

Abstract
Key points
The pancreatic islets of Langerhans maintain glucose homeostasis through insulin secretion, where insulin secretion dynamics are regulated by intracellular Ca2+ signalling and electrical coupling of the insulin producing β‐cells in the islet.
We have previously shown that cytokines decrease β‐cell coupling and that compounds which increase cAMP can increase coupling.
In both mouse and human islets exendin‐4, which increases cAMP, protected against cytokine‐induced decreases in coupling and in mouse islets preserved glucose‐stimulated calcium signalling by increasing connexin36 gap junction levels on the plasma membrane.
Our data indicate that protein kinase A regulates β‐cell coupling through a fast mechanism, such as channel gating or membrane organization, while Epac2 regulates slower mechanisms of regulation, such as gap junction turnover.
Increases in β‐cell coupling with exendin‐4 may protect against cytokine‐mediated β‐cell death as well as preserve insulin secretion dynamics during the development of diabetes.
Abstract
The pancreatic islets of Langerhans maintain glucose homeostasis. Insulin secretion from islet β‐cells is driven by glucose metabolism, depolarization of the cell membrane and an influx of calcium, which initiates the release of insulin. Gap junctions composed of connexin36 (Cx36) electrically couple β‐cells, regulating calcium signalling and insulin secretion dynamics. Cx36 coupling is decreased in pre‐diabetic mice, suggesting a role for altered coupling in diabetes. Our previous work has shown that pro‐inflammatory cytokines decrease Cx36 coupling and that compounds which increase cAMP can increase Cx36 coupling. The goal of this study was to determine if exendin‐4, which increases cAMP, can protect against cytokine‐induced decreases in Cx36 coupling and altered islet function. In both mouse and human islets, exendin‐4 protected against cytokine‐induced decreases in coupling and preserved glucose‐stimulated calcium signalling. Exendin‐4 also protected against protein kinase Cδ‐mediated decreases in Cx36 coupling. Exendin‐4 preserved coupling in mouse islets by preserving Cx36 levels on the plasma membrane. Exendin‐4 regulated Cx36 coupling via both protein kinase A (PKA)‐ and Epac2‐mediated mechanisms in cytokine‐treated islets. In mouse islets, modulating Epac2 had a greater impact in mediating Cx36 coupling, while in human islets modulating PKA had a greater impact on Cx36 coupling. Our data indicate that PKA regulates Cx36 coupling through a fast mechanism, such as channel gating, while Epac2 regulates slower mechanisms of regulation, such as Cx36 turnover in the membrane. Increases in Cx36 coupling with exendin‐4 may protect against cytokine‐mediated β‐cell dysfunction to insulin secretion dynamics during the development of diabetes.
Keywords: Islet of Langerhans, Exendin‐4, Connexin36 Gap Junctions
Key points
The pancreatic islets of Langerhans maintain glucose homeostasis through insulin secretion, where insulin secretion dynamics are regulated by intracellular Ca2+ signalling and electrical coupling of the insulin producing β‐cells in the islet.
We have previously shown that cytokines decrease β‐cell coupling and that compounds which increase cAMP can increase coupling.
In both mouse and human islets exendin‐4, which increases cAMP, protected against cytokine‐induced decreases in coupling and in mouse islets preserved glucose‐stimulated calcium signalling by increasing connexin36 gap junction levels on the plasma membrane.
Our data indicate that protein kinase A regulates β‐cell coupling through a fast mechanism, such as channel gating or membrane organization, while Epac2 regulates slower mechanisms of regulation, such as gap junction turnover.
Increases in β‐cell coupling with exendin‐4 may protect against cytokine‐mediated β‐cell death as well as preserve insulin secretion dynamics during the development of diabetes.
Introduction
Blood glucose homeostasis is maintained by insulin secretion from the pancreatic islets of Langerhans. Insulin secretion from β‐cells in the islet is driven by glucose metabolism, ATP generation, membrane depolarization via closure of ATP‐sensitive potassium channels (KATP), calcium influx (Ca2+) via voltage‐gated Ca2+ channels, and exocytosis of insulin secretory granules (Rutter et al. 2015). Electrical coupling between β‐cells plays a critical role in regulating islet function and insulin secretion (Meda, 2012; Farnsworth & Benninger, 2014). Gap junctions, composed of connexin 36 (Cx36) in β‐cells, provide both physical and electrical coupling, allowing selective passage of cations, such as Ca2+ and K+, coordinating changes in membrane potential across the islet (Perez‐Armendariz et al. 1991; Benninger et al. 2008). Under basal glucose levels, Cx36 gap junction coupling coordinates membrane polarization, silencing electrically active cells at non‐stimulatory glucose concentrations (Rocheleau et al. 2006; Speier et al. 2007; Benninger et al. 2011). Under stimulatory glucose levels, Cx36 gap junctions coordinate glucose‐induced membrane depolarization and Ca2+ signalling, thereby regulating the dynamics of insulin secretion from the islet (Ravier et al. 2005; Benninger et al. 2008; Head et al. 2012).
Diabetes is characterized by the progressive loss of functional pancreatic islet mass resulting in a loss of blood glucose homeostasis (Robertson, 2009). Pro‐inflammatory cytokines, such as tumour necrosis factor α (TNF‐α), interleukin‐1β (IL‐1β) and interferon γ (IFN‐γ), contribute to the destruction and decline in β‐cell function during the development of both type 1 and type 2 diabetes (Verrilli et al. 2011; Imai et al. 2013; Padgett et al. 2013). In vitro, pro‐inflammatory cytokines impair Ca2+ signalling (Ramadan et al. 2011), disrupt Cx36 gap junction coupling (Farnsworth et al. 2015), decrease insulin secretion (Eizirik et al. 1994; Andersson et al. 2001) and initiate β‐cell apoptosis (Grunnet et al. 2009). Previous studies have reported decreased Cx36 gap junction coupling in islets from pre‐diabetic mice (Carvalho et al. 2012) and decreased coupling with sustained hyperglycaemia (Haefliger et al. 2013). Together, this suggests that Cx36 coupling may be altered during the onset of diabetes. Cx36 gap junction coupling has also been shown to protect against pro‐inflammatory cytokine‐induced β‐cell apoptosis (Klee et al. 2011; Allagnat et al. 2013). However, little is known about the mechanisms regulating Cx36 coupling in the islet (Farnsworth & Benninger, 2014).
Previously, we have shown that increasing intracellular cAMP using 3‐isobutyl‐1‐methylxanthine (IBMX) increases Cx36 gap junction coupling in mouse islets (Farnsworth et al. 2014). Cx36 expression has been linked to changes in islet Ca2+ responses to increases in cAMP via glucagon‐like peptide‐1 (GLP‐1) in human islets (Hodson et al. 2013); however, modulation of Cx36 gap junction coupling by GLP‐1 in healthy and pathogenic conditions has not been studied. Therefore, the goal of this study was to determine if GLP‐1 receptor (GLP‐1R) activation can prevent decreases in Cx36 gap junction coupling and altered Ca2+ signalling in cytokine‐treated islets. Exendin‐4, a GLP‐1R agonist which regulates cAMP, has been successfully used to improve glycaemic control in patients with type 2 diabetes (DeFronzo et al. 2005). In the retina, cAMP has been shown to regulate Cx36 gap junction coupling via activation of protein kinase A (PKA; Urschel et al. 2006), while in neuronal cells Epac2 has been shown to mediate cAMP regulation of Cx36 coupling (Li et al. 2012a). In addition to investigating exendin‐4 and cAMP protection against cytokine‐induced islet dysfunction, this study also determined the respective roles of PKA and Epac2 in regulating Cx36 gap junction coupling.
Methods
Ethical approval
The authors have read and understood the policies and regulations of The Journal of Physiology as outlined by Grundy (2015) and have ensured that all experiments comply with these regulations. In compliance with federal regulation, the Institutional Animal Care and Use Committee at the University of Colorado Anschutz Medical Campus has approved the procedures and experiments outline in this study (No. B‐95817(05)1D).
Animal care
Mice were given food and water ad libitum and housed in 12 h light–dark cycles in a temperature‐controlled facility.
Islet isolation, culture and treatments
Pancreata were isolated from C57BL/6NHsd mice (The Jackson Laboratory, Bar Harbour, ME, USA) aged 8–16 weeks. Mice were first anaesthetized by intra‐peritoneal injection of 80 mg kg−1 ketamine and 20 mg kg−1 xylazine and the pancreas was isolated immediately following euthanasia by exsanguination. Islets were isolated by enzymatic digestion of the isolated pancreas as previously reported (Koster et al. 2002). Islets were incubated overnight in 1640 RPMI Medium (Sigma‐aldrich, St Louis, MO, USA) with 10% fetal bovine serum, 10,000 U mL−1 penicillin and 10,000 μg mL−1 streptomycin at 37 °C with 5% CO2.
Human islets were received from the Integrated Islet Distribution Program (Donor IDs: AELM182, AEI1395, AEHI491, AEHW247, AEDG094, AEAN289, ADE1444, ADDQ143, ADBW449B, ADBB290, ADAU462B) with a mean viability of 93% and were cultured overnight in CMRL medium 1066 (Thermo Fisher Scientific, Waltham, MA, USA).
