

Abstract
Chemerin and its G protein-coupled receptor [chemerin receptor 23 (ChemR23)] have been associated with endothelial dysfunction, inflammation, and insulin resistance. However, the role of chemerin on insulin signaling in the vasculature is still unknown. We aimed to determine whether chemerin reduces vascular insulin signaling and whether there is interplay between chemerin/ChemR23, insulin resistance, and vascular complications associated with type 2 diabetes (T2D). Molecular and vascular mechanisms were probed in mesenteric arteries and cultured vascular smooth muscle cells (VSMCs) from C57BL/6J, nondiabetic lean db/m, and diabetic obese db/db mice as well as in human microvascular endothelial cells (HMECs). Chemerin decreased insulin-induced vasodilatation in C57BL/6J mice, an effect prevented by CCX832 (ChemR23 antagonist) treatment. In VSMCs, chemerin, via oxidative stress- and ChemR23-dependent mechanisms, decreased insulin-induced Akt phosphorylation, glucose transporter 4 translocation to the membrane, and glucose uptake. In HMECs, chemerin decreased insulin-activated nitric oxide signaling. AMP-activated protein kinase phosphorylation was reduced by chemerin in both HMECs and VSMCs. CCX832 treatment of db/db mice decreased body weight, insulin, and glucose levels as well as vascular oxidative stress. CCX832 also partially restored vascular insulin responses in db/db and high-fat diet-fed mice. Our novel in vivo findings highlight chemerin/ChemR23 as a promising therapeutic target to limit insulin resistance and vascular complications associated with obesity-related diabetes.
NEW & NOTEWORTHY Our novel findings show that the chemerin/chemerin receptor 23 axis plays a critical role in diabetes-associated vascular oxidative stress and altered insulin signaling. Targeting chemerin/chemerin receptor 23 may be an attractive strategy to improve insulin signaling and vascular function in obesity-associated diabetes.
Keywords: adipokines, endothelial cells, insulin, type 2 diabetes, vascular smooth muscle
INTRODUCTION
Obesity, characterized by hypertrophy and hyperplasia of adipose tissue, is a critical risk factor for hypertension, dyslipidemia, cardiovascular disease, and type 2 diabetes (T2D) (33). Obesity is also linked to decreased sensitivity to the biological actions of insulin, a condition identified as insulin resistance (48). In addition to its central energy storage function, adipose tissue secretes several bioactive hormones and cytokines, called adipokines (51). Adipokines have autocrine/paracrine effects that influence not only adipose tissue development and function (21) but also energy homeostasis, glucose and lipid metabolism, food intake, inflammation, and vascular function (26, 51). Dysregulation of adipokine production and secretion contributes to the pathogenesis of obesity and its associated vascular complications (51).
Chemerin, also known as retinoic acid receptor responder protein 2 (RARRES2) or tazarotene-induced gene 2 protein, is highly expressed in the placenta, liver, and white adipose tissue (WAT), with a lower expression in tissues such as the lung, brown adipose tissue, heart, ovary, kidney, skeletal muscle, and pancreas (4, 14). It is secreted as an 18-kDa inactive proprotein and undergoes extracellular serine protease cleavage to generate 16-kDa active chemerin (50). Chemerin is a chemoattractant protein that binds to the G protein-coupled receptor chemokine-like receptor 1 (CMKLR1), also known as chemerin receptor 23 (ChemR23), which is expressed in macrophages, dendritic cells, and adipocytes (50) as well as in endothelial cells and vascular smooth muscle cells (VSMCs), as recently reported (46). Although chemerin also activates G protein-coupled receptor 1 (GPCR1) with similar affinity to CMKLR1 and is a ligand for a third receptor, chemokine receptor-like 2, which does not seem to activate intracellular responses, essentially all known responses to chemerin have been attributed to the activation of ChemR23 (1).
Chemerin is currently described as a biomarker for adiposity in humans. Circulating chemerin levels were shown to be strongly associated with multiple components of metabolic syndrome, including body mass index, triglycerides, high-density lipoprotein-cholesterol, and hypertension (4), and are also linked to adipogenesis (14, 34). Circulating chemerin levels are increased in numerous diseases associated with chronic inflammation (47). Serum levels of chemerin correlate with levels of proinflammatory cytokines, such as TNF-α, IL-6, and C-reactive protein (24, 47). Importantly, ChemR23 knockout mice present reduced adiposity and body mass (9), and chemerin levels are reduced by weight loss and fat reduction (9). Chemerin expression is upregulated in adipocytes of diet-induced obese mice (10, 34). Increased chemerin expression in WAT, skeletal muscle, and the liver has been reported in mouse models of obesity/diabetes, and chemerin exacerbates glucose intolerance, lowers serum insulin levels, and decreases tissue glucose uptake in obese diabetic db/db mice (10, 34). More recently, reactive oxygen species (ROS) have been demonstrated to play an important role in chemerin signaling in vascular cells. Through ROS, chemerin stimulates mitogenic and proinflammatory signaling pathways, promoting vascular damage and remodeling (45).
Although chemerin has been shown to impair insulin signaling and to induce insulin resistance in skeletal muscle cells (41) and cardiomyocytes (52), the role of chemerin in vascular insulin resistance, particularly in the context of diabetes, has not been fully elucidated. Therefore, the present study aimed to determine whether chemerin influences vascular insulin signaling and whether there is interplay between chemerin/ChemR23, insulin resistance, and vascular complications associated with T2D. We hypothesized that chemerin, through ChemR23, decreases vascular insulin signaling and that ChemR23 antagonism attenuates abnormal vascular responses to insulin in obese diabetic db/db mice.
MATERIALS AND METHODS
All experimental protocols on mice were performed in accordance with the Ethical Principles in Animal Experimentation adopted by the West of Scotland Research Ethics Service and in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals and the National Council for Animal Experimentation Control and were approved by the Ethics Committee on Animal Use from the University of São Paulo (protocol no. 12.1.1593.53.0).
Animals.
Ten- to twelve-week-old male C57BL/6J, lean nondiabetic db/m, and obese diabetic db/db mice were housed in individual cages in a room with controlled humidity and temperature (22–24°C) and 12:12-h light-dark cycles. Animals had free access to food and tap water. Animals were treated with vehicle (PEG400/cremophor) or CCX832, a ChemR23 antagonist (a gift from ChemoCentryx, Mountain View, CA, 75 mg·kg−1·day−1 for 3 wk, by oral gavage). Animals were separated into the following four groups: db/m + vehicle, db/m + CCX832, db/db + vehicle, and db/db + CCX832. In initial experiments, to confirm that the vehicle had no effects itself, the following two additional groups were included and maintained for the same 3-wk period: db/m and db/db mice without any treatment (i.e., untreated db/m and db/db mice). Because no differences were observed between the untreated and vehicle-treated groups, the remaining protocols were performed in animals treated with vehicle or CCX832. In another set of experiments, 6-wk-old male C57BL/6J mice were maintained either on a control diet (protein 22%, carbohydrate 70%, and fat 8% of energy, PragSolucoes, Jau, Brazil) or on a high-fat diet (HFD; protein 10%, carbohydrate 25%, and fat 65% of energy, PragSolucoes) for 18 wk.
Insulin sensitivity was calculated using the homeostasis model assessment-insulin resistance (HOMA-IR) index, which takes into account insulin and fasting blood glucose levels, using the following mathematical formula: HOMA-IR = fasting insulin × fasting glucose/22.5. Additional nutritional and metabolic information from the mouse models can be found in previous studies (6, 7, 42). At the end of treatment, animals were maintained under anesthesia with 2.5% isoflurane for blood collection and then culled by CO2 inhalation.
