 (m, 1FAr, 2-F). HRMS m/z (ESI): calcd for C19H20FN2O2S (M + H)+ 359.12240, found 359.12223.
3-(1,4-Diazabicyclo[3.2.2]nonan-4-yl)-6-fluorodibenzo[b,d]thiophene 5,5-dioxide (10b). In a procedure similar to the preparation of 10a, compound 8b (0.14 g, 0.45 mmol) and amine 9a (0.059 g, 0.47 mmol) were reacted in the presence of Cs2CO3 (0.44 g, 1.35 mmol) and a catalyst made from Pd2(dba)3 (10.3 mg, 0.011 mmol) and BINAP (14 mg, 0.022 mmol). For purification the same protocol as for 10a was followed to afford the title compound 10b (0.077 g, 0.21 mmol, 48% yield, Rf = 0.19, CHCl3/MeOH/NH3 (aq), 100:10:1) as yellow crystals, m.p. 272.5–274 °C (dec.). 1H-NMR (300 MHz, CDCl3): δH 1.77 (qd-like, J = 9.7, 4.6 Hz, 2H, 6′-Ha, 9′-Ha), 2.11 (ddtd, J = 12.4, 9.7, 5.0, 2.6 Hz, 2H, 6′-Hb, 9′-Hb), 3.05–2.92 (m, 2H, 7′-Ha, 8′-Ha), 3.20–3.06 (m, A-part of AA′BB′, 4H, 7′-Hb, 8′-Hba, 3′-H2), 3.61 (t-like, J = 5.7 Hz, B-part of AA′BB′, 2H, 3′-H2), 4.08 (m, not resolved, 1H, 5′-H), 6.89 (dd, J = 8.8, 2.5 Hz, 1HAr, 2-H), 6.97 (t, J = 8.4 Hz, 1HAr, 7-H), 7.09 (d, J = 2.5 Hz, 1HAr, 4-H), 7.35 (d, J = 7.6 Hz, 1HAr, 9-H), 7.50 (dt, J = 8.0, 5.2 Hz, 1HAr, 8-H), 7.53 (d, J = 8.7 Hz, 1HAr, 1-H). 13C-NMR (75 MHz, CDCl3): δC 26.78 (s, 2 CH2), 44.70 (s, CH2), 46.52 (s, 2 CH2), 51.76 (s, CH), 57.06 (s, CH2), 105.09 (s, CArH), 115.17 (d, 2JCF = 19.4 Hz, CArH), 115.85 (d, 4JCF = 3.4 Hz, CArH), 117.09 (s, CArH), 117.65 (d, 4JCF = 2.8 Hz, CAr), 123.25 (s, CArH), 123.63 (d, 2JCF = 17.7 Hz, CAr), 135.83 (d, 3JCF = 2.7 Hz, CAr), 136.17 (d, 3JCF = 7.9 Hz, CArH), 140.42 (s, CAr), 151.12 (s, CAr), 158.05 (d, 1JCF = 257.4 Hz, CAr). 19F-NMR (282.4 MHz, CDCl3): δF −115.66 (m, FAr, 6-F). HRMS m/z (ESI): calcd for C19H20FN2O2S (M + H)+ 359.12240, found 359.12235.
3-(1,4-Diazabicyclo[3.2.2]nonan-4-yl)-2-fluorodibenzo[b,d]thiophene 5,5-dioxide (10c). In a procedure similar to the preparation of 10a, compound 8c (0.126 g, 0.4 mmol) and 9a (0.053 g, 0.42 mmol) were reacted in the presence of Cs2CO3 (0.39 g, 1.2 mmol) and a catalyst mixture made from Pd2(dba)3 (11 mg, 0.012 mmol) and BINAP (14.9 mg, 0.024 mmol). For purification the same protocol for 10a was followed to afford the title compound 10c (0.063 g, 0.18 mmol, 44% yield, Rf = 0.20, CHCl3/MeOH/NH3 (aq), 100:10:1) as pale yellow crystals, m.p. 305–309 °C (dec.). 1H-NMR (300 MHz, CDCl3): δH 1.75 (m, 2H, 6′-Ha, 9′-Ha), 2.11 (m, 2H, 6′-Hb, 9′-Hb), 3.04 (m, 4H, 7′-H2, 8′-H2), 3.16 (t, J = 5.6 Hz, A-part of AA′BB′, 2H, 2′-H2), 3.44 (t, J = 5.6 Hz, B-part of AA′BB′, 2H, 3′-H2), 3.80 (m, 1H, 5′-H), 7.32 (s, 1HAr, 4-H), 7.36 (d, J = 5.0 Hz, 1HAr, 1-H), 7.43 (ddd, J = 7.7, 5.1, 3.4 Hz, 1HAr, 7-H), 7.55–7.63 (m, 2HAr, 8-H, 9-H), 7.75 (d, J = 7.6 Hz, 1HAr, 6-H). 13C-NMR (75 MHz, CDCl3): δC 27.64 (s, 2 CH2), 46.70 (s, 2 CH2), 47.50 (d, 4JCF = 1.2 Hz, CH2), 55.83 (d, 4JCF = 6.0 Hz, CH), 56.30 (s, CH2), 109.97 (d, 2JCF = 24.9 Hz, CH), 111.82 (d, 3JCF = 5.5 Hz, CH), 120.89 (s, CH), 122.19 (s, CH), 123.72 (d, 3JCF = 9.4 Hz, C), 129.25 (s, CH), 131.40 (d, 4JCF = 2.3 Hz, C), 133.98 (d, 4JCF = 2.9 Hz, C), 134.05 (s, CH), 137.87 (s, C), 142.90 (d, 2JCF = 10.5 Hz, C), 157.62 (d, 1JCF = 252.1 Hz, C). 19F-NMR (282.4 MHz, CDCl3): δF −112.95 (m, 1 FAr, 2-F). HRMS m/z (ESI): calcd for C19H20FN2O2S (M+H)+ 359.12240, found 359.12235.
6-Fluoro-3-(9-methyl-3,9-diazabicyclo[3.3.1]nonan-3-yl)dibenzo[b,d]thiophene 5,5-dioxide (12b). In a procedure similar to the preparation of 10a, compound 8b (0.13 g, 0.42 mmol) and amine 9b (0.065 g, 0.46 mmol) were reacted in the presence of Cs2CO3 (0.28 g, 0.84 mmol) and a catalyst made from Pd2(dba)3 (11.5 mg, 0.013 mmol) and BINAP (15.7 mg, 0.0252 mmol). For purification the same protocol for 10a was followed to afford the title compound 12b (0.065 g, 0.175 mmol, 42% yield, Rf = 0.31, CHCl3/MeOH/NH3 (aq), 100:10:1) as yellow crystals, m.p. 264–272 °C (dec.). 1H-NMR (300 MHz, CDCl3): δH 1.52–1.64 (m, 3H, 7′-Ha, 6′-Ha, 8′-Ha), 1.93–2.15 (m, 3H, 7′-Hb, 6′-Hb, 8′-Hb), 2.59 (s, 3H, 9′-NCH3), 3.01 (s, 2H, 1′-H, 5′-H), 3.39–3.51 (m, 4H, 2′-H2, 4′-H2), 6.97–7.03 (m, 2HAr, 7-H, 2-H), 7.22 (d, J = 2.4 Hz, 1HAr, 4-H), 7.39 (dd, J = 7.7, 0.5 Hz, 1HAr, 9-H), 7.52 (ddd, J = 8.2, 7.8, 5.2 Hz, 1HAr, 8-H), 7.59 (d, J = 8.7 Hz, 1HAr, 1-H). 13C-NMR (75 MHz, CDCl3): δC 18.99 (s, 1 Csec), 27.93 (s, 2Csec), 40.62 (s, CH3), 47.40 (s, 2CH2), 53.03 (s, 2CH), 105.41 (s, CArH), 115.46 (d, 2JCF = 19.4 Hz, CArH), 116.05 (d, 4JCF = 3.4 Hz, CArH), 117.22 (s, CArH), 118.83 (d, 4JCF = 2.6 Hz, CAr), 122.99 (s, CArH), 123.77 (d, 2JCF = 18.1 Hz, CAr), 135.76 (d, 3JCF = 2.5 Hz, CAr), 136.20 (d, 3JCF = 7.8 Hz, 1CArH), 140.10 (s, CAr), 152.27 (s, CAr), 158.06 (d, 1JCF = 257.4 Hz, CAr). 19F-NMR (282 MHz, CDCl3): δ −115.56 (dd-like, J = 8.4, 5.1 Hz, 1F, 6-F). HRMS m/z (ESI): calcd for C20H21FN2O2S (M + H)+ 373.13805, found 373.13773.
