

Abstract
In humans, vitamin B12 deficiency causes peripheral and CNS manifestations. Loss of myelin in the peripheral nerves and the spinal cord (SC) contributes to peripheral neuropathy and motor deficits. The metabolic basis for the demyelination and brain disorder is unknown. The transcobalamin receptor–knockout mouse (Cd320−/−) develops cobalamin (Cbl) deficiency in the nervous system, with mild anemia. A decreased S-adenosylmethionine:S-adenosylhomocysteine ratio and increased methionine were seen in the brain with no significant changes in neurotransmitter metabolites. The structural pathology in the SC presented as loss of myelin in the axonal tracts with inflammation. The sciatic nerve (SN) showed increased nonuniform, internodal segments suggesting demyelination, and remyelination in progress. Consistent with these changes, the Cd320−/− mouse showed an increased latency to thermal nociception. Further, lower amplitude of compound action potential in the SN suggested that the functional capacity of the heavily myelinated axons were preferentially compromised, leading to loss of peripheral sensation. Although the metabolic basis for the demyelination and the structural and functional alterations of the nervous system in Cbl deficiency remain unresolved, the Cd320−/− mouse provides a unique model to investigate the pathologic consequences of vitamin B12 deficiency. —Arora, K., Sequeira, J. M., Alarcon, J. M., Wasek, B., Arning, E., Bottiglieri, T., Quadros, E. V. Neuropathology of vitamin B12 deficiency in the Cd320−/− mouse.
Keywords: CNS, neuropathy, demyelination, spinal cord
Vitamin B12 (cobalamin, Cbl) is an essential water-soluble micronutrient. The recommended daily dietary intake for humans is 2–3 μg (1). The human body stores 2–3 mg Cbl, with the liver and kidneys the primary organs of storage (2). Although Cbl uptake in the liver can occur via uptake of haptocorrin-bound Cbl by the asialoglycoprotein receptor, the kidneys can salvage Cbl in the process of renal tubular reabsorption of transcobalamin (TC)-bound Cbl (3–5). Two proteins are involved in the cellular uptake of Cbl: the plasma protein TC secreted by the vascular endothelium (6) and the TC receptor (TCblR), with high affinity for TC saturated with Cbl (7, 8). In addition to dietary deficiency, Cbl deficiency can result from defects in the absorption, transport, cellular uptake, and intracellular processing of the vitamin (9).
Cbl participates in 2 biochemical reactions. These involve conversion of homocysteine (HCY) to methionine, and N5-methyl-tetrahydrofolate to tetrahydrofolate by the cytosolic enzyme methionine synthase (MS), which requires methyl-Cbl as a cofactor (10) and the conversion of methylmalonyl-coenzyme A (MMCoA) to succinyl-CoA by the enzyme methylmalonyl-CoA mutase which uses 5′-deoxyadenosyl-Cbl as its cofactor in the mitochondria (11). Succinyl-CoA is further converted to acetyl-CoA in the tricarboxylic acid (TCA) cycle for the synthesis of lipids that are an integral part of myelin (12). Therefore, Cbl deficiency causes elevation of HCY, N5-methyl-tetrahydrofolate, and, MMA (13). Lack of tetrahydrofolate affects the folate-dependent pathways required for the synthesis of purines and pyrimidines, leading to defective DNA synthesis and megaloblastic anemia (14). Besides the anemia, Cbl deficiency is also known to cause neurologic deficits and neuropsychiatric manifestations. In humans, these include myelopathy and neuropathy in the form of paresthesia, ataxia, proprioception and vibration loss, abnormal reflexes, weakness, pain, and loss of sensation in hands and feet (15–17). The clinical presentation is associated with loss of myelin in the peripheral nerves and the spinal cord (SC) (18, 19). Neuropsychiatric manifestations, such as depression, mania, delirium, and cognitive impairment, are also frequently observed in Cbl-deficient patients, but the metabolic basis for the neuropathologic changes contributing to the clinical phenotype remains unexplained (20, 21).