Cultured vascular cells.
VSMCs from mesenteric arteries of C57BL/6J mice were isolated and characterized as previously described (45). Subconfluent cell cultures were rendered quiescent by serum deprivation for 24 h before experimentation. Low-passage cells (passages 4–6) from different primary cultures were used in our experiments.
Human microvascular endothelial cells (HMECs; Life Technologies, Carlsbad, CA) were also studied. Endothelial cells were cultured in medium 131 supplemented with microvascular growth supplement (25 ml), gentamicin (50 µg/ml), and amphotericin B (0.25 µg/ml). For functional experiments, confluent cells were quiescent for 4 h in low-serum medium containing 0.5% FBS and subsequently stimulated according to the experimental protocol. Four to six different batches of endothelial cells were studied for each experiment. Cells were stimulated with recombinant chemerin (0.5 ng/ml, R&D Systems, Minneapolis, MN). When inhibitors were used in any protocol, parallel experiments were performed to determine the effects of the inhibitor itself. Data were included in the graphics only if the inhibitor by itself produced a significant effect.
Plasma biochemistry.
Mice were fasted for 12 h, and blood was collected immediately before euthanization in tubes containing heparin. After collection, plasma was separated by centrifugation (2,000 rpm, 10 min). Plasma was aliquoted, snap frozen, and stored at −80°C. Glucose, cholesterol, and triglycerides were determined by an automated analyzer (Roche/Hitachi cobas c systems, cobas c 311 Autoanalyser, Roche Diagnostics, Minneapolis, MN).
Chemerin and insulin levels.
Chemerin and insulin plasma levels were determined by ELISA according to instructions from the manufacturer (catalog no. MCHM00, R&D Systems, and 10-1247-01, Mercodia, Winston-Salem, NC, respectively).
Functional experiments in mesenteric arteries.
First- and second-order mesenteric resistance arteries from C57BL/6J, db/m, and db/db mice as well as from mice treated with control diet or HFD for 18 wk were cut into 2-mm ring segments and mounted in a wire myograph, as previously described (16). Myograph chambers were filled with 5 ml of physiological solution [containing (in mmol/l) 130 NaCl, 14.9 NaHCO3, 4.7 KCl, 1.18 KH2PO4, 1.17 MgSO4·7H2O, 5.5 glucose, 1.56 CaCl2·2H2O, and 0.026 EDTA] and continuously gassed with a mixture of 95% O2-5% CO2 at a temperature of 37°C. After 30 min of stabilization, the contractile ability of the preparations was assessed by adding KCl (120 mmol/l) to the organ baths. Endothelial integrity was verified by relaxation induced by ACh (1 µmol/l) in vessels precontracted with phenylephrine (2 µmol/l). Concentration-effect curves to human regular insulin (Eli Lilly, São Paulo, Brazil) were performed in arteries from all animal groups. In some experiments, vascular preparations were incubated with Tiron (ROS scavenger, 100 µmol/l), CCX832 (ChemR23 antagonist, 10 nmol/l), or YS-49 (PI3K activator, 1 µmol/l). For HFD-fed mice, CCX832 was added to the chamber 30 min before measurements.
Immunoblot analysis.
Western blot analysis was developed in cultured VSMCs from C57BL/6J mice and aortas from db/m and db/db mice treated with vehicle or CCX832, as previously described (45). The mesenteric arteries were not further explored for other experiments due to the limitation of the amount of tissue. Briefly, tissues were homogenized in lysis buffer [containing (in mmol/l) 50 sodium pyrophosphate, 50 NaF, 5 NaCl, 5 EDTA, 5 EGTA, 10 HEPES, 2 Na3VO4, and 50 PMSF with 0.5% Triton 100 and 1 mg/ml leupeptin-aprotinin-pepstatin] and then sonicated for 5 s. Proteins (50–60 µg) extracted from each lysate were separated by electrophoresis on SDS polyacrylamide gels and transferred to a nitrocellulose membrane. Nonspecific binding sites were blocked with 5% milk in Tris-buffered saline solution with Tween 20 (TBS-T) for 1 h at room temperature. Primary antibodies were diluted in TBS-T containing 3% BSA and incubated overnight at 4°C. After an incubation for 1 h with secondary antibodies in room temperature, signals were revealed with chemiluminescence, visualized by autoradiography, and quantified densitometrically with the open-source software ImageJ (https://imagej.nih.gov/ij/). Results were normalized by β-actin, α-tubulin, or respective total proteins. The antibodies used were as follows: anti-endothelial nitric oxide (NO) synthase (eNOS; no. 9572), anti-phospho-eNOS (no. 9571), anti-phospho-Akt (no. 9271), anti-Akt (no. 9272), anti-phospho-phosphatidylinositol 3-kinase (PI3K; no. 4228), anti-phospho-AMP-activated protein kinase (AMPK; no. 2531), anti-AMPK (no. 2532), anti-glucose transporter 4 (GLUT4; no. 07-1404, 1:1,000, Millipore, Burlington, MA), anti Na+-K+-ATPase (no. 3010, 1:10,000, Cell Signaling, Danvers, MA), anti-α-tubulin (T6074), and anti-β-actin (no. 2228, 1:10,000, Sigma, St. Louis, MO).
Isolation of membrane and cytosolic fractions.
After stimulation, VSMCs were treated with ice-cold hypotonic lysis buffer [10 mmol/l Tris (pH 7.4), 1.5 mmol/l MgCl2, 5 mmol/l KCl, 1 mmol/l DTT, 0.2 mmol/l sodium vanadate, 1 mmol/l PMSF, 1 g/ml aprotinin, and 1 g/ml leupeptin]. After the lysate was passed through a 25-gauge syringe needle with several rapid strokes, samples were centrifuged at 2,000 g at 4°C for 5 min. The supernatant was recentrifuged at 100,000 g at 4°C for 60 min. The pellet was resuspended in lysis buffer containing 0.1% Triton X-100 and served as the membrane fraction. Proteins were measured by the Bradford method using BSA as the standard. The membrane fraction was used to perform Western blot analysis for GLUT4 translocation.
Immunofluorescence.
Paraffin sections of the aorta (4 µm) were deparaffinized in xylene, rehydrated through graded ethanol, and washed in water. All sections were incubated in EDTA (pH 8) and boiled for 15 min at 95°C for antigen unmasking. Slides were cooled to room temperature, permeabilized in 0.5% Triton X-100 in PBS for 5 min, and blocked with 10% donkey serum and 1% BSA in 1× TBS-T for 1 h at room temperature in a humidified chamber. For 8-hydroxyguanosine immunostaining, slides were incubated overnight with anti-8-hydroxyguanosine goat polyclonal antibody (ab10802, 1:200, Abcam, Cambridge, MA) in a humidified chamber. Alexa-fluor-488-conjugated donkey anti-goat secondary antibody (A-11055, 1:300, Molecular Probes, Eugene, OR) was used after primary antibody incubation for 1 h at room temperature in the dark. Slides were treated with 0.1% Sudan black B (no. 199664, Sigma-Aldrich, St. Louis, MO) in methanol for 10 min to remove lipofuscin-mediated autofluorescence. Nuclei were counterstained with DAPI (100 µg/ml) for 10 min. Sections were mounted with a coverslip using ProLong gold antifade mounting media containing DAPI (P-36931, Molecular Probes) in the dark. Fluorescence images were captured using an Axiovert ×200 microscope with a laser-scanning module (LSM 510, Carl Zeiss, Heidelberg, Germany). Fluorescence quantification was performed using the open-source software ImageJ and determined by the average of five different fields captured from each vessel. For negative controls, goat IgG-matched isotype controls were used (sc-2028, Santa Cruz Biotechnology, Santa Cruz, CA).