Islets were incubated in either RPMI medium for 24 h treatments or Hanks’ balanced salt solution (HBSS; Thermo Fisher Scientific) for 1 h treatments. Islets were treated with a cytokine cocktail mixture containing 1 ng mL−1 of recombinant TNF‐α (R&D Systems, Minneapolis, MN, USA), 0.5 ng mL−1 of recombinant IL‐1β (R&D Systems) and 10 ng mL−1 of recombinant IFN‐γ (R&D Systems). Mouse and human recombinant cytokines were used with mouse and human islets, respectively. Islets were also treated with 10 nM of exendin‐4 (E4,Tocris Bioscience, Minneapolis, MN, USA), 100 μM of IBMX, 300 μM of the Epac2 activator 8‐(4‐chlorophenylthio)‐2′‐O‐methyladenosine 3′,5′‐cyclic monophosphate monosodium hydrate (8‐Me, Sigma‐Aldrich), 10 μM of the Epac2 inhibitor ESI‐05 (ESI; Sigma‐Aldrich), 300 μM of the PKA activator N 6‐benzoyladenosine‐3′,5′‐cyclic monophosphate sodium salt (6‐Bnz; Tocris), 100 μM of the PKA inhibitor Rp‐adenosine 3′,5′‐cyclic monophosphorothioate triethylammonium salt (Rp‐cAMP; Sigma‐Aldrich), 300 nM of the protein kinase C δ (PKCδ) activator phorbol 12‐myristate 13‐acetate (PMA; Sigma‐Aldrich), or 5μM of the nitric oxide (NO) donor molecule S‐nitroso‐N‐acetyl‐d,l‐penicillamine (SNAP; Sigma‐Aldrich) alone or in combination with cytokines. The selection of activators and inhibitors of PKA was based on previous studies in mouse islets which found 100 μM Rp‐cAMP and 300 μM 6‐Bnz to selectively inhibit and activate PKA, respectively (Benninger et al. 2011). The selection of activators and inhibitors of Epac2 was based on previous studies in mouse islets which found 10 μM ESI and 300 μM 8‐Me to selectively inhibit and activate Epac2 respectively (Benninger et al. 2011; Henquin & Nenquin, 2014).
Fluorescence recovery after photobleaching
Islets were cultured on MatTek dishes (MatTek Corp., Ashland, MA, USA) coated with CellTak (BD Biosciences, San Jose, CA, USA) overnight before 1 h or 24 h treatments. Islets were stained with 12.5 μM rhodamine 123 (R123; Sigma‐Aldrich) for 30 min before imaging and islets were imaged as previously described (Farnsworth et al. 2014). Briefly, half of each islet was photobleached for 45 s, and the recovery of fluorescence in the bleached area was measured. Recovery rates were calculated from the inverse exponential fluorescence recovery curve (I(t) = (I ∞ − I p) (1 − e− kt) + I p) for the entire bleached area. Videos of fluorescence recovery for the representative islets are available as Supporting information. Islets were imaged on a Zeiss LSM 510 Meta or a Zeiss LSM 800 confocal microscope with a 40 × 1.0 NA water immersion objective (Zeiss, Thornwood, NY, USA). R123 was excited with a 488 nm Ar+ laser with half of the islet area photobleached with the fluorescence recovery measured in the bleached area.
Intracellular Ca2+ imaging and analysis
After 24 h treatments as indicated, islets were incubated with 4 μM Fluo‐4 AM (Thermo Fisher Scientific) in imaging buffer containing 125 mM NaCl, 5.7 mM KCl, 2.5 mM CaCl2, 1.2 mM MgCl2, 10 mM HEPES, 0.1% bovine serum albumin (BSA) and 2 mM glucose, protected from light for 2 h at room temperature. Islets were imaged at 37°C on a Zeiss 800 LSM confocal microscope with a ×40, 1.0 NA water objective. Images were taken every second for 2.5 min prior to and after stimulation with 11 mM glucose for 10 min. The fraction of the islet area which responded to glucose stimulation with pulsatile increases in intracellular Ca2+ (active area), the coordination of the active area, and the oscillation frequency were determined using MATLAB (The MathWorks, Natick, MA, USA) as previously described (Hraha et al. 2014).
Insulin secretion analysis
After 24 h treatments as indicated, islets were incubated in Krebs–Ringer buffer with 0.1% BSA and 2 mM glucose for 1 h at 37 °C. Samples were collected by incubating five islet equivalents in 500 μl Krebs buffer at either 2 or 20 mM glucose for 1 h. Supernatant and islet lysate were collected for analysis of insulin secretion and content respectively. Islets were lysed by freezing in 2% Triton X‐100 in deionized water. Insulin was measured with a mouse ultrasensitive insulin enzyme‐linked immunosorbent assay kit (ALPCO, Salem, NH, USA) as per the manufacturer's instructions, and is reported as secretion at 20 mM glucose normalized to 2 mM glucose (stimulation index).
Intracellular cAMP imaging and analysis
Freshly isolated islets were transduced with an adenovirus encoding a Förster resonance energy transfer (FRET)‐based sensor for cAMP levels, ICUE3 (Addgene, plasmid#61623, Watertown, MA, USA), as previously described (DiPilato & Zhang, 2009). Islets were incubated with virus for 24 h in serum‐free RPMI medium with or without cytokines. Islets were imaged at 37°C in imaging buffer (125 mM NaCl, 5.7 mM KCl, 2.5 mM CaCl2, 1.2 mM MgCl2, 10 mM HEPES, pH 7.4) with 11 mM glucose and 0.1% BSA on an Eclipse‐Ti widefield microscope (Nikon, Melville, NY, USA) and a ×20, 0.75 NA Plan Apo objective. Cyan fluorescent protein (CFP) was excited at 430/24 nm and images were collected with filters for CFP (470/24 nm), yellow fluorescent protein (YFP; 530/24 nm) fluorescence. Images were acquired every 15 s continuously for 30 min. After 5 min of imaging, 10 nM E4 was added to the imaging solution of both control and cytokine‐treated islets. After 35 min of imaging, 100 μM IBMX and 50 μM forskolin were added to maximally increase intracellular cAMP. As the FRET ratio of YFP/CFP for ICUE3 decreases with increasing cAMP levels, we used the inverse ratio of CFP normalized to YFP to analyse FRET. FRET ratios were corrected for photobleaching by normalizing to a linear fit of the data and normalized to the initial fluorescence intensity for each islet. Exendin‐4‐induced changes in cAMP levels were calculated as a percentage increase from baseline (0–5 min) after 25 min of E4 treatment. Forskolin‐ and IBMX‐induced increases in cAMP levels were calculated as percentage increase from baseline (35 min) after 1–2 min of forskolin+IBMX treatment.
Immunofluorescence and image analysis
After 24 h or 1 h treatment, as described above, islets (50–75 per treatment) were fixed on ice with 4% paraformaldehyde for 10 min. Islets were cryosectioned (20 μm thick) and slices were blocked in 5% normal donkey serum (NDS) in phosphate‐buffered saline (PBS) for 30–60 min and then incubated with rabbit‐anti‐connexin36 primary antibody (NBP1‐59254, Novus Biologicals, Littleton, CO, USA) diluted 1:500 and goat‐anti‐glut2 primary antibody (Ab111117, Abcam, Cambridge, MA, USA) diluted 1:200 or mouse‐anti‐neural cell adhesion molecule (NCAM) primary antibody (3576S, Cell Signaling Technology, Danvers, MA, USA) diluted 1:100. After three washes with PBS, slices were incubated with AlexaFluor 488 donkey anti‐rabbit secondary antibody (711‐545‐152, Jackson ImmunoResearch Laboratories, West Grove, PA, USA) diluted 1:1000 and AlexaFluor 568 donkey anti‐goat secondary antibody (ab175474, Abcam) diluted 1:100 or AlexaFluor 647 donkey anti‐mouse secondary antibody (715‐605‐150, Jackson ImmunoResearch) diluted 1:1000. All primary and secondary antibodies were diluted in 5% NDS in PBS. Slides were incubated with primary and secondary antibodies either overnight at 4°C or for at least 2 h at room temperature. After three washes with PBS, slides were mounted with Fluoromount containing 4′,6‐diamidino‐2‐phenylindole (DAPI; Southern Biotech, Birmingham, AL, USA).
Images of Cx36, NCAM and DAPI were collected on either a Zeiss LSM 510 META or a Zeiss LSM 800 confocal microscope with a ×63, 1.4 NA oil immersion objective, obtaining six to eight images at increasing depths per islet at 1 μm thickness. Image analysis was done using ImageJ to quantify the number of nuclei per islet slice by manual counting and a MATLAB program to identify and quantify the number of Cx36 gap junction plaques and total area of Cx36 staining, as previously reported (Farnsworth et al. 2015). Contiguous regions of Cx36 positive staining were automatically identified and plaques that did not colocalize with membrane NCAM staining were removed, ensuring only Cx36 on the cell surface was included in further analysis.
Statistical analysis
All data represents the averages of all experiments for each treatment and error bars represent the standard error of the mean (SEM). Sets of treatments were analysed with analysis of variance (ANOVA) with or without repeated measures and Tukey's post hoc analysis with α = 0.05, paired sample Student's t‐test with α = 0.05, or 95% confidence intervals as indicated. P‐values are presented where a significant difference was determined. Differences which were not significant are noted as n.s.
Results
Exendin‐4 overcomes cytokine‐induced decreases in Cx36 gap junction coupling and Ca2+ coordination
As previously reported (Farnsworth et al. 2015), pro‐inflammatory cytokines significantly decreased Cx36 gap junction coupling in both mouse islets (Fig. 1 A and B, P = 0.010; Videos S1–S4) and human islets (Fig. 1 C and D, P < 0.001; Videos S5–S8). Treatment with exendin‐4 (E4) for 24 h prevented cytokine‐induced decreases in gap junction coupling in both mouse (Fig. 1 A and B, P < 0.001) and human (Fig. 1 C and D, P < 0.001) islets. Treatment with E4 alone was sufficient to increase gap junction coupling in mouse islets (Fig. 1 A and B, P = 0.002); however, in human islets E4 treatment alone did not affect gap junction coupling (Fig. 1 C and D). In mouse islets, treatment with IBMX, which inhibits degradation of cAMP by phosphodiesterase and increases intracellular cAMP levels, was also sufficient to increase gap junction coupling (Fig. 1 E and F, P = 0.010) and protected against cytokine‐induced decreases in gap junction coupling (Fig. 1 E and F, P < 0.001).