7-(1,4-Diazabicyclo[3.2.2]nonan-4-yl)-2-nitrodibenzo[b,d]thiophene 5,5-dioxide (11a). In a procedure similar to the preparation of 10a, compound 6a (0.17 g, 0.5 mmol) and amine 9a (0.064 g, 0.51 mmol) were reacted in the presence of Cs2CO3 (0.49 g, 1.5 mmol) and a catalyst mixture made from Pd2(dba)3 (18.3 mg, 0.02 mmol) and BINAP (25 mg, 0.04 mmol). For purification the same protocol for 10a was followed to afford the title compound 11a (0.10 g, 0.18 mmol, 53% yield, Rf = 0.21, CHCl3/MeOH/NH3 (aq), 100:10:1) as dark red crystals, m.p. 291–294 °C (dec.). 1H-NMR (300 MHz, CDCl3): δH 1.79 (qd-like, J = 9.6, 4.6 Hz, 2H, 6′-Ha, 9′-Ha), 2.12 (m, 2H, 6′-Hb, 9′-Hb), 2.99 (m, 2H, 7′-Ha, 8′-Ha), 3.10 (A-part of AA′BB′, 2H, 2′-H2), 3.13 (m, 2H, 7′-Hb, 8′-Hba), 3.65 (B-part of AA′BB′, 2H, 3′-H2), 4.11 (m, not resolved, 1H, 5′-H), 6.94 (dd, J = 8.8, 2.6 Hz, 1HAr, 8-H), 7.11 (d, J = 2.5 Hz, 1HAr, 6-H), 7.63 (d, J = 8.8 Hz, 1HAr, 9-H), 7.85 (d, J = 8.3 Hz, 1HAr, 4-H), 8.15 (dd, J = 8.3, 2.0 Hz, 1HAr, 3-H), 8.35 (d, J = 1.9 Hz, 1HAr, 1-H). 13C-NMR (75 MHz, CDCl3): δC 26.70 (2 CH2), 44.71 (CH2), 46.47 (2 CH2), 51.72 (CH), 57.02 (CH2), 105.18 (CH), 115.16 (CH), 116.04 (C), 117.33 (CH), 122.59 (CH), 123.18 (CH), 123.67 (CH), 135.15 (C), 140.17 (C), 141.86 (C), 151.46 (C), 151.82 (C). HRMS m/z (ESI): calcd for C19H20N3O4S (M + H)+ 386.11690, found 386.11704.
3.3. Manual Radiosynthesis of [18F]10a
No-carrier-added (n.c.a.) [18F]fluoride was produced via the 18O(p,n)18F nuclear reaction by irradiation of a [18O]H2O target (Hyox 18 enriched water; Rotem Industries Ltd, Mishor Yamin, Israel) on a Cyclone®18/9 (IBA RadioPharma Solutions, Louvain-la-Neuve, Belgium) with 18 MeV proton beam using a Nirta® [18F]fluoride XL target or [18O]H2O recycled by the established in-house method [63]. Starting with 1–2 GBq of n.c.a. 18F-fluoride, [18F]F−-containing anion resin was eluted with a 20 mg mL−1 aqueous solution of K2CO3 (1.78 mg, 12.9 mmol) and added to a 5 mL V-vial in the Discover PET wave microwave CEM® (CEM Corporation, Matthews, NC, USA) cavity in the presence of Kryptofix®222 (11.2 mg, 29.7 mmol) in 1 mL MeCN. The aqueous [18F]fluoride was dried under vacuum and argon flow in the microwave cavity (75 W, 20 cycles) at 50–60 °C for 10–12 min. Additional aliquots of MeCN (2 × 1.0 mL) were added for azeotropic drying. Thereafter, the precursor (1 mg) was added to the reactive anhydrous K[18F]F-Kryptofix®222/K2CO3-complex. The reaction parameters were optimized varying the reaction time, temperature, solvent (MeCN, DMF, DMSO), and heating condition (thermal vs. microwave-assisted). Thereafter, the crude reaction mixture was diluted with H2O/MeCN (4:1, v/v) and directly applied onto an isocratic semi-preparative HPLC for isolation of [18F]10a. Fractions were collected, diluted with 30 mL of H2O and sodium hydroxide was added to neutralize the solution (~100–200 μL of 1 M NaOH). Final purification was performed using a Sep-Pak® C18 light cartridge (Waters, Milford, MA, USA) followed by elution with 1 mL of ethanol. To obtain an injectable solution the solvent was concentrated under a gentle argon stream and [18F]10a was formulated in a sterile isotonic saline solution (5%–10% EtOH, v/v). The identity of [18F]10a was verified by radio-HPLC analysis of a sample of the radiotracer solution spiked with the non-radioactive reference 10a. Radiochemical and chemical purities were assessed by radio-TLC and analytical HPLC. The mass determination for specific activity was determined on the base of a calibration curve carried out under the same analytical HPLC conditions.
3.4. Automated Radiosynthesis of [18F]10a and [18F]10b ([18F]ASEM)
Remote controlled radiosynthesis was performed using a TRACERLAB™ FXFN synthesizer (GE Healthcare, Waukesha, WI, USA) equipped with a PU-980 pump (JASCO, Gross-Umstadt, Germany), WellChrom K-2001 UV detector (KNAUER GmbH, Berlin, Germany), NaI(Tl)-counter and automated data acquisition (NINA software version 4.8 rev. 4; Nuclear Interface GmbH, Dortmund, Germany). For transfer to the automated module we started with activities in the range of 5–12 GBq of n.c.a. 18F-fluoride. Based upon the conditions optimized manually, the nitro-to-fluoro displacement was achieved by adding the respective nitro precursor (1 mg) dissolved in anhydrous DMF (1 mL) to the K[18F]F-Kryptofix®222/K2CO3-complex. The reaction mixture was stirred at 120 °C for 10 min. After cooling, the reaction mixture was diluted with 3 mL of H2O/MeCN (4:1) and directly applied onto the semi-preparative Reprosil-Pur 120 C18-AQ HPLC column (250 × 20 mm, 10 μm) using a solvent composition of 35% MeCN/H2O/0.05%TFA as eluent and a flow rate of 10 mL·min−1. The desired product was collected in 40 mL of H2O, neutralized with 1 M NaOH and directly transferred to a pre-activated Sep-Pak® C18 light cartridge. The cartridge was washed with 2 mL of water and [18F]10a or [18F]10b was obtained after elution with 1 mL of EtOH. Injectable solutions were obtained by partial evaporation of the solvent under a gentle argon stream at 70 °C and dilution in isotonic saline (5%–10% of EtOH, v/v). Radiochemical purity and specific activity were assessed following the chromatographic methods described in quality control.