NO production.
Production of NO was determined with the NO fluorescent probe diacetate 4-amino-5-methylamino-2′,7′-difluorofluorescein diacetate (DAF-FM; Life Technologies, Carlsbad, CA), as previously described (45). Endothelial cells were incubated with DAF-FM diacetate (5 µmol/l, 30 min) in serum-free media, kept in the dark, and maintained at 37°C. Cells were washed to remove excess probe, and an additional incubation for 10 min was performed to allow complete deesterification of the intracellular diacetates. Cells were stimulated with chemerin (0.5 ng/ml, R&D Systems) for 1 h and insulin (100 nmol/l), CCX832 (10 nmol/l), N-acetyl-cysteine (NAC; 10 µmol/l) and 740Y-P (PI3K/Akt signaling activator, 1 µmol/l) for 30 min. After stimulation, cells were washed with PBS and harvested by mild trypsinization (trypsin: 0.025%). Trypsin was inactivated with soybean trypsin inhibitor (0.025%) in PBS (1:1). Additional PBS (37°C) was added to a final volume of 10 ml. Cells were centrifuged for 3 min at 3,000 g. After centrifugation, the supernatant was discarded, and the cell pellet was reconstituted in PBS (250 µl). Two hundred microliters of the cell suspension were transferred to black 96-well microplates (Nunc 436034). DAF-FM nitrosylation was assessed by a fluorimeter at excitation/emission wavelengths of 495/515 nm. Fluorescence intensity was adjusted to protein concentration.
Glucose uptake measurement.
VSMCs were exposed to chemerin or vehicle, CCX832, Tiron, 5-aminoimidazole-4-carboxamide ribonucleotide (AICAR; AMPK signaling activator; 1 mmol/l), or 740Y-P in the presence of insulin stimulation and then incubated with Krebs-Ringer-HEPES buffer [containing (in mmol/l) 15 HEPES (pH 7.4), 105 NaCl, 5 KCl, 1.4 CaCl2, 1 KH2PO4, 1.4 MgSO4, and 10 NaHCO3] for 2 h. Cells were incubated with insulin (100 nmol/l) for 30 min and 0.2 mmol/l glucose 2-deoxy-D-glucose containing 1 µCi/ml of 2-deoxy-d-[3H]glucose was added over 30 min. The buffer was rapidly removed followed by three washes with ice-cold PBS. Cells were lysed with 500 µl of 0.4 mol/l NaOH for 5 min and neutralized with 500 µl HCl. The amount of radiolabeled glucose associated with the lysed cells was determined by liquid scintillation counting. To control for changes in osmotic pressure, the effect of insulin on [14C]mannitol uptake was also determined.
Data and statistical analysis.
All data are expressed as means ± SE; n represents the number of animals or experiments used. Relaxation responses are expressed as the percentage of contraction in response to phenylephrine. Individual concentration-response curves were fitted into a curve by nonlinear regression analysis. pD2 (defined as the negative logarithm of EC50 values) and maximal response (Emax) values were compared by one-way ANOVA followed by the Tukey post hoc test. Prism software (version 5.0, GraphPad Software, San Diego, CA) was used to analyze these parameters as well as to fit the sigmoidal curves. To analyze the difference between the two groups, an unpaired t-test was used. P values of <0.05 were considered significant.
RESULTS
Chemerin decreases insulin-induced dilation of mesenteric resistance arteries.
As shown in Fig. 1A, chemerin decreased insulin-induced relaxation of mesenteric arteries from C57BL/6 mice, an effect blocked by the ChemR23 antagonist CCX832 (Fig. 1B). An activator of PI3K signaling (YS-49) and a ROS scavenger (Tiron) reversed the effects of chemerin on insulin-induced vasodilatation (Fig. 1, C and D).
Fig. 1.

Chemerin (Chem) decreases insulin-induced vasodilatation via chemerin receptor 23 (ChemR23)-, Akt/nitric oxide (NO)-, and oxidative stress-dependent mechanisms. Endothelium-intact mesenteric arteries from C57BL/6J mice were incubated with chemerin (0.5 ng/ml) or vehicle for 1 h. Relaxation response to insulin was evaluated in the absence (A) and presence (B) of CCX832 (CCX; ChemR23 antagonist), YS-49 [phosphatidylinositol 3-kinase activator (PI3K); C] or Tiron (ROS scavenger; D), which were added 30 min before vehicle or chemerin. Vehicle and chemerin curves in A–D were originated from the same sets of experiments. E: phosphorylation of endothelial NO synthase (eNOS; Ser1177) was determined by Western blot analysis in endothelial cells stimulated with chemerin (0.5 ng/ml, 1 h) and insulin (30 min) in the absence or presence of CCX832, N-acetyl-cysteine (NAC; ROS scavenger), or 740Y-P (PI3K activator). peNOS, phosphorylated eNOS; teNOS, total eNOS. Values were normalized by total NOS expression. F: NO production was determined by diacetate 4-amino-5-methylamino-2′,7′-difluorofluorescein diacetate fluorescence in endothelial cells, and values were normalized by the protein amount. Results are means ± SE of 5−6 experiments. Data were analyzed using one-way ANOVA followed by a post hoc Tukey test. *P < 0.05 vs. vehicle; #P < 0.05 vs. chemerin; σP < 0.05 vs. insulin; φP < 0.05 vs. chemerin + insulin.
Chemerin decreases insulin-induced NO signaling activation in endothelial cells.
To verify whether chemerin affects NO signaling, HMECs were exposed to chemerin (0.5 mg/ml) and probed for eNOS phosphorylation. Chemerin decreased basal and insulin-induced eNOS phosphorylation of the active site (Ser1177; Fig. 1E). These effects were attenuated both by a ROS scavenger (NAC) and a PI3K activator (740Y-P; Fig. 1E), indicating that oxidative stress mechanisms and insulin downstream signaling are involved in chemerin-induced decreased NO bioavailability, leading to endothelial dysfunction.
To determine whether reduced eNOS phosphorylation was associated with decreased NO production, levels of NO were estimated in insulin and chemerin-stimulated HMECs. Chemerin decreased NO levels in HMECs. Insulin increased NO levels in these cells, and in the presence of chemerin, insulin-induced NO production was significantly reduced. This effect involves ChemR23-, PI3K signaling-, and ROS-dependent mechanisms since CCX832, 740Y-P, and NAC partially inhibited the effects of chemerin (Fig. 1F).
Chemerin decreases insulin signaling in VSMCs.
In addition to its effects on endothelial cells, chemerin decreased insulin signaling in VSMCs. Insulin increased Akt phosphorylation in VSMCs, and chemerin decreased insulin-induced Akt phosphorylation via ChemR23- and ROS-dependent mechanisms (Fig. 2A).
Fig. 2.