Figure 1. Exendin‐4 overcomes cytokine‐induced decreases in gap junction coupling.
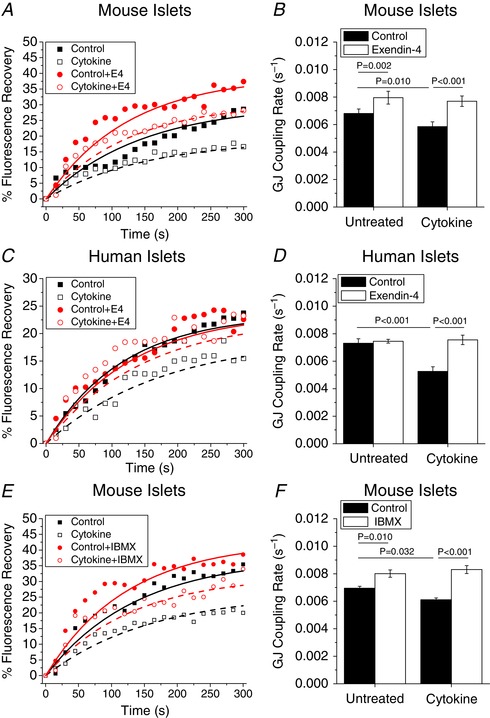
A and B, representative fluorescence recovery (A) and mean gap junction (GJ) coupling rate (B) based on fluorescence recovery rate (s−1) in mouse islets treated with or without cytokines and with or without 10 nM exendin‐4 (E4) for 24 h (n = 5). C and D, representative fluorescence recovery (C) and mean gap junction coupling rate (D) based on fluorescence recovery rate (s−1) in human islets treated with or without cytokines and with or without 10 nM E4 for 24 h (n = 7). E and F, representative fluorescence recovery (E) and mean gap junction coupling rate (F) based on fluorescence recovery rate (s−1) in mouse islets treated with or without cytokines and with or without 100 μM IBMX for 24 h (n = 4). B–D show means ± standard error of the mean (SEM). B and D, P < 0.05 indicates a significant difference based on repeated measures ANOVA. F, P < 0.05 indicates a significant difference based on one‐way ANOVA. [Color figure can be viewed at wileyonlinelibrary.com]
As electrical coupling regulates intracellular Ca2+ in the islet, we next determined if E4 could recover cytokine‐induced impairment of Ca2+ signalling. As previously reported (Farnsworth et al. 2015) cytokine treatment decreased glucose‐stimulated (11 mM glucose) Ca2+ pulsatility, oscillation coordination (P = 0.007, Fig. 2 A and B, Fig. 2 D) and peak amplitude (Fig. 2 E) compared to control islets. Treatment with E4 blunted the effects of cytokine treatment resulting in improved oscillation coordination (P = 0.041, Fig. 2 C and D) and improved peak amplitude (Fig. 2 E) compared to cytokine treatment alone. Cytokine treatment alone significantly increased Ca2+ oscillation frequency (Fig. 2 F, P = 0.043); however, concurrent treatment with cytokines and E4 further increased oscillation frequency (Fig. 2 F, P = 0.004).
Figure 2. Exendin‐4 overcomes cytokine‐induced dysfunction of Ca2+ signalling.
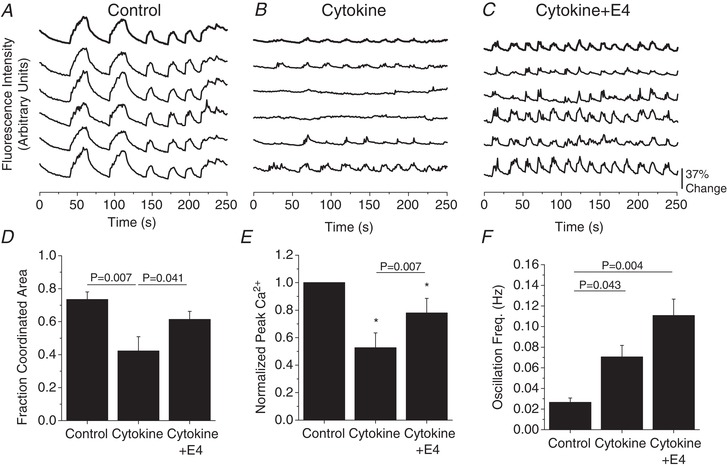
A–C, representative intracellular Ca2+ oscillations at 11 mM glucose in control (A), cytokine‐treated (B), and cytokine treatment with 10 nM E4 (C). D–F, fraction islet area with coordinated Ca2+ oscillations (n = 3) (D), average Ca2+ oscillation peak normalized to control islets (n = 3) (E), and average oscillation frequency (Hz, n = 3) (F) in mouse islets with cytokines, and with or without E4 as indicated. D–F show means ± SEM. D and F, P < 0.05 indicates a significant difference based on repeated measures ANOVA. E, *significant difference from controls based on 95% confidence intervals; P < 0.05 indicates significance based on paired sample t‐test.
To further understand how changes in Cx36 gap junction coupling and Ca2+ signalling affect islet function, insulin secretion and intracellular cAMP levels were analysed. As previously reported (Farnsworth et al. 2015), cytokine treatment decreased the stimulation index (Fig. 3 A, P = 0.036). E4 blunted the effects of cytokine treatment resulting in similar insulin secretion and stimulation index (Fig. 3 A, P = 0.005) compared to control islets. In control islets, both E4 and F+IBMX induced robust increases in cAMP levels as measured by the ICUE3 FRET sensor (Fig. 3 B and C); however, treatment with cytokines for 24 h significantly dampened both the E4‐ and the F+IBMX‐induced increases in cAMP (Fig. 3 D, P = 0.047 and 0.022, respectively).
Figure 3. Cytokines impair insulin secretion and islet response to cAMP elevating agents.
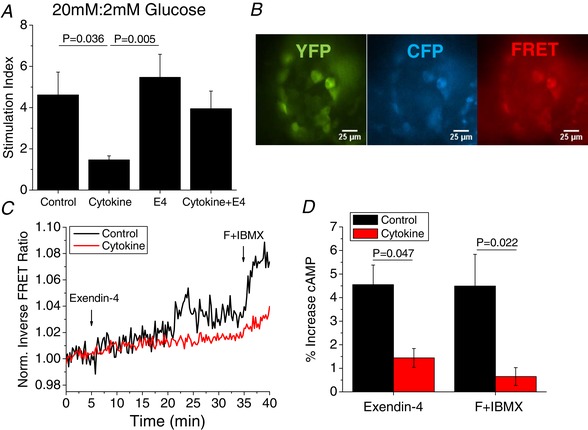
A, insulin secretion at 20 mM glucose normalized to 2 mM glucose (stimulation index) for islets treated with or without cytokines and with or without 10 nM exendin‐4 for 24 h (n = 5–6). B, representative images of YFP (green), CFP (blue) and FRET ratio (red) (CFP/YFP) in an islet transduced with the ICUE3 FRET sensor for intracellular cAMP. C, representative FRET ratio over time for control (black) or cytokine‐treated islets (red) treated with E4 (5 min) and F+IBMX (35 min). D, quantification of the percentage increase in FRET signal indicating increased cAMP in control (black) and cytokine‐treated islets (red) after treatment with E4 for 25 min and F+IBMX for 5 min (n = 3). A and D show the means ± SEM. P < 0.05 indicates a significant difference based on repeated measures ANOVA. [Color figure can be viewed at wileyonlinelibrary.com]
To investigate the mechanism by which E4 preserves gap junction coupling in cytokine‐treated islets, we determined changes in Cx36 protein on the β‐cell membrane using immunohistochemical analysis of fixed mouse islets. Islets were costained for Cx36, neural cell adhesion molecule (NCAM) as a marker of the β‐cell membrane, and DAPI staining of nuclei (Fig. 4 A). Clusters of Cx36 channels, termed plaques, were identified as shown in Fig. 4 A. As previously reported (Farnsworth et al. 2015), islets treated with cytokines have significantly less Cx36 stained area on the β‐cell membrane; however, treatment with E4 maintained Cx36 area in cytokine‐treated islets to levels similar to that of untreated islets (Fig. 4 B). Similarly, the number of Cx36 plaques per cell was significantly reduced in cytokine‐treated islets and treatment with E4 maintained Cx36 plaque number per cell in cytokine‐treated islets (Fig. 4 C).
Figure 4. Exendin‐4 regulates gap junction coupling through increases in Cx36 on the β‐cell membrane.
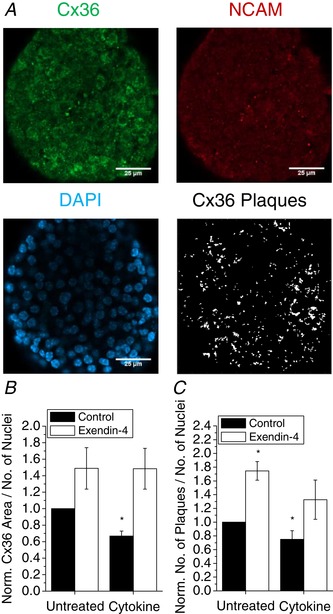
A, immunohistochemical staining of Cx36 gap junctions (green), NCAM (red), nuclei (DAPI, blue), and Cx36 gap junction plaques identified from Cx36 and NCAM staining in fixed mouse islets. B and C, quantification of Cx36 plaque area per number of nuclei (B) and number of Cx36 plaques per number of nuclei (C) in mouse islets treated with cytokines and 10 nM exendin‐4 as indicated for 24 h, normalized to control islets from each experiment (n = 3). B and C show means ± SEM. *Significant difference from control islets determined with 95% confidence intervals. [Color figure can be viewed at wileyonlinelibrary.com]
Exendin‐4 protects against NO‐activated PKCδ‐mediated decreases in gap junction coupling
We have previously reported that pro‐inflammatory cytokines decrease Cx36 gap junction coupling through NO‐activated PKCδ (Farnsworth et al. 2015). We sought to determine if E4 treatment could protect against PKCδ‐mediated decreases in gap junction coupling. Treatment with the PKCδ activator PMA for 1 h significantly decreased gap junction coupling (P = 0.005, Fig. 5 A). Concurrent treatment with PMA and E4 for 1 h significantly improved gap junction coupling compared to untreated cytokine treatment alone (P = 0.006, Fig. 5 A). Similarly, treatment with the NO donor molecule SNAP for 1 h significantly decreased gap junction coupling (P = 0.004, Fig. 5 B), while concurrent treatment with SNAP and E4 for 1 h significantly improved gap junction coupling compared to untreated cytokine treatment alone (P = 0.003, Fig. 5 B).