3.5. Quality Control
To control the quality of the K[18F]F-Kryptofix®222/K2CO3-complex, reactions using ethylene glycol ditosylate were performed randomly. Radio-TLC of [18F]10a and [18F]10b was performed on Polygram® ALOX N/UV254 plates (Macherey-Nagel GmbH & Co. KG) with dichloromethane/methanol (9:1, v/v). The spots of the references were visualized using UV light at 254 nm. Radiochemical purity and specific activity were determined using analytical radio-HPLC with a Reprosil-Pur C18-AQ column (250 × 4.6 mm, 5 μm) and 26% MeCN/H2O/0.05% TFA as eluent at a flow rate of 1 mL·min−1. Analytical radio-HPLC profiles for the in vivo metabolism analysis in plasma was assessed by using a gradient mode (0–10 min: 10% MeCN/20 mM NH4OAcaq., 10–40 min: 10%→90% MeCN/20 mM NH4OAcaq., 40–50 min: 90%→10% MeCN/20 mM NH4OAcaq., 50–60 min: 10% MeCN/20 mM NH4OAcaq.) on a Reprosil-Pur C18-AQ (250 × 4.6 mm, 5 μm) column at a flow rate of 1 mL·min−1.
3.6. Determination of in Vitro Stability and Lipophilicity (Log D7.2)
in vitro radiochemical stability of [18F]10a was investigated in 0.9% NaCl solution, PBS (pH 7.2), and 0.01 M Tris-HCl (pH 7.4 at 21 °C) at 40 °C and in EtOH at room temperature for up to 90 min. Samples were taken at 15, 30, 60, and 90 min of incubation time and analyzed by radio-TLC and radio-HPLC. Log D7.2 of [18F]10a was experimentally determined in n-octanol/phosphate-buffered saline (PBS; 0.01 M, pH 7.2) at room temperature by the shake-flask method in multiple distribution. Measurement was performed twice in triplicate.
3.7. Biological Evaluation
3.7.1. Animals
Male Sprague-Dawley rats (275–300 g) were obtained from Charles River Laboratories, and female piglets (German Landrace pigs DL × Large White/Pietrain; mean weight 15.8 ± 0.8 kg, mean age 8 weeks) as well as female CD-1 mice (10–12 weeks, 22–26 g) were obtained from Medizinisch-Experimentelles Zentrum, Universität Leipzig, and all animals were provided with food and water ad libitum. All of the animal experiments were performed in accordance with the European Communities Council Directive of 24th November 1986 (86/609/EEC), and experiments using female piglets and female CD-1 mice were approved by the Animal Care and Use Committee of Saxony (TVV 08/13). Imaging experiments in rhesus monkeys were performed in accordance with a protocol approved by the Yale Institutional Animal Care and Use Committee.
3.7.2. Binding Assays
Affinity for Human α7 nAChR
The affinity of the test compounds for human α7 nAChR was determined by radioligand displacement experiments as previously described [12]. In brief, SH-SY5Y cells stably expressing human α7 nAChR [64] were grown in DMEM/Ham’sF-12, supplemented with 10% FCS, stable glutamine, geneticin (100 μg∙mL−1), penicillin/streptomycin (100 U∙mL−1, 100 μg∙mL−1) at 37 °C in an atmosphere containing 5% CO2. Cells were harvested by scraping, sedimented (800 rpm, 3 min), diluted with 50 mM TRIS–HCl, pH 7.4, and stored at −25°C until use. For determination of α7 nAChR affinity of reference compounds, frozen cell suspensions were thawed, homogenized by a 27-gauge needle, and diluted with incubation buffer (50 mM TRIS–HCl, pH 7.4, 120 mM NaCl, 5 mM KCl). Membrane suspension was incubated with [3H]methyllycaconitine ([3H]MLA; ~0.3 nM final concentration; specific activity of 2.220 GBq∙mmol−1; NEN Life Sciences Products, Boston, MA, USA) and various concentrations of the test compound. Nonspecific binding was determined by co-incubation with 300 μM (−)-nicotine tartrate. The incubation was performed at room temperature for 120 min and terminated by rapid filtration using Whatman GF/B glass-fibre filters, presoaked in 0.3% polyethyleneimine, and a 48-channel harvester (Biomedical Research and Development Laboratories, Gaithersburg, MD, USA), followed by 4× washing with ice-cold 50 mM TRIS-HCl, pH 7.4. Filter-bound radioactivity was quantified by liquid scintillation counting. The 50% inhibition concentrations (IC50) were estimated from the competition curves by nonlinear regression using GraphPadPrism software and the Ki values calculated according to the Cheng-Prusoff equation [65].
Affinity for Human α4β2 and α3β4 nAChR
The affinity of the test compounds for human α4β2 and α3β4 nAChR was determined as described above. In brief, HEK293 cells stably expressing human α4β2 or α3β4 nAChR [66] were cultured. The cells were harvested and processed as described above, and membrane suspension was incubated with (±)-[3H]epibatidine (0.4 to 0.6 nM final concentration; specific activity of 1250 GBq∙mmol−1; PerkinElmer Human Health, Rodgau, Germany) and various concentrations of the test compound. Nonspecific binding was determined in the presence of 300 μM (−)-nicotine tartrate. Incubation of the samples, separation of free from receptor-bound radioligand, and data evaluation were performed as described above.
Affinity for Subunit-Specific Antibodies Immobilized Native Rat α6β2* nAChR
The specificity of the anti-α6 antibody was tested as described [67]. The receptors were prepared and immobilized as previously described [68] by the subunit-specific antibody. The inhibition of [3H]epibatidine binding to the immobilized subtypes by the test compounds was measured by pre-incubating increasing concentrations of the compounds for 1 h at room temperature, followed by incubation overnight at 4 °C with 0.1 nM (±)-[3H]epibatidine (specific activity of 1250 GBq∙mmol−1; PerkinElmer Human Health, Rodgau, Germany). Incubations were performed in a buffer containing 50 mM Tris-HCl, pH 7, 150 mM NaCl, 5 mM KCl, 1 mM MgCl2, 2.5 mM CaCl2, 2 mg∙mL−1 BSA, and 0.05% Tween 20. Specific ligand binding was defined as total binding minus the binding in the presence of 100 nM (±)-epibatidine dihydrochloride hydrate. After incubation, the wells were washed seven times with ice-cold PBS containing 0.05% Tween 20, the bound [3H]epibatidine released by adding 200 μL of 2 M NaOH to each well and after incubation for 2 h determined by means of liquid scintillation in a beta counter.
Saturation binding analysis revealed an affinity of (±)-[3H]epibatidine of 25 pM (CV 20%) for the nAChRs immobilized by anti-α6 antibody from rat striatum, which contains two major α6β2* subtypes (α6β2β3 and α6α4β2β3) in very similar proportions.
The experimental data obtained from the binding experiments were analyzed by means of a non-linear least square procedure using the LIGAND program [69]. The Ki values of the test compounds were also determined by means of the LIGAND program by simultaneously fitting the data obtained from three to five independent saturation and competition binding experiments.