Chemerin (Ch) decreases vascular insulin signaling in vascular smooth muscle cells (VSMCs). Phosphorylation of Akt (A) and glucose transporter 4 (GLUT4) translocation to the membrane (B) as well as phosphorylation (p) of AMP-activated protein kinase (AMPK) in VSMCs (C) and human microvascular endothelial cells (ECs; D) were determined by Western blot analysis. Cells were pretreated with CCX832, N-acetyl-cysteine (NAC), or 740Y-P added 30 min before the stimulation with chemerin and/or insulin. Values were normalized by total protein or Na+-K+-ATPase. To keep the same sequence of experimental groups in all graphs, representative Western blot figures for Akt were spliced as indicated. Data are expressed as means ± SE of 4–6 experiments. Data were analyzed using one-way ANOVA followed by a post hoc Tukey test or t-test. *P < 0.05 vs. vehicle (Veh); #P < 0.05 vs. chemerin; σP < 0.05 vs. insulin; φP < 0.05 vs. chemerin + insulin.
To further investigate chemerin effects on insulin signaling, GLUT4 translocation from the intracellular membrane compartments to the membrane was determined. In VSMCs, chemerin decreased insulin-induced GLUT4 translocation to the membrane (Fig. 2B), which was blocked by CCX832 and 740Y-P (Fig. 2B). This suggests that ChemR23 and PI3K activation contributes to the effects of chemerin on GLUT4 translocation in VSMCs.
Chemerin reduces insulin-stimulated glucose uptake by VSMCs.
We evaluated the effects of chemerin on AMPK phosphorylation, another mechanism for glucose uptake. Chemerin decreased AMPK phosphorylation (Thr172) in both VSMCs and HMECs (Fig. 2, C and D). To evaluate whether chemerin also affects insulin-induced glucose uptake, 2-deoxyglucose uptake was measured in VSMCs exposed to 2 μCi/ml 2-[3H]DG in the presence of unlabeled 2-DG. In VSMCs, chemerin decreased insulin-induced glucose uptake (Fig. 3) through ROS generation, AMPK, and PI3K/Akt signaling since Tiron, AICAR, CCX832, and 740Y-P reversed the effects of chemerin on glucose transport (Fig. 3).
Fig. 3.
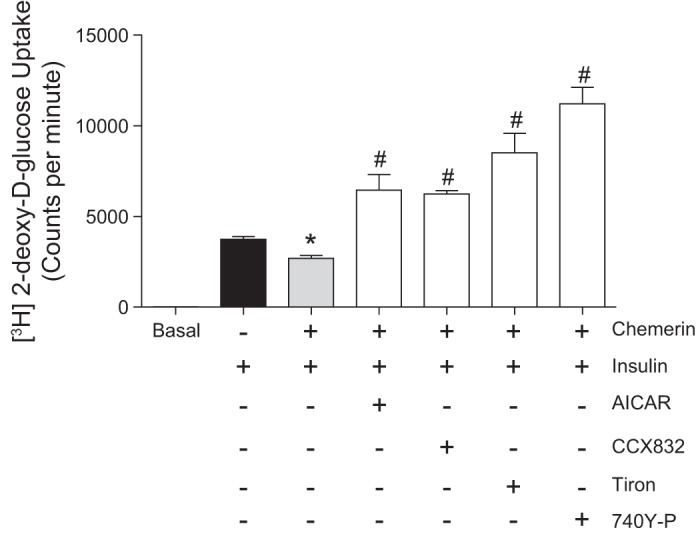
Chemerin reduces insulin-stimulated glucose uptake in vascular smooth muscle cells. 2-Deoxyglucose (2-DG) uptake was measured by exposing vascular smooth muscle cells to 2 μCi/ml 2-[3H]DG in the presence of unlabeled 2-DG. Cells were treated with chemerin, Tiron, 5-aminoimidazole-4-carboxamide ribonucleotide (AICAR; AMP-activated protein kinase activator), CCX832, and 740Y-P before the stimulation with insulin. Values are expressed as means ± SE of 5−6 experiments. Data were analyzed using one-way ANOVA followed by a post hoc Tukey test. *P < 0.05 vs. vehicle; #P < 0.05 vs. chemerin.
ChemR23 antagonism decreases body weight and insulin and glucose levels in db/db mice.
Since chemerin influences vascular insulin signaling, we investigated whether ChemR23 antagonism attenuates diabetes-related vascular dysfunction and insulin resistance. Plasma chemerin levels were significantly increased in diabetic obese db/db mice (Fig. 4A). CCX832 treatment slightly reduced body weight of db/db mice compared with vehicle-treated db/db mice (Fig. 4B). In addition, CCX-treated db/db mice exhibited decreased plasma levels of glucose and insulin versus vehicle-treated db/db mice (Fig. 4, C and D). The HOMA index indicated that CCX832 improved insulin sensitivity in db/db mice (Fig. 4E). The increased cholesterol and triglyceride levels in db/db mice were reduced by CCX832. No differences in the lipid profile were observed in db/m mice treated with vehicle or CCX832 (Fig. 4, F and G).
Fig. 4.

Chemerin receptor 23 antagonism decreases body weight and insulin and glucose levels in db/db mice. A: plasma levels of chemerin were measured by ELISA in db/m and db/db mice. B: body weights of untreated, vehicle-treated, and CCX832-treated db/m and db/db mice were measured through 3 wk of treatment. C and D: glucose (C) and insulin (D) levels in vehicle- and CCX832-treated db/m and db/db mice. E: insulin sensitivity, calculated using the homeostasis model assessment of insulin resistance (HOMA-IR) index, as well as cholesterol (F) and triglyceride (G) levels in vehicle- and CCX832-treated db/m and db/db mice. Values are expressed as means ± SE of 5−8 experiments. Data were analyzed using one-way ANOVA followed by a post hoc Tukey test or t-test. *P < 0.05 vs. db/m mice; ∞P < 0.05 vs. vehicle-treated db/db mice.
CCX832 partially reverses dysfunctional insulin responses in vessels from db/db mice.
As shown in Fig. 5A, insulin-induced dilation was decreased in mesenteric arteries from vehicle-treated db/db mice versus vehicle-treated db/m mice. CCX832 treatment partially improved insulin-induced vasodilation in db/db mice (Fig. 5A). Moreover, Akt (Ser473) phosphorylation was reduced in aortas from vehicle-treated db/db mice versus vehicle-treated db/m mice and CCX832-treated db/db mice (Fig. 5B), indicating beneficial effects of in vivo treatment with CCX832 on insulin signaling.
Fig. 5.

CCX832 attenuates vascular insulin dysfunction in db/db and high-fat diet (HFD)-fed mice. A: concentration-effect curves to insulin determined in mesenteric arteries from vehicle- and CCX832-treated db/m and db/db mice. B: phosphorylation of Akt in aortas from vehicle- and CCX832-treated db/m and db/db mice determined by Western blot analysis. Values were normalized by total protein expression. pAkt, phosphorylated Akt; tAkt, total Akt. C: plasma levels of chemerin measured by ELISA in control diet- or HFD-fed mice. D: concentration-effect curves to insulin determined in mesenteric arteries from control diet- and HFD-treated mice. Curves were performed in the presence and absence of CCX832 added 30 min before measurements. Phe, phenylephrine. Results are expressed as means ± SE of 5−8 experiments. Data were analyzed using one-way ANOVA followed by a post hoc Tukey test or t-test. *P < 0.05 vs. db/m mice; ∞P < 0.05 vs. vehicle-treated db/db mice; θP < 0.05 vs. the control diet; †P < 0.05 vs. the HFD.
Additionally, as shown in Fig. 5C, increased levels of chemerin were also observed in HFD-fed mice. The ChemR23 antagonist CCX832 attenuated impaired insulin-induced dilatation in mesenteric arteries from HFD-fed mice (Fig. 5D).
CCX832 reduces vascular oxidative stress in db/db mice.