Figure 5. Exendin‐4 attenuates NO‐activated PKCδ‐mediated decreases in gap junction coupling.
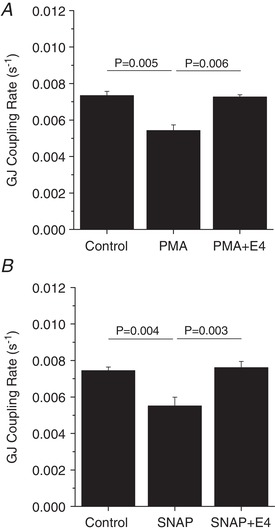
Gap junction coupling (GJ coupling rate) based on fluorescence recovery rate (s−1) in mouse islets treated with 300 μM of the PKCδ activator PMA with or without 10 nM E4 (n = 4) (A) or 5 mM of the nitric oxide donor molecule SNAP with or without 10 nM E4 (n = 4) (B) for 1 h. A and B show means ± SEM. P < 0.05 indicates a significant difference based on repeated measures ANOVA.
Exendin‐4 protects against cytokine‐induced decreases in Cx36 gap junction coupling via both PKA and Epac2
As both PKA and Epac2 are activated by E4‐induced increases in cAMP (Tengholm & Gylfe, 2017), we sought to determine the respective roles of PKA and Epac2 in mediating the protection of gap junction coupling with E4 in cytokine‐treated islets. Selective inhibition of cAMP‐activated PKA with Rp‐cAMP did not affect E4‐mediated maintenance of gap junction coupling in cytokine‐treated mouse islets (Fig. 6 A); however, in human islets gap junction coupling was significantly attenuated (P = 0.026, Fig. 6 B). Treatment with the PKA inhibitor alone for 24 h did not significantly affect gap junction coupling in mouse islets (Fig. 6C). In contrast, addition of the cAMP‐dependent PKA activator 6‐Bnz was sufficient to recover cytokine‐induced decreases in gap junction coupling in both mouse (P < 0.001) and human (P = 0.003) islets (Fig. 6 D and E). Treatment with the PKA activator alone for 24 h did not significantly affect gap junction coupling in mouse islets (Fig. 6 F).
Figure 6. Role of PKA in mediating exendin‐4 regulation of gap junction coupling.
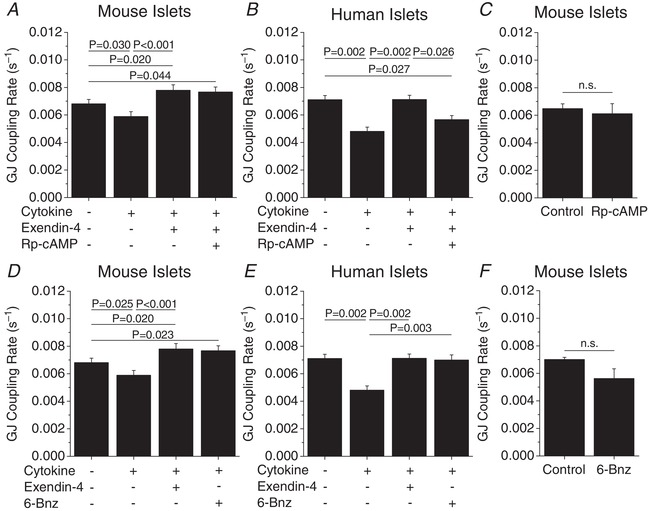
A and B, gap junction coupling (GJ coupling rate) based on fluorescence recovery rate (s−1) in mouse (n = 5) (A) and human (n = 4) (B) islets treated with cytokines, 10 nM exendin‐4 and 100 μM of the PKA inhibitor Rp‐cAMP as indicated for 24 h. C, gap junction coupling in mouse islets treated with the PKA inhibitor Rp‐cAMP alone for 24 h (n = 4). D and E, gap junction coupling in mouse (n = 5) (D) and human (n = 4) (E) islets treated with cytokines, 10 nM exendin‐4 and 300 μM of the PKA activator 6‐Bnz as indicated for 24 h. F, gap junction coupling in mouse islets treated with the PKA activator 6‐Bnz alone for 24 h (n = 4). Data represent means ± SEM. P < 0.05 indicates a significant difference based on repeated measures ANOVA.
Selective inhibition of Epac2 activation with ESI attenuated E4‐mediated preservation of gap junction coupling in cytokine‐treated mouse islets (P = 0.008, Fig. 7 A); however in human islets gap junction coupling was only slightly reduced (P = 0.08, Fig. 7 B). Treatment with the Epac2 inhibitor ESI alone for 24 h did not significantly affect gap junction coupling in mouse islets (Fig. 7 C). Addition of the cAMP‐dependent Epac2 activator 8‐Me preserved gap junction coupling in cytokine‐treated mouse (P < 0.001, Fig. 7 D) and human islets (P = 0.003, Fig. 7 E) to levels similar to those of islets treated with cytokines and E4. Treatment with the Epac2 activator 8‐Me alone for 24 h slightly increased gap junction coupling (P = 0.05, Fig. 7 F).
Figure 7. Role of EPAC2 in mediating exendin‐4 regulation of gap junction coupling.
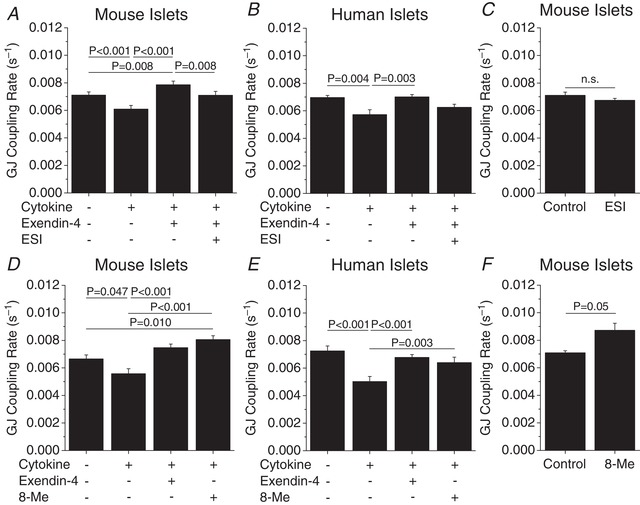
A and B, gap junction coupling (GJ coupling rate) based on fluorescence recovery rate (s−1) in mouse (n = 5) (A) and human (n = 5) (B) islets treated with cytokines, 10 nM exendin‐4 and 10 μM of the EPAC inhibitor ESI as indicated for 24 h. C, gap junction coupling in mouse islets treated with the EPAC2 inhibitor ESI alone for 24 h (n = 5). D and E, gap junction coupling in mouse (n = 5) (D) and human (n = 5) (E) islets treated with cytokines, 10 nM exendin‐4 and 300 μM of the EPAC activator 8‐Me as indicated for 24 h. F, gap junction coupling in mouse islets treated with the EPAC2 activator 8‐Me alone for 24 h (n = 5). Data represent means ± SEM. P < 0.05 indicates a significant difference based on repeated measures ANOVA.
To determine the mechanism of PKA and Epac2 regulation of Cx36 coupling, Cx36 protein levels on the cell membrane were quantified. Activation of either PKA or Epac2 in islets treated for 24 h with cytokine preserved Cx36 area (Fig. 8 A) and plaque number (Fig. 8 B). Treatment with either the PKA activator or Epac2 activator with cytokine treatments did not significantly improve insulin secretion compared to cytokine treatment alone (data not shown). These results indicate that both PKA and Epac2 play a role in E4 regulation of Cx36 gap junction coupling in cytokine‐treated islets with 24 h treatments.
Figure 8. Exendin‐4 regulates Cx36 levels on the cell membrane via both PKA and Epac2.
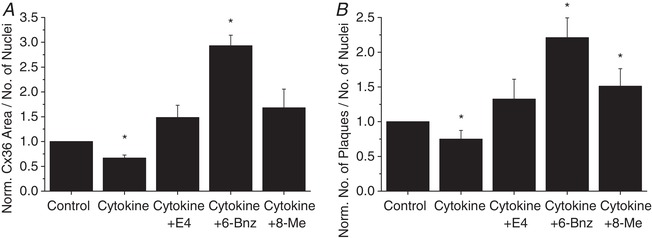
Quantification of Cx36 plaque area per number of nuclei (A) and number of Cx36 plaques per number of nuclei (B) in mouse islets treated with cytokines, 10 nM exendin‐4, the PKA activator 6‐Bnz, or the Epac2 activator 8‐Me as indicated for 24 h, normalized to control islets from each experiment (n = 3). Data represent means ± SEM. *Significant difference from control islets determined with 95% confidence intervals.