Affinity for Human 5-HT3 Receptor
The affinity of the test compounds for human 5-HT3 receptor was evaluated by a contract research organization (Eurofins Panlabs, Inc., Taipei, Taiwan). All compounds were tested at 1 μM in radioligand binding assays using human recombinant HEK-293 cells for the percentage inhibition of the binding by the 5-HT3 specific radioligand [3H]GR-65630 (working concentration = 0.69 nM, KD = 0.20 nM). In addition, for compound 10a a Ki value was estimated based on the inhibition of the radioligand binding at seven concentrations of the test compound (20 μM–5 nM).
Affinity for Other Target Proteins
The activity of compound 10a was evaluated by a contract research organization (Eurofins Panlabs, Inc.). The test concentration was 1 μM and radioligand binding assays for dopamine transporter (DAT), serotonin transporter (SERT), norepinephrine transporter (NET), monoamine transporter (VMAT), choline transporter, and α1 nAChR performed according to the manufacturer’s protocols. Responses were judged as significant at ≥50% inhibition or stimulation of the binding of the specific radioligand.
3.7.3. PET Studies in Piglets
Six female animals were used in this study with anesthesia and surgery performed as described previously [70]. In brief, the animals received initially a premedication with midazolam (1 mg∙kg−1 i.m.) followed by induction of anesthesia with 3% isoflurane in 70% N2O/30% O2. All incision sites were infiltrated with 1% lidocaine, and the anesthesia was maintained with 1.5% isoflurane throughout the surgical procedure. A central venous catheter was introduced through the left external jugular vein and used for the administration of the radiotracer and drugs, and for volume substitution with heparinized (50 IE∙mL−1) lactated Ringer’s solution (2 mL∙kg−1∙h−1). An endotracheal tube was inserted by tracheotomy for artificial ventilation (Servo Ventilator 900C, Siemens-Elema, Solna, Sweden) after immobilization with pancuronium bromide (0.2 mg∙kg−1∙h−1). The artificial ventilation was adjusted to obtain normoxia and normocapnia (Radiometer ABL 500, Copenhagen, Denmark). Polyurethane catheters (i.d., 0.5 mm) were advanced through the left and the right femoral arteries into the abdominal aorta to withdraw arterial blood samples for regular monitoring of blood gases and for radiotracer input function measurements and metabolite analyses. The body temperature was monitored by a rectal temperature probe and maintained at ~38 °C by a heating pad. After the surgical procedure has been completed, anesthesia was maintained with 0.5% isoflurane in 70% N2O/30% O2, and the animals were allowed to stabilize for 1 h before imaging procedure.
PET Scanning Protocol
PET imaging was performed according to the protocol described recently [21]. In brief, a clinical tomograph (ECAT Exact HR+; Siemens Healthcare GmbH, Erlangen, Germany) was used with animals lying prone with the head placed in a custom-made head holder. For attenuation and scatter correction, transmission scans were acquired using three rotating 68Ge rod sources. The radiotracer was injected as an intravenous (i.v.) bolus (10 mL saline with 340–494 MBq [18F]10a) by a syringe pump within 2 min followed by flushing with 10 mL heparinized saline (50 IE∙mL−1). PET scanning started at the time of injection (0 min) and dynamic emission data were acquired for a total of 240 min. Three animals were investigated under baseline conditions, and another three under blocking conditions with administration of 3 mg∙kg−1 i.v. of the α7 nAChR partial agonist NS6740 at 3 min before radiotracer injection followed by a continuous infusion throughout the scan (1 mg∙kg−1∙h−1).
Arterial blood was sampled using a peristaltic pump during the first 20 min of the scan followed by manual sampling at 25, 30, 40, 50, 60, 90, 120, 150, 180, 210, and 240 min after injection. Plasma was obtained by centrifugation (13,000 rpm, 2 min at 21 °C) and total radioactivity concentration was measured using a gamma-counter (1470 Wizard; PerkinElmer, Shelton, CT, USA) cross-calibrated to the PET scanner. In addition, arterial whole blood was sampled manually at 4, 16, 30, 60, and 120 min p.i. and plasma obtained for metabolite analyses to determine the fraction of non-metabolized [18F]10a.
Arterial Input Function
The plasma input function was corrected for the presence of radioactive metabolites by determination of the parent fraction as described previously [12]. Plasma samples were de-proteinized by addition of ice-cold acetonitrile (500 μL MeCN/100 μL plasma) followed by centrifugation (13,000 rpm, 10 min). Aliquots of the original plasma sample and of the supernatant obtained after centrifugation were counted for radioactivity, and the supernatant was analyzed by radio-HPLC following the chromatographic methods described in quality control. The parent fraction as a function of time was fitted iteratively with the Levenberg-Marquardt algorithm, including decay correction, to calculate metabolite corrected input function [71].
Quantification of PET Data
After correction for attenuation, scatter, decay and scanner-specific dead time, images were reconstructed by filtered back-projection using a Hanning-filter of 4.9 mm FWHM into 40 frames of increasing length. A summed PET image of the 240-min scan was reconstructed for each animal and used for alignment with a T1-weighed MR image of a 6-week-old farm-bred pig as described previously [72]. The time-activity curves (TACs) were calculated for the following volumes of interest (VOIs): frontal cortex, temporal cortex, parietal cortex, occipital cortex, hippocampus, striatum (defined as mean radioactivity in caudate and putamen), cerebellum, thalamus, middle cortex, ventral cortex, midbrain, pons, and colliculi. Radioactivity in all VOIs was calculated as the average of radioactivity concentration (kBq∙cm−3) in the left and right sides. To generate standardized uptake values (SUVs) the TACs of the individual VOIs were normalized to the injected dose and corrected for animal weight (in kBq∙g−1). Kinetic analysis of the time-activity curves (TACs) was performed using an “in house” data analysis tool [73].
3.7.4. PET Study in Rhesus Monkey
One male rhesus monkey (Macaca mulatta, 6 years old, body weight = 10 kg) underwent two PET imaging sessions with [18F]10a and [18F]10b, respectively. The scans were conducted 19 days apart. The monkey was initially sedated by intramuscular ketamine at 10 mg∙kg−1 at ~2 h before the PET scan and anesthetized by 1.5%–3% isoflurane throughout the imaging session. PET scan was performed using a FOCUS-220 PET scanner (Siemens Preclinical Solutions, Knoxville, TN, USA). After a transmission scan, 167 MBq of [18F]10a (specific activity of 362 GBq∙μmol−1) or 185 MBq of [18F]10b (specific activity of 159 GBq·μmol−1) were administered intravenously. Emission data were acquired in list mode for 240 min. Raw list-mode PET data was histogrammed (frames of 6 × 30 s; 3 × 1 min; 2 × 2 min; 46 × 5 min to scan termination) and reconstructed with 2D filtered back projection, using a 0.15 cm−1 Shepp filter and including corrections for scanner normalization, detector dead time, randoms, and radiation scatter and attenuation. The PET images were then registered to MR images, acquired with a Siemens 3 T Trio scanner, with a 6-parameter rigid body registration. The MR native space was then normalized using nonlinear affine registration to a high-resolution rhesus monkey atlas using BioImage Suite 3.01 (http://www.bioimagesuite.org/index.html). Time-activity curves were extracted by mapping atlas-defined regions to PET native space using the optimal transformation matrices calculated in the registration and normalization steps. Regions extracted included amygdala, caudate, cerebellum, cingulate, cortical regions (frontal, occipital, and temporal cortex), hippocampus, putamen, thalamus, and pons.