Considering that chemerin increases ROS generation (45) and that oxidative stress accounted for most of the effects of chemerin upon vascular insulin signaling, we investigated whether ChemR23 antagonism attenuates ROS generation in db/db mice. Diabetic mice presented increased vascular levels of DNA oxidation products compared with vehicle- and CCX832-treated db/m mice (Fig. 6, A and B). This was reduced by treatment of db/db mice with CCX832 (Fig. 6, A and B).
Fig. 6.

CCX832 reduces vascular oxidative stress in db/db mice. A and B: representative images (A) and quantitative analysis (B) of 8-hydroxyguanosine (8-OHG)-positive nuclei in endothelium-intact aortas from vehicle- and CCX832-treated db/m and db/db mice. Scale bar = 20 µm. Magnification: ×40. Results are means ± SE of 5 experiments. Data were analyzed using one-way ANOVA followed by a post hoc Tukey test or t-test. *P < 0.05 vs. db/m mice; ∞P < 0.05 vs. vehicle-treated db/db mice.
DISCUSSION
Our results demonstrated that 1) chemerin decreases insulin-induced vasodilation by mechanisms involving ChemR23 activation, disruption of PI3K/Akt signaling, and oxidative stress; 2) chemerin decreases insulin-induced eNOS phosphorylation and NO production in endothelial cells through mechanisms that affect ROS generation and PI3K/Akt signaling; 3) chemerin decreases activation of insulin signaling pathways via ChemR23 and oxidative stress in VSMCs; 4) ChemR23 antagonism with CCX832 decreases glucose and insulin levels in db/db mice; 5) CCX832 partially restores insulin-induced vasodilatation and improves insulin signaling in diabetic obese mice; and 6) CCX832 decreases vascular oxidative stress in db/db mice. These novel findings show that the chemerin/ChemR23 axis plays a critical role in diabetes-associated vascular oxidative stress and altered insulin signaling.
Chemerin was first described as a chemoattractant agent, promoting chemotaxis of leukocyte populations that express ChemR23. Later on, chemerin was associated with regulation of key effectors of the glucose and lipid metabolism in mature adipocytes (11) as well as a regulator of adipogenesis in 3T3-L1 cells (14). Regarding vascular homeostasis, chemerin has been shown to promote proliferation of endothelial cells and release of matrix metalloproteinase -2 and matrix metalloproteinase-9, suggesting a contribution of the chemerin/ChemR23 system in angiogenesis. In cocultures of fibroblast and endothelial cells, chemerin promotes the formation of endothelial tubules in a MAPK-dependent manner (8). On the other hand, in obesity, production of adipokines triggers chronic inflammation and leads to increased levels of proinflammatory cytokines (20), which can modify insulin signaling. TNF-α and IL-6, for example, are good predictors of the development of T2D (13). Of importance, chemerin has been associated with upregulation of IL-1β, TNF-α, IL-6, and monocyte chemoattractant protein-1 (2, 45), an additional and potential mechanism, whereby chemerin signals may modulate vascular oxidative stress and altered insulin signaling. TNF-α, for example, has been identified as a regulator of insulin sensitivity (40). In 3T3-L1 adipocytes, IL-6 reduces expression of insulin receptor substrate (IRS-1) and GLUT4 (36). Similarly, increased levels of TNF-α reduce IRS-1 phosphorylation and decrease insulin signal transduction in skeletal muscle and adipose tissue from obese rats (18). Accordingly, our data show that chemerin, a proinflammatory adipokine, decreases insulin-induced phosphorylation of Akt as well as reduces GLUT4 translocation to the membrane and insulin-induced glucose uptake in VSMCs from control mice. Molecular effects of chemerin in vascular insulin signaling are associated with a decrease in insulin-induced vasodilation, by mechanisms that affect Akt signaling and oxidative status. Together, these findings suggest that the adipokine chemerin confers an insulin-resistant phenotype to VSMCs.
Corroborating our data, a recent study (52) has demonstrated that chemerin decreases glucose uptake and Akt phosphorylation in insulin-stimulated cardiomyocytes. Inhibition of ERK1/2 MAPK partially prevented chemerin-induced impairment of Akt phosphorylation and glucose uptake (52). In addition, Jager et al. (19) have shown that IL-1β reduces insulin-induced Akt phosphorylation (Thr308). Similarly, IL-1β decreases IRS-1 and Akt protein expression in adipocytes, which leads to lower insulin-induced glucose uptake.
Furthermore, db/db mice treated with ChemR23 antagonist (CCX832) present a slight decrease in body weight, supporting the suggestion that chemerin regulates adipogenesis via ChemR23 (34). However, the effects of depletion or inhibition of ChemR23 upon body weight are still controversial. Whereas Rouger et al. (37) did not observe changes in body weight or fat mass in ChemR23 knockout mice, Ernst et al. (9) showed that CMKLR1−/− mice have lower food intake, total body mass, and percent of body fat compared with wild-type control mice.
In addition to considering ChemR23 blockade effects on body weight, it is important to consider metabolic aspects, such as glucose and insulin levels. CCX832-treated db/db mice exhibited a significant decrease in plasma glucose and insulin levels and HOMA index, strengthening the notion that chemerin contributes to insulin resistance in this model of obesity related to T2D. In addition, cholesterol and triglyceride levels in db/db mice were also reduced by CCX832. Of importance, significantly increased levels of chemerin were observed both in db/db and HFD-treated mice, reinforcing the idea that this adipokine is linked to glucose homeostasis and metabolic disorders in obesity and T2D (10). Ernst et al. (9) demonstrated that CMKLR1−/− mice fed with a HFD present reduced blood glucose and serum insulin. However, CMKLR1 loss is associated with glucose intolerance, which was linked to decreased glucose-stimulated insulin secretion. On the other hand, Rouger et al. (37) verified that glucose tolerance was not affected in young ChemR23 knockout mice.
Classically, insulin metabolic signaling results in vasodilatation via increased NO production and production of prostaglandins and EDHFs (30). In addition to reducing insulin-induced vasodilatation in nonobese mice by mechanisms involving the PI3K/Akt pathway and oxidative stress, chemerin also reduces eNOS phosphorylation (Ser1177) and decreases insulin-stimulated NO production in endothelial cells, also by oxidative stress-mediated events. These effects of chemerin may be directly involved in vascular insulin dysfunction associated with diabetes and obesity. It has been proposed that resistance to the vascular effects of insulin selectively involves the PI3K/Akt/NO signaling pathway (29). In endothelial cells, blockade of the PI3K pathway induces insulin resistance, which, in turn, blunts production of NO (28). As demonstrated in this study, most of chemerin effects are mediated by ChemR23, since its antagonism partially restores the decreased insulin-induced vasodilatation in both obesity models, db/db and HFD-fed mice, and also chemerin decreases vascular Akt phosphorylation in db/db mice.
Oxidative stress contributes to insulin resistance, particularly in skeletal muscle, as well as dysfunction of pancreatic β cells (12). Cellular and systemic disorders that may contribute to overproduction of ROS, including hyperglycemia, dyslipidemia, endoplasmic reticulum stress, advanced glycation end products, NOS, and lipid peroxides, can activate factors associated with insulin resistance (43). In humans, there is a positive association between serum markers of oxidative stress and the degree of insulin resistance (32). In experimental animal models, oxidative stress interrupts IRS-1 and PI3K activation induced by insulin and impairs the translocation of GLUT4 in 3T3-L1 adipocytes (44). In adipose tissue from healthy men with excessive caloric intake, oxidative stress induces insulin resistance partially via carbonylation and oxidation-induced inactivation of GLUT4 (3). Hence, increased ROS generation in vessels from db/db mice may be associated with the decreased insulin signaling observed in those animals. In addition, chemerin/ChemR23 axis may contribute to insulin resistance since ChemR23 antagonism attenuates ROS generation and also restores insulin signaling in vessels from db/db mice. Furthermore, our findings clearly show the involvement of oxidative stress upon chemerin-induced insulin signaling impairment since antioxidants attenuate the reduced Akt phosphorylation as well as the decreased glucose uptake and insulin-induced vasodilatation observed in VSMCs and arteries challenged with chemerin.