PKA mediates E4 protection of Cx36 gap junction coupling with acute cytokine treatment
We next tested E4‐mediated protection from cytokine‐induced decreases in Cx36 gap junction coupling on an acute 1 h time scale. Similar to 24 h treatments, 1 h cytokine treatment significantly reduced gap junction coupling (P < 0.001, Fig. 9 A), as previously reported (Farnsworth et al. 2015). E4 treatment prevented cytokine‐induced decreases in gap junction coupling over the 1 h treatment (P < 0.001); however, E4 treatment alone for 1 h was not sufficient to increase gap junction coupling (Fig. 9 A). One hour treatment with the PKA inhibitor Rp‐cAMP did not significantly attenuate the E4‐mediated preservation of gap junction coupling (Fig. 9 B), similar to 24 h treatment. One hour treatment with the PKA activator 6‐Bnz showed similar protection against cytokine‐mediated decreases in gap junction coupling as in E4‐treated islets (Fig. 9 C). In contrast, 1 h treatment with the Epac2 inhibitor ESI did not significantly attenuate the E4‐mediated preservation of gap junction coupling (Fig. 9 D). One hour treatment with the Epac2 activator 8‐Me did not significantly protect against cytokine‐mediated decreases in gap junction coupling with 1 h treatment (Fig. 9 E).
Figure 9. Exendin‐4 acutely mediates gap junction coupling via PKA in mouse islets.
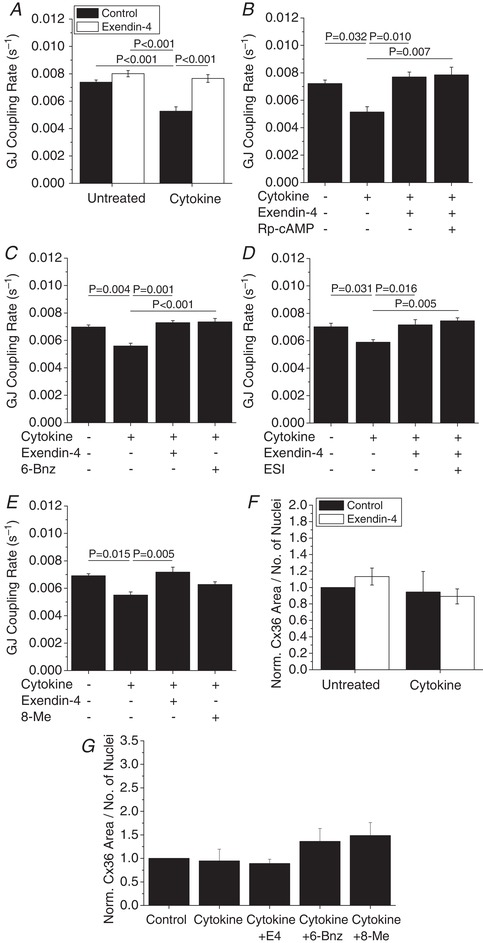
A–E, gap junction coupling (GJ coupling rate) based on fluorescence recovery rate (s−1) in mouse islets treated with cytokines and 10 nM exendin‐4 (n = 4–5) (A–E), 100 μM of the PKA inhibitor Rp‐cAMP (n = 4) (B), 300 μM of the PKA activator 6‐Bnz (n = 4) (C), 10 μM of the EPAC inhibitor ESI (n = 4) (D), or 300 μM of the EPAC activator 8‐Me (n = 4) (E) as indicated for 1 h. Data represent means ± SEM. P < 0.05 indicates a significant difference based on repeated measures ANOVA. F, quantification of Cx36 plaque area per number of nuclei in mouse islets treated with cytokines and 10 nM exendin‐4 as indicated for 1 h, normalized to control islets from each experiment (n = 3). G, quantification of Cx36 plaque area per number of nuclei in mouse islets treated with cytokines, 10 nM exendin‐4, the PKA activator 6‐Bnz, or the Epac2 activator 8‐Me as indicated for 1 h, normalized to control islets from each experiment (n = 3). Data represent means ± SEM. *Significant difference from control islets determined with 95% confidence intervals.
To further elucidate the mechanism of PKA and Epac2 regulation of Cx36 coupling, Cx36 protein levels on the cell membrane were quantified. Acute (1 h) treatment with cytokines with and without E4 had no effect on Cx36 area (Fig. 9 F) or plaque number (data not shown). Acute treatment with cytokines alone or in combination with PKA or Epac2 activation did not affect Cx36 area (Fig. 9 G) or plaque number (data not shown). These results indicate that both PKA and Epac2 regulate Cx36 levels and coupling with chronic E4 treatment, but PKA can regulate Cx36 coupling with acute E4 treatment.
Discussion
Pro‐inflammatory cytokines contribute to β‐cell death and dysfunction in both type 1 and type 2 diabetes. In vitro, cytokines alter islet Ca2+ signalling and gap junction coupling, similar to what has been observed in models of pre‐diabetes. Our previous study has shown that factors that increase intracellular cAMP increase Cx36 gap junction coupling, and in other cell types cAMP‐activated PKA and Epac2 play a role in regulating gap junctions. The goal of this study was to determine if increasing cAMP with E4 can overcome cytokine‐induced decreases in Cx36 gap junction coupling and altered Ca2+ signalling and to determine the role of both PKA and Epac2 in cAMP regulation of Cx36 coupling.
Exendin‐4 protects against cytokine‐induced islet dysfunction via increased plasma membrane Cx36
In this study we found that E4 increased Cx36 coupling in mouse islets following 24 h treatment. E4 alone did not affect Cx36 coupling in human islets. Differential effects of cAMP increases on human islets have been reported with donor age, including impaired E4‐induced proliferation in donors >35 years of age (Tian et al. 2011). While age‐based comparisons of E4 regulation of Cx36 were not possible due to the lack of diversity in donor age, the age of the donors (median ∼44 years) may partially explain the difference in E4‐induced increases in coupling in mouse compared to human islets. Regardless, in both mouse and human islets, treatment with cytokines decreased Cx36 gap junction coupling, as we have previously reported (Farnsworth et al. 2015). Johnston et al. similarly found that treatment with IL‐1β and TNF‐α decreased hub cell coupling with neighbouring β‐cells, reduced Cx36 gene expression and reduced Cx36 staining on the cell surface as measured by immunohistochemistry (Johnston et al. 2016). In our study, concurrent treatment with cytokines and E4 prevented losses in Cx36 gap junction coupling. Our results combined with the findings of Johnston et al. suggest that E4 may protect against loss of hub cell coupling. Increases in Cx36 coupling with IBMX, a phosphodiesterase inhibitor which raises intracellular cAMP independent of GLP‐1R activation, under both normal and cytokine‐stressed conditions indicates that GLP‐1R regulation of Cx36 coupling is likely mediated by increased cAMP alone, rather than in combination with other signalling cascades associated with GLP‐1R activation such as mitogen‐activated protein kinase, phosphoinositide 3‐kinase (Doyle & Egan, 2007), and PKC (Shigeto et al. 2015).
While changes in Cx36 gap junction coupling as measured by FRAP reflect changes in Cx36 protein levels, changes in Cx36 channel conductance and electrical coupling as measured by dye coupling are only correlative (Benninger et al. 2008). Therefore, to assess changes in electrical coupling and Cx36 channel conductance, we measured coordination of glucose‐induced intracellular Ca2+ oscillations. E4 protection against cytokine‐induced decreases in coupling is further supported by the observed improvement in Ca2+ oscillation coordination and peak amplitude in islets treated with cytokines and E4, compared to cytokines alone. While peak Ca2+ amplitude was improved with E4 treatment compared to cytokine‐treated islets, the amplitude was still significantly reduced compared to controls. Tong et al. (2015) have shown that cytokines decrease levels of SERCAb2, an ER Ca2+ channel, resulting in altered Ca2+ signalling. Taken together with the results of this study, this indicates that cytokines affect multiple regulators of islet Ca2+ signalling, and E4 rescues Cx36 electrical coupling‐induced changes in Ca2+ signalling, but other mechanisms likely remain unaltered. Treatment with cytokines significantly increased Ca2+ oscillation frequency, which is consistent with the study by Dickerson et al. (2018). Ca2+ oscillation frequency was further increased compared to control and cytokine‐treated islets. As E4 regulates KATP channel activity, where KATP channel closure mediates glucose‐stimulated Ca2+ oscillations (Light et al. 2002), this could potentially explain the E4‐mediated increases in oscillation frequency. However, further experiments are required to fully explore the role of E4 in regulating Ca2+ oscillations.
Further analysis of islet function revealed that E4 protected against cytokine‐induced decreases in insulin secretion. While E4 is a rapidly acting insulin secretagogue, its effects are not prolonged (Alarcon et al. 2006), as supported by our data where E4 treatment alone prior to analysis of insulin secretion did not significantly affect the stimulation index. This suggests that any recovery in insulin secretion with cytokines and E4 combined may be due to increased Cx36 coupling rather than increased amplification of insulin secretion. While cytokine treatment significantly decreased both E4 and F+IBMX‐mediated increases in intracellular cAMP, E4 did still increase cAMP levels, likely resulting in the improved glucose‐stimulated insulin secretion observed.
To determine the mechanism of E4 regulation of Cx36, we analysed immunohistochemical staining of Cx36. Cx36 connexons, or functional channels, aggregate in the cellular membrane at tight junctions forming plaques, or gap junctions (Duffy et al. 2002). While cAMP has been shown to increase gap junction coupling in Novikoff hepatoma cells (Paulson et al. 2000) and smooth muscle cells (Begandt et al. 2013), the mechanisms of cAMP regulation of Cx36 coupling in β‐cells has not been directly studied. Changes in Cx36 turnover may reflect a number of regulatory mechanisms, including Cx36 gene expression, Cx36 trafficking to the membrane, recruitment and assembly of Cx36 into gap junction plaques, and endocytosis of connexons to be recycled (Segretain & Falk, 2004; Laird, 2006; Farnsworth & Benninger, 2014). Treatment with E4 protected against cytokine‐induced decreases in Cx36 plaque area and number, suggesting that E4 regulates Cx36 turnover at the cell membrane in cytokine‐treated islets. Experiments with 1h treatments did not show significant changes in Cx36 levels, further supporting a role for E4 in mediating Cx36 turnover as the half‐life of Cx36 is ∼4 h (Laird, 2006).