3.7.5. Toxicity Studies in Rats
The acute toxicity of 10a was assessed when administered by a single intravenous injection to rats followed by an observation period of 2 or 15 day (Hameln rds, Modra, Slovak Republic). The study was performed with 4 test groups, including 1 control and 3 dose groups (6.2, 62 and 620 μg∙kg−1), with 60 males and 60 females Wistar rats divided into two experiment cohorts: Day 2 (40 males and 40 females) and Day 15 (20 males and 20 females). The animals were weighed and allocated to the test groups based on their body weights. The animals were sacrificed in two time periods (day 2 and day 15 after test item administration) and then examined macroscopically and histopathologically. The following parameters were evaluated: mortality, clinical observation, body weights, food consumption, haematology (white blood cells, red blood cells, hemoglobin, hematocrit, mean corpuscular volume, mean corpuscular hemoglobin, platelets, lympohcytes, neutrophils, eosinophils, basophils, monocytes), clinical chemistry (alkaline phosphatase, aspartate aminotransferase, alanine aminotransferase, glucose, cholesterol, triacylglycerols, creatinine, urea, bilirubin, albumin, calcium, phosphorus, sodium, potassium chloride), pathology and histopathology.
4. Conclusions
We have designed and synthesized a number of compounds based on a new pharmacophore and tested their in vitro binding affinity and selectivity for α7 nAChR over other receptor subtypes. Among the new ligands, compound 10a was found to have high α7 nAChR binding affinity and selectivity, and was selected for radiolabeling with 18F and evaluation as PET imaging radioligand for α7 nAChR in vivo. [18F]10a was produced in high radiochemical yield and purity both manually and in an automated synthesis. PET imaging experiments in piglets and monkey demonstrated that [18F]10a possessed appropriate pharmacokinetic properties and displayed specific and displaceable binding to α7 nAChR in the brain. Taken together, the new radioligand [18F]10a ([18F]DBT-10) appears to be a promising agent for PET imaging and quantification of α7 nAChR availability in the primate brain.
Acknowledgments
The Deutsche Forschungsgemeinschaft is acknowledged for financial support (Project DE 1165/2-3). We thank the staff of the Institute of Analytical Chemistry, Department of Chemistry and Mineralogy of the University of Leipzig, for the NMR spectra, Karsten Franke, Helmholtz-Zentrum Dresden-Rossendorf (HZDR), and the cyclotron staff of the Department of Nuclear Medicine of the University Hospital Leipzig for providing [18F]fluoride, as well as Tina Spalholz, HZDR, and Juliane Schaller, HZDR, for technical assistance. Support to DanPET AB from CCJobs (Creating Competitive Jobs), FBÖ TransTechTrans and NRU, COGNITO, Copenhagen, is gratefully acknowledged.
Supplementary Materials
Supplementary materials including protocols for the preparation of intermediate compounds and NMR spectra of compounds 10, 10a, 10b, 10c, 11a, 12a, 12b, 13b and 14b can be accessed at: http://www.mdpi.com/1420-3049/20/10/18387/s1.
Author Contributions
Rodrigo Teodoro, Barbara Wenzel, Jörg Steinbach, Marianne Patt, Ming-Qiang Zheng and Yiyun Huang designed and conducted the radiochemical experiments. Matthias Scheunemann and Dan Peters conceived and performed the chemical syntheses. Francesca Maria Fasoli, Cecilia Gotti and Winnie Deuther-Conrad conducted the in vitro binding studies. Winnie Deuther-Conrad, Cornelius K. Donat, Peter Brust, Osama Sabri, Ming-Qiang Zheng, Ansel Hillmer and Yiyun Huang designed and conducted the PET imaging experiments. Rodrigo Teodoro, Barbara Wenzel, Peter Brust, Mathias Kranz, Winnie Deuther-Conrad, Cecilia Gotti, Ansel Hillmer and Yiyun Huang analyzed the data. Rodrigo Teodoro, Matthias Scheunemann, Winnie Deuther-Conrad, Peter Brust, Osama Sabri, Ansel Hillmer, Ming-Qiang Zheng and Yiyun Huang wrote and revised the manuscript.
Conflicts of Interest
The authors declare no conflict of interest.
Footnotes
Sample Availability: Samples of the compounds 10a,b and 14b are available from the authors.
References
- 1.Dineley K.T., Pandya A.A., Yakel J.L. Nicotinic ACh receptors as therapeutic targets in CNS disorders. Trends Pharmacol. Sci. 2015;36:96–108. doi: 10.1016/j.tips.2014.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Weiland S., Bertrand D., Leonard S. Neuronal nicotinic acetylcholine receptors: From the gene to the disease. Behav. Brain Res. 2000;113:43–56. doi: 10.1016/S0166-4328(00)00199-6. [DOI] [PubMed] [Google Scholar]
- 3.Briggs C.A., Gronlien J.H., Curzon P., Timmermann D.B., Ween H., Thorin-Hagene K., Kerr P., Anderson D.J., Malysz J., Dyhring T., et al. Role of channel activation in cognitive enhancement mediated by α7 nicotinic acetylcholine receptors. Br. J. Pharmacol. 2009;158:1486–1494. doi: 10.1111/j.1476-5381.2009.00426.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Papke R.L. Merging old and new perspectives on nicotinic acetylcholine receptors. Biochem. Pharmacol. 2014;89:1–11. doi: 10.1016/j.bcp.2014.01.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Palma E., Conti L., Roseti C., Limatola C. Novel approaches to study the involvement of α7-nAChR in human diseases. Curr. Drug Targets. 2012;13:579–586. doi: 10.2174/138945012800398838. [DOI] [PubMed] [Google Scholar]
- 6.Horenstein N.A., Leonik F.M., Papke R.L. Multiple pharmacophores for the selective activation of nicotinic α7-type acetylcholine receptors. Mol. Pharmacol. 2008;74:1496–1511. doi: 10.1124/mol.108.048892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mazurov A., Hauser T., Miller C.H. Selective α7 nicotinic acetylcholine receptor ligands. Curr. Med. Chem. 2006;13:1567–1584. doi: 10.2174/092986706777442011. [DOI] [PubMed] [Google Scholar]