This study also demonstrates that chemerin decreases AMPK phosphorylation both in HAECs and VSMCs. AMPK is a serine-threonine kinase that has emerged as an important mediator of glucose metabolism, and AMPK-activating drugs are potentially useful in T2D treatment. It has already been established that acute AMPK activation stimulates glucose uptake by skeletal muscle via both GLUT1 and GLUT4 (39). In cardiomyocytes, AMPK activation increases GLUT4 expression in a PKC-dependent manner (31). Moreover, AMPK activation by AICAR increases heart and skeletal muscle glucose uptake (27, 38), possibly by increasing GLUT4 expression (17). Most importantly, overexpression of the human GLUT4 gene protects db/db mice from insulin resistance and diabetes (5). Similarly, our findings show that AICAR (and also PI3K/Akt activation) reverses the effects of chemerin on insulin-induced glucose uptake in VSMCs from normal mice, in accordance with the diminished AMPK phosphorylation in response to chemerin.
Cross-talk between GPCRs and receptor tyrosine kinases (RTKs) is increasingly being recognized as a major mechanism regulating complex signaling pathways (25). ChemR23 is a GPCR that acts through Gi to activate various intracellular signaling pathways, such as ERK1/2 (49). The insulin receptor belongs to the family of RTKs, which plays a critical role in development, cell division, and metabolism. Activation of RTK family members results in autophosphorylation and phosphorylation of selective protein substrates (IRS) exclusively on tyrosine residues. Recently, various likely mechanisms for the transactivation between GPCRs and RTKs have emerged. Molecules such as PKC, Src, and ROS as well as adaptor/scaffold proteins such as Gab1, IRS-1, and GIT1 have been implicated as mediators of GPCR ligand-induced RTK transactivation (23). This study shows that chemerin, through ChemR23, modulates IRS-1 serine phosphorylation and the subsequent signaling cascade. Recently, chemerin has been shown to stimulate bone marrow-derived mesenchymal stromal cells migration via ERK1/2, p38 MAPK, and JNK-II kinases in a PKC-dependent manner (22). A limitation of the present study is that mechanisms underlying ChemR23 and insulin receptor/IRS cross-talk have not been explored. Thus, further studies are needed to understand and dissect specific proteins linking ChemR23 and insulin receptor/IRS signaling.
In conclusion, our data indicate that the chemerin/ChemR23 system plays an important role in diabetes-associated impaired vascular insulin signaling and oxidative stress, suggesting its involvement in the pathogenesis of vascular insulin resistance. Antagonism of the system ameliorates vascular dysfunction and normalizes insulin signaling in a T2D animal model. Targeting chemerin/ChemR23 may be an attractive strategy to improve insulin signaling and vascular function in obesity-associated diabetes.
Perspectives and Significance
Adipokines participate in many physiological processes implicated in cardiovascular complications associated with obesity and diabetes. Advances in the understanding of the role of adipocyte-derived factors in obesity-associated vascular dysfunction may uncover mechanisms involved in adiposity-related diseases. The chemerin/ChemR23 system plays an important role in diabetes-associated impaired vascular insulin signaling and oxidative stress, suggesting its involvement in the pathogenesis of vascular insulin resistance. Antagonism of the system ameliorates vascular dysfunction and normalizes insulin signaling in a T2D animal model. We believe that targeting chemerin/ChemR23 may be an attractive therapeutic strategy to improve insulin signaling and vascular function in obesity-associated diabetes.
GRANTS
This work was supported by Fundação de Amparo à Pesquisa do Estado de São Paulo Grants 2012/13144-8 and 2015/01630-3 (to K. B. Neves) and 2013/08216-2 (from the Center of Research in Inflammatory Diseases), Coordenação de Aperfeiçoamento de Pessoal de Nível Superior Grant 2053-13-6, and British Heart Foundation Grant RE/13/5/30177 (British Heart Foundation Chair CH/12/429762].
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
K.B.N., A.N.D.C., R.A.-L., K.Y.H., R.M.d.C., N.S.L., A.C.M., A.M.d.O., R.M.T., and R.C.T. conceived and designed research; K.B.N., R.A.-L., K.Y.H., and R.M.d.C. performed experiments; K.B.N., R.A.-L., and R.M.d.C. analyzed data; K.B.N., A.N.D.C., N.S.L., A.C.M., and R.C.T. interpreted results of experiments; K.B.N. prepared figures; K.B.N. drafted manuscript; K.B.N., A.N.D.C., R.A.-L., N.S.L., A.C.M., A.M.d.O., R.M.T., and R.C.T. edited and revised manuscript; K.B.N., A.N.D.C., R.A.-L., K.Y.H., R.M.d.C., N.S.L., A.C.M., A.M.d.O., R.M.T., and R.C.T. approved final version of manuscript.
ACKNOWLEDGMENTS
We thank ChemoCentryx (Mountain View, CA) for providing the CCX832 compound and consultation, Carla Pavan for technical support with the animals, and Aikaterini Anagnostopoulou for immunofluorescence support.