PKCδ is activated by pro‐inflammatory cytokines in the β‐cell (Carpenter et al. 2001) and has been shown to mediate free fatty acid‐induced β‐cell apoptosis (Eitel et al. 2003). Our previous study has shown that pro‐inflammatory cytokines decrease Cx36 gap junction coupling through NO‐activated PKCδ (Farnsworth et al. 2015). Our results indicate that E4 protects against NO‐ and PKCδ‐mediated decreases in Cx36 gap junction coupling. In the brain, E4 has been shown to activate PKCδ (Cabou et al. 2011); however, no link between E4 or cAMP and PKCδ has been identified in the β‐cell. Our previous study found that cytokines decrease Cx36 plaque area per cell via PKCδ; therefore, we propose that E4 protects against NO‐activated PKCδ‐induced decreases in Cx36 coupling by preserving functional Cx36 plaque area, rather than directly interacting with PKCδ. As Cx36 gap junction coupling has been shown to protect against cytokine‐induced β‐cell apoptosis (Allagnat et al. 2013), increases in Cx36 coupling may contribute to the mechanism by which E4 treatment protects against decline in islet mass in a mouse model of type 1 diabetes (Hadjiyanni et al. 2008).
PKA mediates acute cAMP‐induced regulation of Cx36 gap junction coupling
To determine the mechanism of cAMP regulation of Cx36 coupling, we investigated the roles of cAMP‐activated PKA and Epac2, which mediate cAMP‐induced augmentation of glucose stimulated insulin secretion (Tengholm & Gylfe, 2017). Our results indicate that under the stress of cytokine treatment, PKA plays a role in mediating cAMP regulation of Cx36 in both mouse and human islets. As PKA alone did not significantly alter coupling in healthy islets, this may suggest that PKA may differentially regulate Cx36 coupling under normal and stressed environments. Under acute (1 h) cytokine treatment, E4 protected against cytokine‐induced decreases in Cx36 coupling with PKA dependence, while Cx36 levels at the cell membrane were not altered with acute activation of PKA. This implies that PKA regulates Cx36 coupling via a fast mechanism, such as Cx36 phosphorylation and channel gating, under inflammatory conditions. In the retina, PKA phosphorylation of serine293 has been shown to increase Cx36 coupling without affecting trafficking or distribution of Cx36 on the cell membrane, supporting this potential mechanism in the islet (Kothmann et al. 2009; Ivanova et al. 2015).
In cardiomyocytes, PKA is anchored to the cell membrane by A‐kinase anchoring protein (AKAP), which colocalizes with gap junctions and mediates increases in gap junction coupling initiated by localized increases in cAMP (Pidoux & Taskén, 2015). Similar machinery exists in β‐cells to localize PKA to tight junctions (Lester et al. 1997), where Cx36 plaques are localized in the plasma membrane (Serre‐Beinier et al. 2009), further supporting a role for PKA in directly mediating protection against cytokine‐induced decreases in Cx36 coupling via channel gating. Changes in PKA localization, and therefore action, with sequestering by different members of the AKAP family may explain the differential role for PKA under normal and cytokine‐stressed conditions as found in this study.
Overall, our results indicate that PKA plays a role in mediating E4 protection against cytokine‐induced decreases in Cx36 gap junction coupling. Based on our findings, we propose that E4‐activated PKA regulates Cx36 coupling via gating of gap junction channel conductance as shown in Fig. 10.
Figure 10. cAMP‐activated PKA and Epac2 mediate exendin‐4 regulation of Cx36 gap junction coupling.
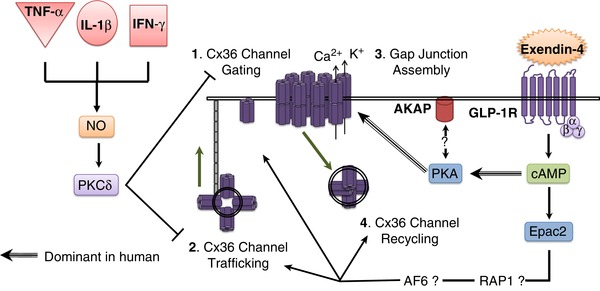
Cytokines decrease Cx36 coupling via NO and PKCδ‐mediated decreases in Cx36 trafficking and channel gating. Exendin‐4 counteracts cytokine‐induced decreases in coupling via both PKA and Epac2. In human islets (striped arrows), PKA played a greater role in mediating Cx36 coupling; however, in mouse islets (solid arrows) Epac2 played a greater role in mediating Cx36 coupling. PKA mediates Cx36 coupling under cell stress conditions through gating of Cx36 channel conductance likely mediated by AKAP sequestering near the plasma membrane (1). Epac2 mediates Cx36 coupling through channel turnover by Cx36 channel trafficking (2), gap junction assembly (3), or Cx36 channel recycling (4). Epac2 regulation of gap junction turnover likely occurs via Ras‐related protein 1 (Rap1) activation of afadin (AF6), which colocalizes with Cx36. [Color figure can be viewed at wileyonlinelibrary.com]
Epac2 mediates cAMP‐induced Cx36 turnover
Our studies indicate a role for Epac2 alone in mediating Cx36 gap junction coupling as well as protecting against cytokine‐induced decreases in Cx36 gap junction coupling in mouse islets. While Epac2 plays a significant role in E4 protection against cytokine‐induced decreases in Cx36 coupling and Cx36 levels under 24 h treatment, Epac2‐mediated protection was minimal under acute (1 h) treatment and no changes in Cx36 levels at the cell membrane were detected. This suggests that Epac2 regulates Cx36 coupling via slower mechanisms which occur on a time scale >1 h, such as trafficking, assembly, or turnover of Cx36 channels which occurs over ∼4 h (Wang et al. 2015). The robust increase in Cx36 plaque area per cell in 24 h E4 and cytokine‐treated islets compared to untreated controls supports this hypothesis.
In neurons and cardiomyocytes Epac2 regulates gap junction coupling via Ras‐related protein 1 (Rap1), a GTP‐binding protein, and afadin (AF6), which are proposed to mediate gap junction assembly and turnover (Somekawa et al. 2005; Li et al. 2012b). While cAMP mediates priming of insulin secretion granules via Rap1 (Leech et al. 2010), it is unknown if AF6 is expressed in β‐cells. In general, cell–cell adhesion via adherens and tight junctions where Cx36 plaques are localized is mediated by occludins, cadherins and nectins, which recruit zonula occludin 1 (ZO1), α‐ and β‐catenin and AF6, respectively, to the plasma membrane (Segretain & Falk, 2004; Campbell et al. 2017). As ZO1 has been shown to colocalize with Cx36 in several cell types (Giepmans, 2004; Li et al. 2004), AF6 may also be present at these junctions in the β‐cell and could mediate Epac2 regulation of Cx36 turnover.
We observed species‐dependent differences in the respective roles of PKA and Epac2 in mediating E4 protection against cytokines. In human islets, PKA appeared to strongly mediate E4 protection, while in mouse islets Epac2 strongly mediated E4 protection. One study in human islets has shown that Epac2‐mediated potentiation of glucose‐stimulated insulin secretion is dependent on activation of PKA, suggesting that activation of PKA may dominantly mediate E4 signalling in human islets, as seen in this study (Chepurny et al. 2010). RNA sequencing of human and mouse islets has also shown decreased expression of RAPGEF4, the gene encoding Epac2, in human islets compared to mouse islets (Benner et al. 2014). This may also explain the minimal role for Epac2 in regulating Cx36 coupling in human islets compared to mouse islets.
Overall, our results suggest that in healthy islets Epac2 plays a role in E4‐induced increases in Cx36 coupling by regulation of Cx36 trafficking, gap junction assembly, or connexon endocytosis in the β‐cell, as shown in Fig. 10. Under the stress of cytokine treatment, both PKA and Epac2 play a role in E4‐mediated protection against cytokine‐induced decreases in Cx36 coupling, where PKA likely regulates channel gating.
Conclusion
This study has built upon our previous work, which found pro‐inflammatory cytokines decreased Cx36 gap junction coupling and that increases in cAMP increase Cx36 coupling. Our results indicate that the GLP‐1R agonist exendin‐4 can protect against cytokine‐induced decreases in Cx36 coupling in both mouse and human islets by preserving Cx36 gap junction area on the β‐cell surface. Our results implicate that both cAMP‐activated PKA and Epac2 play a role in mediating this protection, where PKA plays a greater role in human islets and Epac2 plays a greater role in mouse islets. Specifically, we propose that PKA mediates Cx36 channel gating and Epac2 mediates Cx36 gap junction trafficking, assembly and turnover. Future studies will be needed to further define the molecular mechanisms underlying Epac2 and PKA regulation of Cx36 gap junctions under normal and stress conditions. As Cx36 coupling protects against cytokine‐mediated apoptosis in vitro and is important for regulating insulin secretion dynamics, treatments which can increase Cx36 coupling in vivo may protect against β‐cell death and dysfunction during the development of diabetes.
Additional information
Competing interests
Authors declare no conflict of interest.
Author contributions
N.L.F. designed the experiments, researched the data and wrote the manuscript; R.W. designed the experiments, researched the data and wrote the manuscript; R.P. researched the data and edited the manuscript; W.E.S. researched the data and edited the manuscript; R.K.P.B. designed the experiments and edited the manuscript. All authors have read and approved the final version of this manuscript and agree to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved. All persons designated as authors qualify for authorship, and all those who qualify for authorship are listed.
Funding
This study was primarily supported by NIH grant R01 DK102950, NIH grant R01 DK106412 and JDRF grant 5‐CDA‐2014‐198‐A‐N to R.K.P.B. and NIH grant F32 DK102276 to N.L.F.