- 8.Kim S.W., Ding Y.S., Alexoff D., Patel V., Logan J., Lin K.S., Shea C., Muench L., Xu Y., Carter P., et al. Synthesis and positron emission tomography studies of C-11-labeled isotopomers and metabolites of GTS-21, a partial α7 nicotinic cholinergic agonist drug. Nucl. Med. Biol. 2007;34:541–551. doi: 10.1016/j.nucmedbio.2007.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Toyohara J., Ishiwata K., Sakata M., Wu J., Nishiyama S., Tsukada H., Hashimoto K. In vivo evaluation of α7 nicotinic acetylcholine receptor agonists [11C]A-582941 and [11C]A-844606 in mice and conscious monkeys. PLoS ONE. 2010;5:e8961. doi: 10.1371/journal.pone.0008961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hashimoto K., Nishiyama S., Ohba H., Matsuo M., Kobashi T., Takahagi M., Iyo M., Kitashoji T., Tsukada H. [11C]CHIBA-1001 as a novel PET ligand for α7 nicotinic receptors in the brain: A PET study in conscious monkeys. PLoS ONE. 2008;3:e3231. doi: 10.1371/journal.pone.0003231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ettrup A., Mikkelsen J.D., Lehel S., Madsen J., Nielsen E.Ø., Palner M., Timmermann D.B., Peters D., Knudsen G.M. 11C-NS14492 as a novel PET radioligand for imaging cerebral α7 nicotinic acetylcholine receptors: In vivo evaluation and drug occupancy measurements. J. Nucl. Med. 2011;52:1449–1456. doi: 10.2967/jnumed.111.088815. [DOI] [PubMed] [Google Scholar]
- 12.Deuther-Conrad W., Fischer S., Hiller A., Stergaard Nielsen E., Brunicardi Timmermann D., Steinbach J., Sabri O., Peters D., Brust P. Molecular imaging of α7 nicotinic acetylcholine receptors: Design and evaluation of the potent radioligand [18F]NS10743. Eur. J. Nucl. Med. Mol. Imaging. 2009;36:791–800. doi: 10.1007/s00259-008-1031-7. [DOI] [PubMed] [Google Scholar]
- 13.Rötering S., Scheunemann M., Fischer S., Hiller A., Peters D., Deuther-Conrad W., Brust P. Radiosynthesis and first evaluation in mice of [18F]NS14490 for molecular imaging of α7 nicotinic acetylcholine receptors. Bioorg. Med. Chem. 2013;21:2635–2642. doi: 10.1016/j.bmc.2013.02.018. [DOI] [PubMed] [Google Scholar]
- 14.Briggs C.A., Schrimpf M.R., Anderson D.J., Gubbins E.J., Grønlien J.H., Håkerud M., Ween H., Thorin-Hagene K., Malysz J., Li J., et al. α7 nicotinic acetylcholine receptor agonist properties of tilorone and related tricyclic analogues. Br. J. Pharmacol. 2008;153:1054–1061. doi: 10.1038/sj.bjp.0707649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Schrimpf M.R., Sippy K.B., Briggs C.A., Anderson D.J., Li T., Ji J., Frost J.M., Surowy C.S., Bunnelle W.H., Gopalakrishnan M., et al. SAR of α7 nicotinic receptor agonists derived from tilorone: Exploration of a novel nicotinic pharmacophore. Bioorg. Med. Chem. Lett. 2012;22:1633–1638. doi: 10.1016/j.bmcl.2011.12.126. [DOI] [PubMed] [Google Scholar]
- 16.Teodoro R., Deuther-Conrad W., Rötering S., Scheunemann M., Patt M., Donat C.K., Wenzel B., Peters D., Sabri O., Brust P. Comparative evaluation of two novel fluorine-18 PET radiotracers for the α7 nicotinic acetylcholine receptor. J. Nucl. Med. 2014;55:1099. [Google Scholar]
- 17.Gao Y., Kellar K.J., Yasuda R.P., Tran T., Xiao Y., Dannals R.F., Horti A.G. Derivatives of dibenzothiophene for positron emission tomography imaging of α7-nicotinic acetylcholine receptors. J. Med. Chem. 2013;56:7574–7589. doi: 10.1021/jm401184f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Horti A.G., Gao Y., Kuwabara H., Wang Y., Abazyan S., Yasuda R.P., Tran T., Xiao Y., Sahibzada N., Holt D.P., et al. 18F-ASEM, a radiolabeled antagonist for imaging the α7-nicotinic acetylcholine receptor with PET. J. Nucl. Med. 2014;55:672–677. doi: 10.2967/jnumed.113.132068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Horti A.G. Development of [18F]ASEM, a specific radiotracer for quantification of the α7-nAChR with positron-emission tomography. Biochem. Pharmacol. 2015 doi: 10.1016/j.bcp.2015.07.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wong D.F., Kuwabara H., Pomper M., Holt D.P., Brasic J.R., George N., Frolov B., Willis W., Gao Y., Valentine H., et al. Human brain imaging of α7 nAChR with [18F]ASEM: A new PET radiotracer for neuropsychiatry and determination of drug occupancy. Mol. Imaging Biol. 2014;16:730–738. doi: 10.1007/s11307-014-0779-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Deuther-Conrad W., Fischer S., Hiller A., Becker G., Cumming P., Xiong G., Funke U., Sabri O., Peters D., Brust P. Assessment of α7 nicotinic acetylcholine receptor availability in juvenile pig brain with [18F]NS10743. European J. Nucl. Med. Mol. Imaging. 2011;38:1541–1549. doi: 10.1007/s00259-011-1808-y. [DOI] [PubMed] [Google Scholar]
- 22.Toma L., Quadrelli P., Bunnelle W.H., Anderson D.J., Meyer M.D., Cignarella G., Gelain A., Barlocco D. 6-Chloropyridazin-3-yl derivatives active as nicotinic agents: Synthesis, binding, and modeling studies. J. Med. Chem. 2002;45:4011–4017. doi: 10.1021/jm0208830. [DOI] [PubMed] [Google Scholar]
- 23.Eibl C., Tomassoli I., Munoz L., Stokes C., Papke R.L., Gündisch D. The 3,7-diazabicyclo[3.3.1]nonane scaffold for subtype selective nicotinic acetylcholine receptor (nAChR) ligands. Part 1: The influence of different hydrogen bond acceptor systems on alkyl and (hetero)aryl substituents. Bioorg. Med. Chem. 2013;21:7283–7308. doi: 10.1016/j.bmc.2013.09.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Papke R.L., Schiff H.C., Jack B.A., Horenstein N.A. Molecular dissection of tropisetron, an α7 nicotinic acetylcholine receptor-selective partial agonist. Neurosci. Lett. 2005;378:140–144. doi: 10.1016/j.neulet.2004.12.025. [DOI] [PubMed] [Google Scholar]