REFERENCES
- 1.Barnea G, Strapps W, Herrada G, Berman Y, Ong J, Kloss B, Axel R, Lee KJ. The genetic design of signaling cascades to record receptor activation. Proc Natl Acad Sci USA 105: 64–69, 2008. doi: 10.1073/pnas.0710487105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Berg V, Sveinbjörnsson B, Bendiksen S, Brox J, Meknas K, Figenschau Y. Human articular chondrocytes express ChemR23 and chemerin; ChemR23 promotes inflammatory signalling upon binding the ligand chemerin(21-157). Arthritis Res Ther 12: R228, 2010. doi: 10.1186/ar3215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Boden G, Homko C, Barrero CA, Stein TP, Chen X, Cheung P, Fecchio C, Koller S, Merali S. Excessive caloric intake acutely causes oxidative stress, GLUT4 carbonylation, and insulin resistance in healthy men. Sci Transl Med 7: 304re7, 2015. doi: 10.1126/scitranslmed.aac4765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bozaoglu K, Bolton K, McMillan J, Zimmet P, Jowett J, Collier G, Walder K, Segal D. Chemerin is a novel adipokine associated with obesity and metabolic syndrome. Endocrinology 148: 4687–4694, 2007. doi: 10.1210/en.2007-0175. [DOI] [PubMed] [Google Scholar]
- 5.Brozinick JT Jr, McCoid SC, Reynolds TH, Nardone NA, Hargrove DM, Stevenson RW, Cushman SW, Gibbs EM. GLUT4 overexpression in db/db mice dose-dependently ameliorates diabetes but is not a lifelong cure. Diabetes 50: 593–600, 2001. doi: 10.2337/diabetes.50.3.593. [DOI] [PubMed] [Google Scholar]
- 6.da Costa RM, Fais RS, Dechandt CRP, Louzada-Junior P, Alberici LC, Lobato NS, Tostes RC. Increased mitochondrial ROS generation mediates the loss of the anti-contractile effects of perivascular adipose tissue in high-fat diet obese mice. Br J Pharmacol 174: 3527–3541, 2017. doi: 10.1111/bph.13687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.da Costa RM, Neves KB, Mestriner FL, Louzada-Junior P, Bruder-Nascimento T, Tostes RC. TNF-α induces vascular insulin resistance via positive modulation of PTEN and decreased Akt/eNOS/NO signaling in high fat diet-fed mice. Cardiovasc Diabetol 15: 119, 2016. doi: 10.1186/s12933-016-0443-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dixelius J, Makinen T, Wirzenius M, Karkkainen MJ, Wernstedt C, Alitalo K, Claesson-Welsh L. Ligand-induced vascular endothelial growth factor receptor-3 (VEGFR-3) heterodimerization with VEGFR-2 in primary lymphatic endothelial cells regulates tyrosine phosphorylation sites. J Biol Chem 278: 40973–40979, 2003. doi: 10.1074/jbc.M304499200. [DOI] [PubMed] [Google Scholar]
- 9.Ernst MC, Haidl ID, Zúñiga LA, Dranse HJ, Rourke JL, Zabel BA, Butcher EC, Sinal CJ. Disruption of the chemokine-like receptor-1 (CMKLR1) gene is associated with reduced adiposity and glucose intolerance. Endocrinology 153: 672–682, 2012. doi: 10.1210/en.2011-1490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ernst MC, Issa M, Goralski KB, Sinal CJ. Chemerin exacerbates glucose intolerance in mouse models of obesity and diabetes. Endocrinology 151: 1998–2007, 2010. doi: 10.1210/en.2009-1098. [DOI] [PubMed] [Google Scholar]
- 11.Ernst MC, Sinal CJ. Chemerin: at the crossroads of inflammation and obesity. Trends Endocrinol Metab 21: 660–667, 2010. doi: 10.1016/j.tem.2010.08.001. [DOI] [PubMed] [Google Scholar]
- 12.Evans JL, Goldfine ID, Maddux BA, Grodsky GM. Oxidative stress and stress-activated signaling pathways: a unifying hypothesis of type 2 diabetes. Endocr Rev 23: 599–622, 2002. doi: 10.1210/er.2001-0039. [DOI] [PubMed] [Google Scholar]
- 13.Fain JN, Bahouth SW, Madan AK. TNFα release by the nonfat cells of human adipose tissue. Int J Obes Relat Metab Disord 28: 616–622, 2004. doi: 10.1038/sj.ijo.0802594. [DOI] [PubMed] [Google Scholar]
- 14.Goralski KB, McCarthy TC, Hanniman EA, Zabel BA, Butcher EC, Parlee SD, Muruganandan S, Sinal CJ. Chemerin, a novel adipokine that regulates adipogenesis and adipocyte metabolism. J Biol Chem 282: 28,175–28,188, 2007. doi: 10.1074/jbc.M700793200. [DOI] [PubMed] [Google Scholar]
- 16.Halpern W, Mulvany MJ, Warshaw DM. Mechanical properties of smooth muscle cells in the walls of arterial resistance vessels. J Physiol 275: 85–101, 1978. doi: 10.1113/jphysiol.1978.sp012179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Holmes BF, Kurth-Kraczek EJ, Winder WW. Chronic activation of 5′-AMP-activated protein kinase increases GLUT-4, hexokinase, and glycogen in muscle. J Appl Physiol 87: 1990–1995, 1999. doi: 10.1152/jappl.1999.87.5.1990. [DOI] [PubMed] [Google Scholar]
- 18.Hotamisligil GS, Peraldi P, Budavari A, Ellis R, White MF, Spiegelman BM. IRS-1-mediated inhibition of insulin receptor tyrosine kinase activity in TNF-α- and obesity-induced insulin resistance. Science 271: 665–668, 1996. doi: 10.1126/science.271.5249.665. [DOI] [PubMed] [Google Scholar]
- 19.Jager J, Grémeaux T, Cormont M, Le Marchand-Brustel Y, Tanti JF. Interleukin-1beta-induced insulin resistance in adipocytes through down-regulation of insulin receptor substrate-1 expression. Endocrinology 148: 241–251, 2007. doi: 10.1210/en.2006-0692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Karalis KP, Giannogonas P, Kodela E, Koutmani Y, Zoumakis M, Teli T. Mechanisms of obesity and related pathology: linking immune responses to metabolic stress. FEBS J 276: 5747–5754, 2009. doi: 10.1111/j.1742-4658.2009.07304.x. [DOI] [PubMed] [Google Scholar]
- 21.Kougias P, Chai H, Lin PH, Yao Q, Lumsden AB, Chen C. Effects of adipocyte-derived cytokines on endothelial functions: implication of vascular disease. J Surg Res 126: 121–129, 2005. doi: 10.1016/j.jss.2004.12.023. [DOI] [PubMed] [Google Scholar]
- 22.Kumar JD, Holmberg C, Kandola S, Steele I, Hegyi P, Tiszlavicz L, Jenkins R, Beynon RJ, Peeney D, Giger OT, Alqahtani A, Wang TC, Charvat TT, Penfold M, Dockray GJ, Varro A. Increased expression of chemerin in squamous esophageal cancer myofibroblasts and role in recruitment of mesenchymal stromal cells. PLoS One 9: e104877, 2014. doi: 10.1371/journal.pone.0104877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lee J, Pilch PF. The insulin receptor: structure, function, and signaling. Am J Physiol Cell Physiol 266: C319–C334, 1994. doi: 10.1152/ajpcell.1994.266.2.C319. [DOI] [PubMed] [Google Scholar]
- 24.Lehrke M, Becker A, Greif M, Stark R, Laubender RP, von Ziegler F, Lebherz C, Tittus J, Reiser M, Becker C, Göke B, Leber AW, Parhofer KG, Broedl UC. Chemerin is associated with markers of inflammation and components of the metabolic syndrome but does not predict coronary atherosclerosis. Eur J Endocrinol 161: 339–344, 2009. doi: 10.1530/EJE-09-0380. [DOI] [PubMed] [Google Scholar]
- 25.Lowes VL, Ip NY, Wong YH. Integration of signals from receptor tyrosine kinases and g protein-coupled receptors. Neurosignals 11: 5–19, 2002. doi: 10.1159/000057317. [DOI] [PubMed] [Google Scholar]