Translational perspective.
Pro‐inflammatory cytokines contribute to pancreatic β‐cell death and dysfunction in both type 1 and type 2 diabetes. We have previously shown that cytokines decrease β‐cell coupling, which regulates glucose‐stimulated insulin secretion, while compounds that increase intracellular cAMP can increase coupling. The goal of this study was to determine if increasing cAMP with exendin‐4 (exenatide) can overcome cytokine‐induced decreases in β‐cell coupling and insulin secretion. We found that in mouse and human islets exendin‐4 protected against cytokine‐mediated decreases in β‐cell coupling via distinct mechanism. As Cx36 coupling protects against cytokine‐mediated apoptosis in vitro and is important for regulating insulin secretion dynamics, treatments which can increase Cx36 coupling in vivo may protect against β‐cell death and dysfunction during the development of diabetes. This may partially explain the ability of exendn‐4 to improve glycaemic control in patients with type 2 diabetes and to improve graft viability in mice receiving islet transplants.
Supporting information
Video S1: Mouse Control 24h FRAP
Video S2: Mouse Control + Exendin‐4 24h FRAP
Video S3: Mouse Cytokine 24h FRAP
Video S4: Mouse Cytokine + Exendin‐4 24h FRAP
Video S5: Human Control 24h FRAP
Video S6: Human Control + Exendin‐4 24h FRAP
Video S7: Human Cytokine 24h FRAP
Video S8: Human Cytokine + Exendin‐4 24h FRAP
Acknowledgements
The authors would like to acknowledge the University of Colorado Anschutz Medical Campus Advanced Light Microscopy Core for assistance with imaging on the Zeiss LSM510 microscope, as well as the Barbara Davis Center Islet Preparation Core for assistance with islet isolations.
Biography
Nikki Farnsworth is a Research Instructor at the University of Colorado Denver Anschutz Medical Campus. She received her BS in Chemical Engineering at Rensselaer Polytechnic Institute, and her MS and PhD in Chemical Engineering at the University of Colorado Boulder. Her current research focuses on understanding the events which lead to the onset of type 1 diabetes. Her future research goal is to further the understanding of islet dysfunction and death mechanisms during the pathogenesis of type 1 diabetes and to understand the role of extracellular matrix interactions in the onset and progression of disease.

Edited by: Kim Barrett & Fiona Gribble
N. L. Farnsworth and R. Walter have co‐first authorship and made equal contributions
References
- Alarcon C, Wicksteed B & Rhodes CJ (2006). Exendin 4 controls insulin production in rat islet beta cells predominantly by potentiation of glucose‐stimulated proinsulin biosynthesis at the translational level. Diabetologia 49, 2920–2929. [DOI] [PubMed] [Google Scholar]
- Allagnat F, Klee P, Cardozo AK, Meda P & Haefliger J‐A (2013). Connexin36 contributes to INS‐1E cells survival through modulation of cytokine‐induced oxidative stress, ER stress and AMPK activity. Cell Death Differ 20, 1742–1752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersson AK, Flodstrom M & Sandler S (2001). Cytokine‐induced inhibition of insulin release from mouse pancreatic β‐cells deficient in inducible nitric oxide synthase. Biochem Biophys Res Commun 281, 396–403. [DOI] [PubMed] [Google Scholar]
- Begandt D, Bader A, Dreyer L, Eisert N, Reeck T & Ngezahayo A (2013). Biphasic increase of gap junction coupling induced by dipyridamole in the rate aortic A‐10 vascular smooth muscle cell line. J Cell Commun Signal 7, 151–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benner C, van der Meulen T, Caceres E, Tigyi K, Donaldson CJ & Huising MO (2014). The transcriptional landscape of mouse beta cells compared to human beta cells reveals notable species differences in long non‐coding RNA and protein‐coding gene expression. BMC Genomics 15, 620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benninger RKP, Head WS, Zhang M, Satin LS & Piston DW (2011). Gap junctions and other mechanisms of cell–cell communication regulate basal insulin secretion in the pancreatic islet. J Physiol 589, 5453–5466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benninger RKP, Zhang M, Head WS, Satin LS & Piston DW (2008). Gap junction coupling and calcium waves in the pancreatic islet. Biophys J 95, 5048–5061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cabou C, Vachoux C, Campistron G, Drucker DJ & Burcelin R (2011). Brain GLP‐1 signaling regulates femoral artery blood flow and insulin sensitivity through hypothalmic PKC‐δ. Diabetes 60, 2245–2256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell HK, Maiers JL & DeMali KA (2017). Interpaly between tight junctions and adherens junctions. Exp Cell Res 358, 39–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carpenter L, Cordery D & Biden TJ (2001). Protein kinase Cδ activation by interleukin‐1β stabilizes inducible nitric‐oxide synthase mRNA in pancreatic β‐cells. J Biol Chem 276, 5368–5374. [DOI] [PubMed] [Google Scholar]
- Carvalho CPF, Oliviera RB, Britan A, Santos‐Silva JCR, Boschero AC, Meda P & Collares‐Buzato CB (2012). Impaired β‐cell‐β‐cell coupling mediated by Cx36 gap junctions in prediabetic mice. Am J Physiol Endocrinol Metab 303, E144–E151. [DOI] [PubMed] [Google Scholar]
- Chepurny OG, Kelley GG, Dzhura I, Leech CA, Roe MW, Dzhura E, Li X, Schwede F, Genieser H‐G & Holz GG (2010). PKA‐dependent potentiation of glucose‐stimulated insulin secretion by Epac activator 9‐pCPT‐2′‐O‐Me‐cAMP‐AM in human islets of Langerhans. Am J Physiol Endocrinol Metabol 298, E622–E633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeFronzo RA, Ratner RE, Han J, Kim DD, Fineman MS & Baron AD (2005). Effects of exenatide (exendin‐4) on glycemic control and weight over 30 weeks in metformin‐treated patients with type 2 diabetes. Diabetes Care 28, 1092–1100. [DOI] [PubMed] [Google Scholar]
- Dickerson MT, Bogart AM, Altman MK, Milian SC, Jordan KL, Dadi PK & Jacobson DA (2018). Cytokine‐mediated changes in K+ channel activity promotes an adaptive Ca2+ response that sustains β‐cell insulin secretion during inflammation. Sci Rep 8, 1158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiPilato LM & Zhang J (2009). The role of membrane microdomains in shaping β2‐adrenergic receptor‐mediated cAMP dynamics Mol Biosyst 5, 832–837. [DOI] [PubMed] [Google Scholar]
- Doyle ME & Egan JM (2007). Mechanisms of action of GLP‐1 in the pancreas. Pharmacol Ther 113, 546–593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duffy HS, Delmar M & Spray DC (2002). Formation of the gap junction nexus: Binding partners for connexins. J Physiol 96, 243–249. [DOI] [PubMed] [Google Scholar]
- Eitel K, Staiger H, Rieger J, Mischak H, Brandhorst H, Brendel MD, Bretzel RG, Haring HU & Kellerer M (2003). Protein kinase Cδ activation and translocation to the nucleus are required for fatty acid‐induced apoptosis of insulin‐secreting cells. Diabetes 52, 991–997. [DOI] [PubMed] [Google Scholar]
- Eizirik DL, Sandler S, Welsh N, Cetkovic‐Cvrlje M, Nieman A, Geller DA, Pipeleers DG, Bendtzen K & Hellerstrom C (1994). Cytokines suppress human islet function irrespective of their effects on nitric oxide generation. J Clin Invest 93, 1968–1974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farnsworth NL & Benninger RKP (2014). New insights into the role of connexins in pancreatic islet function and diabetes. FEBS Lett 588, 1278–1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farnsworth NL, Hemmati A, Pozzoli M & Benninger RKP (2014). Fluorescence recovery after photobleaching reveals regulation and distribution of connexin36 gap junction coupling within mouse islets of Langerhans. J Physiol 592, 4431–4446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farnsworth NL, Walter R, Hemmati A, Westacott MJ & Benninger RKP (2015). Low‐level pro‐inflammatory cytokines decrease connexin36 gap junction coupling in mouse and human islets through nitric oxide mediated protein kinase Cδ. J Biol Chem 291, 3184–3196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giepmans BNG ( 2004). Gap junctions and connexin‐interacting proteins. Cardiovasc Res 62, 233–245. [DOI] [PubMed] [Google Scholar]
- Grundy D ( 2015). Principles and standards for reporting animal experiments in The Journal of Physiology and Experimental Physiology . J Physiol 593, 2547–2549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grunnet LG, Aikin R, Tonnesen MF, Paraskevas S, Blaabjerg L, Storling J, Rosenberg L, Billestrup N, Maysinger D & Mandrup‐Poulsen T (2009). Proinflammatory cytokines activate the intrinsic apoptotic pathway in β‐cells. Diabetes 58, 1807–1815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hadjiyanni I, Baggio LL, Poussier P & Drucker DJ (2008). Exendin‐4 modulates diabetes onset in nonobese diabetic mice. Endocrinology 149, 1338–1349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haefliger J‐A, Rohner‐Jeanrenaud F, Caille D, Charollais A, Meda P & Allagnat F (2013). Hyperglycemia downregulates Connexin36 in pancreatic islets via upregulation of ICER‐1/ICER‐1γ. J Mol Endocrinol 51, 49–58. [DOI] [PubMed] [Google Scholar]