- 25.Lehel S., Madsen J., Ettrup A., Mikkelsen J.D., Timmermann D.B., Peters D., Knudsen G.M. [11C]NS-12857: A novel PET ligand for α7-nicotinergic receptors. J. Labelled Compd. Rad. 2009;52:S379. [Google Scholar]
- 26.O’Donnell C.J., Peng L., O’Neill B.T., Arnold E.P., Mather R.J., Sands S.B., Shrikhande A., Lebel L.A., Spracklin D.K., Nedza F.M. Synthesis and SAR studies of 1,4-diazabicyclo[3.2.2]nonane phenyl carbamates—Subtype selective, high affinity α7 nicotinic acetylcholine receptor agonists. Bioorg. Med. Chem. Lett. 2009;19:4747–4751. doi: 10.1016/j.bmcl.2009.06.059. [DOI] [PubMed] [Google Scholar]
- 27.Slowinski F., Ben Ayad O., Vache J., Saady M., Leclerc O., Lochead A. Synthesis of bridgehead-substituted azabicyclo[2.2.1]heptane and -[3.3.1]nonane derivatives for the elaboration of α7 nicotinic ligands. J. Org. Chem. 2011;76:8336–8346. doi: 10.1021/jo201501f. [DOI] [PubMed] [Google Scholar]
- 28.Wawzonek S., Thelen P.J. Preparation of N-methylgranatanine. J. Am. Chem. Soc. 1950;72:2118–2120. doi: 10.1021/ja01161a068. [DOI] [Google Scholar]
- 29.Lambert F.L., Ellis W.D., Parry R.J. Halogenation of aromatic compounds by N-bromo- and N-chlorosuccinimide under ionic conditions. J. Org. Chem. 1965;30:304–306. doi: 10.1021/jo01012a512. [DOI] [Google Scholar]
- 30.Butts C.P., Eberson L., Hartshorn M.P., Radner F., Robinson W.T., Wood B.R. Regiochemistry of the reaction between dibenzothiophene radical cation and nucleophiles or nitrogen dioxide. Acta Chem. Scand. 1997;51:839–848. doi: 10.3891/acta.chem.scand.51-0839. [DOI] [Google Scholar]
- 31.Gilman H., Nobis J.F. Some aminodibenzothiophenes. J. Am. Chem. Soc. 1949;71:274–276. doi: 10.1021/ja01169a070. [DOI] [Google Scholar]
- 32.Cullinane N.M., Davies C.G., Davies G.I. Substitution derivatives of diphenylene sulphide and diphenylenesulphone. J. Chem. Soc. 1936:1435–1437. doi: 10.1039/JR9360001435. [DOI] [Google Scholar]
- 33.Adams D.J., Clark J.H. Nucleophilic routes to selectively fluorinated aromatics. Chem. Soc. Rev. 1999;28:225–231. doi: 10.1039/a808707e. [DOI] [Google Scholar]
- 34.Boechat N., Clark J.H. Fluorodenitrations using tetramethylammonium fluoride. J. Chem. Soc. Chem. Commun. 1993;11:921–922. doi: 10.1039/c39930000921. [DOI] [Google Scholar]
- 35.Manna S., Maity S., Rana S., Agasti S., Maiti D. Ipso-nitration of arylboronic acids with bismuth nitrate and perdisulfate. Org. Lett. 2012;14:1736–1739. doi: 10.1021/ol300325t. [DOI] [PubMed] [Google Scholar]
- 36.Molander G.A., Cavalcanti L.N. Nitrosation of aryl and heteroaryltrifluoroborates with nitrosonium tetrafluoroborate. J. Org. Chem. 2012;77:4402–4413. doi: 10.1021/jo300551m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wu X.F., Schranck J., Neumann H., Beller M. Convenient and mild synthesis of nitroarenes by metal-free nitration of arylboronic acids. Chem. Commun. 2011;47:12462–12463. doi: 10.1039/c1cc15484b. [DOI] [PubMed] [Google Scholar]
- 38.Xiao Y., Hammond P.S., Mazurov A.A., Yohannes D. Multiple interaction regions in the orthosteric ligand binding domain of the α7 neuronal nicotinic acetylcholine receptor. J. Chem. Inf. Model. 2012;52:3064–3073. doi: 10.1021/ci3001953. [DOI] [PubMed] [Google Scholar]
- 39.Brunzell D.H., McIntosh J.M., Papke R.L. Diverse strategies targeting α7 homomeric and α6β2* heteromeric nicotinic acetylcholine receptors for smoking cessation. Ann. N. Y. Acad. Sci. 2006;1327:27–45. doi: 10.1111/nyas.12421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Baddick C.G., Marks M.J. An autoradiographic survey of mouse brain nicotinic acetylcholine receptors defined by null mutants. Biochem. Pharmacol. 2011;82:828–841. doi: 10.1016/j.bcp.2011.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gotti C., Guiducci S., Tedesco V., Corbioli S., Zanetti L., Moretti M., Zanardi A., Rimondini R., Mugnaini M., Clementi F., et al. Nicotinic acetylcholine receptors in the mesolimbic pathway: Primary role of ventral tegmental area α6β2* receptors in mediating systemic nicotine effects on dopamine release, locomotion, and reinforcement. J. Neurosci. 2010;30:5311–5325. doi: 10.1523/JNEUROSCI.5095-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Quik M., Polonskaya Y., Gillespie A., Jakowec M., Lloyd G.K., Langston J.W. Localization of nicotinic receptor subunit mRNAs in monkey brain by in situ hybridization. J. Comp. Neurol. 2000;425:58–69. doi: 10.1002/1096-9861(20000911)425:1<58::AID-CNE6>3.0.CO;2-X. [DOI] [PubMed] [Google Scholar]
- 43.Han Z.Y., Le Novere N., Zoli M., Hill J.A., Jr., Champtiaux N., Changeux J.P. Localization of nAChR subunit mRNAs in the brain of Macaca mulatta. European J. Neurosci. 2000;12:3664–3674. doi: 10.1046/j.1460-9568.2000.00262.x. [DOI] [PubMed] [Google Scholar]
- 44.Exley R., Maubourguet N., David V., Eddine R., Evrard A., Pons S., Marti F., Threlfell S., Cazala P., McIntosh J.M., et al. Distinct contributions of nicotinic acetylcholine receptor subunit α4 and subunit α6 to the reinforcing effects of nicotine. Proc. Natl. Acad. Sci. USA. 2011;108:7577–7582. doi: 10.1073/pnas.1103000108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Faure P., Tolu S., Valverde S., Naudé J. Role of nicotinic acetylcholine receptors in regulating dopamine neuron activity. Neuroscience. 2014;282:86–100. doi: 10.1016/j.neuroscience.2014.05.040. [DOI] [PubMed] [Google Scholar]
- 46.Gotti C., Marks M.J., Millar N.S., Wonnacott S. Nicotinic acetylcholine receptors: α6. [(accessed on 2 October 2015)]. Available online: http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId = 467&familyId=76&familyType=IC.