- 26.Matsuzawa Y. White adipose tissue and cardiovascular disease. Best Pract Res Clin Endocrinol Metab 19: 637–647, 2005. doi: 10.1016/j.beem.2005.07.001. [DOI] [PubMed] [Google Scholar]
- 27.Merrill GF, Kurth EJ, Hardie DG, Winder WW. AICA riboside increases AMP-activated protein kinase, fatty acid oxidation, and glucose uptake in rat muscle. Am J Physiol Endocrinol Metab 273: E1107–E1112, 1997. doi: 10.1152/ajpendo.1997.273.6.E1107. [DOI] [PubMed] [Google Scholar]
- 28.Montagnani M, Golovchenko I, Kim I, Koh GY, Goalstone ML, Mundhekar AN, Johansen M, Kucik DF, Quon MJ, Draznin B. Inhibition of phosphatidylinositol 3-kinase enhances mitogenic actions of insulin in endothelial cells. J Biol Chem 277: 1794–1799, 2002. doi: 10.1074/jbc.M103728200. [DOI] [PubMed] [Google Scholar]
- 29.Muniyappa R, Sowers JR. Role of insulin resistance in endothelial dysfunction. Rev Endocr Metab Disord 14: 5–12, 2013. doi: 10.1007/s11154-012-9229-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Muniyappa R, Yavuz S. Metabolic actions of angiotensin II and insulin: a microvascular endothelial balancing act. Mol Cell Endocrinol 378: 59–69, 2013. doi: 10.1016/j.mce.2012.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nishino Y, Miura T, Miki T, Sakamoto J, Nakamura Y, Ikeda Y, Kobayashi H, Shimamoto K. Ischemic preconditioning activates AMPK in a PKC-dependent manner and induces GLUT4 up-regulation in the late phase of cardioprotection. Cardiovasc Res 61: 610–619, 2004. doi: 10.1016/j.cardiores.2003.10.022. [DOI] [PubMed] [Google Scholar]
- 32.Paolisso G, D’Amore A, Volpe C, Balbi V, Saccomanno F, Galzerano D, Giugliano D, Varricchio M, D’Onofrio F. Evidence for a relationship between oxidative stress and insulin action in non-insulin-dependent (type II) diabetic patients. Metabolism 43: 1426–1429, 1994. doi: 10.1016/0026-0495(94)90039-6. [DOI] [PubMed] [Google Scholar]
- 33.Poirier P, Giles TD, Bray GA, Hong Y, Stern JS, Pi-Sunyer FX, Eckel RH. Obesity and cardiovascular disease: pathophysiology, evaluation, and effect of weight loss. Arterioscler Thromb Vasc Biol 26: 968–976, 2006. doi: 10.1161/01.ATV.0000216787.85457.f3. [DOI] [PubMed] [Google Scholar]
- 34.Roh SG, Song SH, Choi KC, Katoh K, Wittamer V, Parmentier M, Sasaki S. Chemerin—a new adipokine that modulates adipogenesis via its own receptor. Biochem Biophys Res Commun 362: 1013–1018, 2007. doi: 10.1016/j.bbrc.2007.08.104. [DOI] [PubMed] [Google Scholar]
- 36.Rotter V, Nagaev I, Smith U. Interleukin-6 (IL-6) induces insulin resistance in 3T3-L1 adipocytes and is, like IL-8 and tumor necrosis factor-α, overexpressed in human fat cells from insulin-resistant subjects. J Biol Chem 278: 45777–45784, 2003. doi: 10.1074/jbc.M301977200. [DOI] [PubMed] [Google Scholar]
- 37.Rouger L, Denis GR, Luangsay S, Parmentier M. ChemR23 knockout mice display mild obesity but no deficit in adipocyte differentiation. J Endocrinol 219: 279–289, 2013. doi: 10.1530/JOE-13-0106. [DOI] [PubMed] [Google Scholar]
- 38.Russell RR III, Bergeron R, Shulman GI, Young LH. Translocation of myocardial GLUT-4 and increased glucose uptake through activation of AMPK by AICAR. Am J Physiol Heart Circ Physiol 277: H643–H649, 1999. doi: 10.1152/ajpheart.1999.277.2.H643. [DOI] [PubMed] [Google Scholar]
- 39.Rutter GA, Da Silva Xavier G, Leclerc I. Roles of 5′-AMP-activated protein kinase (AMPK) in mammalian glucose homoeostasis. Biochem J 375: 1–16, 2003. doi: 10.1042/bj20030048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sell H, Dietze-Schroeder D, Eckel J. The adipocyte-myocyte axis in insulin resistance. Trends Endocrinol Metab 17: 416–422, 2006. doi: 10.1016/j.tem.2006.10.010. [DOI] [PubMed] [Google Scholar]
- 41.Sell H, Laurencikiene J, Taube A, Eckardt K, Cramer A, Horrighs A, Arner P, Eckel J. Chemerin is a novel adipocyte-derived factor inducing insulin resistance in primary human skeletal muscle cells. Diabetes 58: 2731–2740, 2009. doi: 10.2337/db09-0277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Shalaby F, Rossant J, Yamaguchi TP, Gertsenstein M, Wu XF, Breitman ML, Schuh AC. Failure of blood-island formation and vasculogenesis in Flk-1-deficient mice. Nature 376: 62–66, 1995. doi: 10.1038/376062a0. [DOI] [PubMed] [Google Scholar]
- 43.Solinas G, Karin M. JNK1 and IKKβ: molecular links between obesity and metabolic dysfunction. FASEB J 24: 2596–2611, 2010. doi: 10.1096/fj.09-151340. [DOI] [PubMed] [Google Scholar]
- 44.Tirosh A, Potashnik R, Bashan N, Rudich A. Oxidative stress disrupts insulin-induced cellular redistribution of insulin receptor substrate-1 and phosphatidylinositol 3-kinase in 3T3-L1 adipocytes. A putative cellular mechanism for impaired protein kinase B activation and GLUT4 translocation. J Biol Chem 274: 10,595–10,602, 1999. doi: 10.1074/jbc.274.15.10595. [DOI] [PubMed] [Google Scholar]
- 45.Waltenberger J, Claesson-Welsh L, Siegbahn A, Shibuya M, Heldin CH. Different signal transduction properties of KDR and Flt1, two receptors for vascular endothelial growth factor. J Biol Chem 269: 26,988–26,995, 1994. [PubMed] [Google Scholar]
- 46.Watts SW, Dorrance AM, Penfold ME, Rourke JL, Sinal CJ, Seitz B, Sullivan TJ, Charvat TT, Thompson JM, Burnett R, Fink GD. Chemerin connects fat to arterial contraction. Arterioscler Thromb Vasc Biol 33: 1320–1328, 2013. doi: 10.1161/ATVBAHA.113.301476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Weigert J, Neumeier M, Wanninger J, Filarsky M, Bauer S, Wiest R, Farkas S, Scherer MN, Schäffler A, Aslanidis C, Schölmerich J, Buechler C. Systemic chemerin is related to inflammation rather than obesity in type 2 diabetes. Clin Endocrinol (Oxf) 72: 342–348, 2010. doi: 10.1111/j.1365-2265.2009.03664.x. [DOI] [PubMed] [Google Scholar]
- 48.Wilcox G. Insulin and insulin resistance. Clin Biochem Rev 26: 19–39, 2005. [PMC free article] [PubMed] [Google Scholar]
- 49.Wittamer V, Franssen JD, Vulcano M, Mirjolet JF, Le Poul E, Migeotte I, Brézillon S, Tyldesley R, Blanpain C, Detheux M, Mantovani A, Sozzani S, Vassart G, Parmentier M, Communi D. Specific recruitment of antigen-presenting cells by chemerin, a novel processed ligand from human inflammatory fluids. J Exp Med 198: 977–985, 2003. doi: 10.1084/jem.20030382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wittamer V, Grégoire F, Robberecht P, Vassart G, Communi D, Parmentier M. The C-terminal nonapeptide of mature chemerin activates the chemerin receptor with low nanomolar potency. J Biol Chem 279: 9956–9962, 2004. doi: 10.1074/jbc.M313016200. [DOI] [PubMed] [Google Scholar]
- 51.Yamawaki H. Vascular effects of novel adipocytokines: focus on vascular contractility and inflammatory responses. Biol Pharm Bull 34: 307–310, 2011. doi: 10.1248/bpb.34.307. [DOI] [PubMed] [Google Scholar]
- 52.Zhang R, Liu S, Guo B, Chang L, Li Y. Chemerin induces insulin resistance in rat cardiomyocytes in part through the ERK1/2 signaling pathway. Pharmacology 94: 259–264, 2014. doi: 10.1159/000369171. [DOI] [PubMed] [Google Scholar]