- Head WS, Orseth ML, Nunemaker CS, Satin LS, Piston DW & Benninger RKP (2012). Connexin‐36 gap junctions regulate in vivo first‐ and second‐phase insulin secretion dynamics and glucose tolerance in the conscious mouse. Diabetes 61, 1700–1707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henquin J‐C & Nenquin M (2014). Activators of PKA and Epac distinctly influence insulin secretion and cytosolic Ca2+ in female mouse islets stimulated by glucose and tolbutamide. Endocrinology 155, 3274–3287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodson DJ, Mitchell RK, Bellomo EA, Sun G, Vinet L, Meda P, Li D, Li W‐H, Bugliani M, Marchetti P, Bosco D, Piemonti L, Johnson P, Hughes SJ & Rutter GA (2013). Lipotoxicity disrupts incretin‐regulated human β cell connectivity. J Clin Invest 123, 4182–4194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hraha TH, Bernard AB, Nguyen LM, Anseth KS & Benninger RKP (2014). Dimensionality and size scaling of coordinated Ca2+ dynamics in MIN6 β‐cell clusters. Biophys J 106, 299–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imai Y, Dobrian AD, Morris MA & Nadler JL (2013). Islet inflammation: a unifying target for diabetes treatment? Trends Endocrinol Metab 24, 351–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivanova E, Lee CW & Sagdullaev BT (2015). Increased phosphorylation of Cx36 gap junctions in the AII amacrine cells of the retina. Front Cell Neurosci 9, 390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnston NR, Mitchell RK, Haythorne E, Pessoa MP, Semplici F, Ferrer J, Piemonti L, Marchetti P, Bugliani M, Bosco D, Berishvili E, Duncanson P, Watkinson M, Broichhagen J, Trauner D, Rutter GA & Hodson DJ (2016). Beta cell hubs dictate pancreatic islet responses to glucose. Cell Metabolism 24, 389–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klee P, Allagnat F, Pontes H, Cederroth M, Charollais A, Caille D, Britan A, Haefliger J‐A & Meda P (2011). Connexins protect mouse pancreatic β cells against apoptosis. J Clin Invest 121, 4870–4879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koster JC, Remedi MS, Flagg TP, Johnson JD, Markova KP, Marshall BA & Nichols CG (2002). Hyperinsulinism induced by targeted suppression of beta cell KATP channels. Proc Natl Acad Sci U S A 99, 16992–16997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kothmann WW, Massey SC & O'Brien J (2009). Dopamine‐stimulated dephosphorylation of connexin 36 mediates AII amacrine cell uncoupling. J Neurosci 29, 14903–14911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laird DW ( 2006). Life cycle of connexins in health and disease. Biochem J 394, 527–543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leech CA, Chepurby OG & Holz GG (2010). Epac2‐dependent Rap1 activation and the control of islet insulin secretion by glucagon‐like peptide‐1. Vitam Horm 84, 279–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lester LB, Langeberg LK & Scott JD (1997). Anchoring of protein kinase A facilitates hormone‐mediated insulin secretion. Proc Natl Acad Sci U S A 94, 14942–14947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Lynn BD & Nagy JI (2012a). The effector and scaffolding proteins AF6 and MUPP1 interact wth connexin36 and localize at gap junctions that form electrical synapses in the rodent brain. Eur J Neurosci 35, 166–181. [DOI] [PubMed] [Google Scholar]
- Li X, Lynn BD & Nagy JI (2012b). The effector and scaffolding proteins AF6 anf MUPP1 interact with connexin36 and localize at gap junctions that form electrical synapses in rodent brain Eur J Neurosci 35, 166–181. [DOI] [PubMed] [Google Scholar]
- Li X, Olson C, Lu S, Kamasawa N, Yasumura T, Rash JE & Nagy JI (2004). Neuronal connexin36 association with zonula occludens‐1 protein (ZO‐1) in mouse brain and interaction with the first PDZ domain of ZO‐1. Eur J Neurosci 19, 2132–2146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Light PE, Fox JEM, Riedel MJ & Wheeler MB (2002). Glucagon‐like peptide‐1 inhibits pancreatic ATP‐sensitive potassium channels via a protein kinase A‐ and ADP‐dependent mechanism. Mol Endocrinol 16, 2135–2144. [DOI] [PubMed] [Google Scholar]
- Meda P ( 2012). The in vivo β‐to‐β‐cell chat room: connexin connections matter. Diabetes 61, 1656–1658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Padgett LE, Broniowska KA, Hansen PA & Corbett JA (2013). The role of reactive oxygen species and proinflammatory cytokines in type 1 diabetes pathogenesis. Ann N Y Acad Sci 1281, 16–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paulson AF, Lampe PD, Meyer RA, TenBroek E, Atkinson MM, Walseth TF & Johnson RG (2000). Cyclic AMP and LDL trigger a rapid enhancement in gap junction assembly through a stimulation of connexin trafficking. J Cell Sci 113, 3037–3049. [DOI] [PubMed] [Google Scholar]
- Perez‐Armendariz EM, Roy C, Spray DC & Bennet MV (1991). Biophysical properties of gap junctions between freshly dispersed pairs of mouse pancreatic beta cells. Biophys J 59, 76–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pidoux G & Taskén K (2015). Anchored PKA as a gatekeeper for gap junctions. Commun Integr Biol 8, e1057361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramadan JW, Steiner SR, O'Neill CM & Nunemaker CS (2011). The central role of calcium in the effects of cytokines on beta‐cell function: Implications for type 1 and type 2 diabetes. Cell Calcium 50, 481–490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ravier MA, Guldenagel M, Charollais A, Gjinovci A, Caille D, Sohl G, Wollheim CB, Willecke K, Henquin J‐C & Meda P (2005). Loss of connexin36 channels alters β‐cell coupling, islet synchronization of glucose‐induced Ca2+ and insulin oscillations, and basal insulin release. Diabetes 54, 1798–1807. [DOI] [PubMed] [Google Scholar]
- Robertson RP ( 2009). β‐Cell deterioration during diabetes: what's in the gun? Trends Endocrinol Metab 20, 388–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rocheleau JV, Remedi MS, Granada B, Head WS, Koster JC, Nichols CG & Piston DW (2006). Critical role of gap junction coupled KATP channel activity for regulated insulin secretion. PLoS Biol 4, e26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rutter GA, Pullen TJ, Hodson DJ & Martinez‐Sanchez A (2015). Pancreatic β‐cell identity, glucose sensing and the control of insulin secretion. J Biochem 466, 203–218. [DOI] [PubMed] [Google Scholar]
- Segretain D & Falk MM (2004). Regulation of connexin biosynthesis, assembly, gap junction formation, and removal. Biochim Biophys Acta 1662, 3–21. [DOI] [PubMed] [Google Scholar]
- Serre‐Beinier V, Bosco D, Zulianello L, Charollais A, Caille D, Charpantier E, Gauthier BR, Diaferia GR, Giepmans BN, Lupi R, Marchetti P, Deng S, Buhler L, Berney T, Cirulli V & Meda P (2009). Cx36 makes channels coupling human pancreatic β‐cells, and corrlates with insulin expression. Hum Mol Genet 18, 428–439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shigeto M, Ramracheya R, Tarasov AI, Cha CY, Chibalina MV, Hastoy B, Philippaert K, Reinbothe T, Rorsman N, Salehi A, Sones WR, Vergari E, Weston C, Gorelik J, Katsura M, Nikolaev VO, Vennekens R, Zaccolo M, Galione A, Johnson PR, Kahu K, Ladds G & Rorsman P (2015). GLP‐1 stimulates insulin secretion by PKC‐dependent TRPM4 and TRPM5 activation. J Clin Invest 125, 4714–4728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Somekawa S, Fukuhara S, Nakaoka Y, Fujita H, Saito Y & Mochizuki N (2005). Enhanced functional gap junction neoformation by protein kinase A‐dependent and Epac‐dependent signals downstream of cAMP in cardiac myocytes. Circ Res 97, 655–662. [DOI] [PubMed] [Google Scholar]
- Speier S, Gjinovci A, Charollais A, Meda P & Rupnik M (2007). Cx36‐mediated coupling reduces β‐cell heterogeneity, confines the stimulating glucose concentration range, and affects insulin release kinetics. Diabetes 56, 1078–1086. [DOI] [PubMed] [Google Scholar]
- Tengholm A & Gylfe E (2017). cAMP signaling in insulin and glucagon secretion. Diabetes Obes Metab 19, 42–53. [DOI] [PubMed] [Google Scholar]
- Tian L, Gao J, Yi H, Tian B, O'Brien TD & Guo Z (2011). Comparison of exendin‐4 on beta‐cell replication in mouse and human islet gratfs. Transpl Int 24, 856–864. [DOI] [PubMed] [Google Scholar]
- Tong X, Kono T & Evans‐Molina C (2015). Nitric oxide stress and activation of AMP‐activated protein kinase impair β‐cell sarcoendoplasmic reticulum calcium ATPase 2b activity and protein stability. Cell Death Dis 6, e1790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urschel S, Hoher T, Schubert T, Alev C, Sohl G, Worsdorfer P, Asahara T, Dermietzel R, Weiler R & Willecke K (2006). Protein kinase A‐mediated phosphorylation of connexin36 in mouse retina results in decreased gap junctional communication between AII amacrine cells. J Biol Chem 281, 33163–33171. [DOI] [PubMed] [Google Scholar]
- Verrilli GM, Corbin KL & Nunemaker CS (2011). Circulating cytokines as biomarkers for early stages of type 2 diabetes in the bd/bd mouse. FASEB J 25, 1063–1063. [Google Scholar]
- Wang HY, Lin Y‐P, Mitchell CK, Ram S & O'Brien J (2015). Two‐color fluroescent analysis of connexin 36 turnover: relationship to functional plasticity. J Cell Sci 128, 3888–3897. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Video S1: Mouse Control 24h FRAP
Video S2: Mouse Control + Exendin‐4 24h FRAP
Video S3: Mouse Cytokine 24h FRAP
Video S4: Mouse Cytokine + Exendin‐4 24h FRAP
Video S5: Human Control 24h FRAP
Video S6: Human Control + Exendin‐4 24h FRAP
Video S7: Human Cytokine 24h FRAP
Video S8: Human Cytokine + Exendin‐4 24h FRAP