- 47.Le Novere N., Grutter T., Changeux J.P. Models of the extracellular domain of the nicotinic receptors and of agonist- and Ca2+-binding sites. Proc. Natl. Acad. Sci. USA. 2002;99:3210–3215. doi: 10.1073/pnas.042699699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ding M., Ghanekar S., Elmore C.S., Zysk J.R., Werkheiser J.L., Lee C.M., Liu J., Chhajlani V., Maier D.L. [3H]Chiba-1001(methyl-SSR180711) has low in vitro binding affinity and poor in vivo selectivity to nicotinic alpha-7 receptor in rodent brain. Synapse. 2012;66:315–322. doi: 10.1002/syn.21513. [DOI] [PubMed] [Google Scholar]
- 49.Mazurov A.A., Kombo D.C., Akireddy S., Murthy S., Hauser T.A., Jordan K.G., Gatto G.J., Yohannes D. Novel nicotinic acetylcholine receptor agonists containing carbonyl moiety as a hydrogen bond acceptor. Bioorg. Med. Chem. Lett. 2013;23:3927–3934. doi: 10.1016/j.bmcl.2013.04.058. [DOI] [PubMed] [Google Scholar]
- 50.Girard B.M., Merriam L.A., Tompkins J.D., Vizzard M.A., Parsons R.L. Decrease in neuronal nicotinic acetylcholine receptor subunit and PSD-93 transcript levels in the male mouse MPG after cavernous nerve injury or explant culture. Am. J. Physiol. 2013;305:F1504–F1512. doi: 10.1152/ajprenal.00343.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Glushakov A.V., Voytenko L.P., Skok M.V., Skok V. Distribution of neuronal nicotinic acetylcholine receptors containing different alpha-subunits in the submucosal plexus of the guinea-pig. Auton. Neurosci. Basic Clin. 2004;110:19–26. doi: 10.1016/j.autneu.2003.08.012. [DOI] [PubMed] [Google Scholar]
- 52.Teodoro R., Wenzel B., Oh-Nishi A., Fischer S., Peters D., Suhara T., Deuther-Conrad W., Brust P. A high-yield automated radiosynthesis of the alpha-7 nicotinic receptor radioligand [18F]NS10743. Appl. Radiat. Isot. 2015;95:76–84. doi: 10.1016/j.apradiso.2014.09.016. [DOI] [PubMed] [Google Scholar]
- 53.Jacobson O., Chen X. PET designated flouride-18 production and chemistry. Curr. Top. Med. Chem. 2010;10:1048–1059. doi: 10.2174/156802610791384298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ravert H.T., Holt D.P., Gao Y., Horti A.G., Dannals R.F. Microwave-assisted radiosynthesis of [18F]ASEM, a radiolabeled α7-nicotinic acetylcholine receptor antagonist. J. Labelled Compd. Radiopharm. 2015;58:180–182. doi: 10.1002/jlcr.3275. [DOI] [PubMed] [Google Scholar]
- 55.Egleton R.D., Brown K.C., Dasgupta P. Angiogenic activity of nicotinic acetylcholine receptors: Implications in tobacco-related vascular diseases. Pharmacol. Ther. 2009;121:205–223. doi: 10.1016/j.pharmthera.2008.10.007. [DOI] [PubMed] [Google Scholar]
- 56.Cooke J.P., Bitterman H. Nicotine and angiogenesis: A new paradigm for tobacco-related diseases. Ann. Med. 2004;36:33–40. doi: 10.1080/07853890310017576. [DOI] [PubMed] [Google Scholar]
- 57.Pena V.B., Bonini I.C., Antollini S.S., Kobayashi T., Barrantes F.J. α7-Type acetylcholine receptor localization and its modulation by nicotine and cholesterol in vascular endothelial cells. J. Cell. Biochem. 2011;112:3276–3288. doi: 10.1002/jcb.23254. [DOI] [PubMed] [Google Scholar]
- 58.Innis R.B., Cunningham V.J., Delforge J., Fujita M., Gjedde A., Gunn R.N., Holden J., Houle S., Huang S.C., Ichise M., et al. Consensus nomenclature for in vivo imaging of reversibly binding radioligands. J. Cereb. Blood Flow Metab. 2007;27:1533–1539. doi: 10.1038/sj.jcbfm.9600493. [DOI] [PubMed] [Google Scholar]
- 59.Cunningham V.J., Rabiner E.A., Slifstein M., Laruelle M., Gunn R.N. Measuring drug occupancy in the absence of a reference region: The Lassen plot re-visited. J. Cereb. Blood Flow Metab. 2010;30:46–50. doi: 10.1038/jcbfm.2009.190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Brust P., Peters D., Deuther-Conrad W. Development of radioligands for the imaging of α7 nicotinic acetylcholine receptors with positron emission tomography. Curr. Drug Targets. 2012;13:594–601. doi: 10.2174/138945012800398955. [DOI] [PubMed] [Google Scholar]
- 61.Harwood L.M. “Dry-column” flash chromatography. Aldrichimica Acta. 1985;18:25. [Google Scholar]
- 62.Schrimpf M., Sippy K., Ji J., Li T., Frost J., Briggs C., Bunnelle W. Amino-Substituted Tricyclic Derivatives and Methods of Use. 20,050,234,031 A1. U.S. Patent. 2005 Oct 20;
- 63.Rötering S., Franke K., Zessin J., Brust P., Füchtner F., Fischer S., Steinbach J. Convenient recycling and reuse of bombarded [18O]H2O for the production and the application of [18F]F. Appl. Radiat. Isot. 2015;101:44–52. doi: 10.1016/j.apradiso.2015.03.009. [DOI] [PubMed] [Google Scholar]
- 64.Charpantier E., Wiesner A., Huh K.H., Ogier R., Hoda J.C., Allaman G., Raggenbass M., Feuerbach D., Bertrand D., Fuhrer C. α7 neuronal nicotinic acetylcholine receptors are negatively regulated by tyrosine phosphorylation and Src-family kinases. J. Neurosci. 2005;25:9836–9849. doi: 10.1523/JNEUROSCI.3497-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Cheng Y., Prusoff W.H. Relationship between the inhibition constant (K1) and the concentration of inhibitor which causes 50 per cent inhibition (IC50) of an enzymatic reaction. Biochem. Pharmacol. 1973;22:3099–3108. doi: 10.1016/0006-2952(73)90196-2. [DOI] [PubMed] [Google Scholar]
- 66.Michelmore S., Croskery K., Nozulak J., Hoyer D., Longato R., Weber A., Bouhelal R., Feuerbach D. Study of the calcium dynamics of the human α4β2, α3β4 and α1β1γδ nicotinic acetylcholine receptors. Naunyn-Schmiedeberg’s Arch. Pharmacol. 2002;366:235–245. doi: 10.1007/s00210-002-0589-z. [DOI] [PubMed] [Google Scholar]
- 67.Grady S.R., Moretti M., Zoli M., Marks M.J., Zanardi A., Pucci L., Clementi F., Gotti C. Rodent habenulo-interpeduncular pathway expresses a large variety of uncommon nAChR subtypes, but only the αβ4* and αβ3β4* subtypes mediate acetylcholine release. J. Neurosci. 2009;29:2272–2282. doi: 10.1523/JNEUROSCI.5121-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Pucci L., Grazioso G., Dallanoce C., Rizzi L., De Micheli C., Clementi F., Bertrand S., Bertrand D., Longhi R., De Amici M., et al. Engineering of α-conotoxin MII-derived peptides with increased selectivity for native α6β2* nicotinic acetylcholine receptors. FASEB J. 2011;25:3775–3789. doi: 10.1096/fj.10-179853. [DOI] [PubMed] [Google Scholar]
- 69.Munson P.J., Rodbard D. Ligand: A versatile computerized approach for characterization of ligand-binding systems. Anal. Biochem. 1980;107:220–239. doi: 10.1016/0003-2697(80)90515-1. [DOI] [PubMed] [Google Scholar]
- 70.Brust P., Patt J.T., Deuther-Conrad W., Becker G., Patt M., Schildan A., Sorger D., Kendziorra K., Meyer P., Steinbach J., et al. In vivo measurement of nicotinic acetylcholine receptors with [18F]norchloro-fluoro-homoepibatidine. Synapse. 2008;62:205–218. doi: 10.1002/syn.20480. [DOI] [PubMed] [Google Scholar]
- 71.Gillings N.M., Bender D., Falborg L., Marthi K., Munk O.L., Cumming P. Kinetics of the metabolism of four PET radioligands in living minipigs. Nucl. Med. Biol. 2001;28:97–104. doi: 10.1016/S0969-8051(00)00187-6. [DOI] [PubMed] [Google Scholar]
- 72.Brust P., Zessin J., Kuwabara H., Pawelke B., Kretzschmar M., Hinz R., Bergman J., Eskola O., Solin O., Steinbach J., et al. Positron emission tomography imaging of the serotonin transporter in the pig brain using [11C](+)-McN5652 and S-[18F]fluoromethyl-(+)-McN5652. Synapse. 2003;47:143–151. doi: 10.1002/syn.10163. [DOI] [PubMed] [Google Scholar]
- 73.Brust P., Hinz R., Kuwabara H., Hesse S., Zessin J., Pawelke B., Stephan H., Bergmann R., Steinbach J., Sabri O. In vivo measurement of the serotonin transporter with (S)-([18F]fluoromethyl)-(+)-McN5652. Neuropsychopharmacology. 2003;28:2010–2019. doi: 10.1038/sj.npp.1300281. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.