To understand the cause of the neurologic deficits, we studied TCblR-knockout mouse (Cd320−/−). Lack of TCblR limits Cbl uptake and causes Cbl deficiency in the CNS and elevated HCY and MMA in the blood of this mouse (22). The TCblR/Cd320−/− mouse exhibits behavioral deficits, including cognitive deficits, also seen in the human condition. This mouse model is associated with decreased expression of the α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid synaptic receptor subunit glutamate receptor-1 with a significant decrease in hippocampal pyramidal cell nuclei and brain mass (23). Because the Cd320−/− mouse fed a Cbl replete diet develops Cbl deficiency in the nervous system, in this study, we examined the effects of Cbl deficiency by dietary restriction and by ablating the receptor for cellular uptake of the vitamin in the C57BL/6J strain and examined changes in metabolites, the SC, and the peripheral nervous system, to identify structural changes responsible for the clinical phenotype of neurologic manifestations, such as the loss of peripheral sensation and motor deficits that is seen in humans. To date, the metabolic basis for the nervous system pathology in Cbl deficiency remains unexplained, and therefore we examined the metabolites of Cbl-dependent pathways and pathologic changes in the SC and the sciatic nerve (SN).
MATERIALS AND METHODS
Six groups of C57BL/6J wild-type (Cd320+/+) and TCblR/Cd320-knockout (Cd320−/−) [C57BL/6J-Cd320Gt(pGt01xft2)Qua] mice, 5-mo-old males and females (1:1 ratio), backcrossed twice on a C57BL/6J background, were used. The age of the mice for the study was based on the observation of severe CNS Cbl deficiency in the Cd320−/− mice and systemic Cbl deficiency after 3 mo of consuming a Cbl-deficient diet and the observation of behavioral deficits in 5-mo-old mice (23, 24). The wild-type C57BL/6J mice (Cd320+/+) used in the study were derived from a wild-type litter, and the Cd320−/− were generated by breeding homozygous mice. Hematologic parameters, metabolite analysis related to Cbl pathways, and histologic changes were determined.
Dietary information
Two groups of mice (Cd320+/+; n = 8 and Cd320−/−; n = 7) were fed a normal diet (Picolab rodent diet 20) containing 50 μg vitamin B12 and 3 mg folic acid per kilogram (5001; LabDiet, St. Louis, MO, USA). Two additional groups identified as Cd320+/+-Cbl− (n = 13) and Cd320−/−-Cbl− (n = 4) were fed a modified Cbl-deficient rodent diet containing l-amino acids and 1% succinyl sulfathiazole, supplemented with 4 mg/kg folic acid, with Dyetrose to enable pelleting (517927; Dyets, Inc., Bethlehem, PA, USA). Two additional groups fed a Cbl-deficient diet (n = 7) also received 0.5 mg/ml folic acid in their drinking water ad libitum. Folate supplementation was included in the study because folate utilization can place additional demand on Cbl and potentially correct the anemia but worsen the neuropathology (25). All the mice were started on their respective diets at birth by feeding the mothers the diets and were fed the diets for the duration of the study. The Institutional Animal Care and Use Committee of the State University of New York Downstate Medical Center, approved the protocol.
Blood profile and metabolite analysis
Blood was collected immediately after the mice were euthanized by cardiac puncture with a tuberculin syringe with a 23-gauge needle. The samples were transferred to tubes containing an equal volume of heparin (1000 U/ml; Elkins-Sinn, Cherry Hill, NJ, USA) and were used for the complete blood count (CBC), performed in a Coulter counter (Beckman Coulter, Brea, CA, USA).
Total Cbl levels were measured in the serum and tissues, as previously described (26). In brief, an aliquot of the serum or homogenized tissue was mixed in a glass tube with 3–5 volumes of acetate buffer (pH 4.5), containing 100 μg/ml potassium cyanide, placed in a boiling water bath for 30 min, and cooled, and the precipitate was separated by centrifugation. The supernatant was assayed for Cbl concentration by a competitive radioassay, with 57CoB12 used as the tracer and recombinant human TC as the binding protein. Analysis of levels of S-adenosylmethionine (SAM), S-adenosylhomocysteine (SAH), methionine, cystathionine, choline, and betaine in various brain regions was performed with stable-isotope dilution liquid chromatography-electrospray ionization (ESI)-tandem mass spectrometry (27). Tissue samples were prepared by deproteinization in 0.1 M perchloric acid containing stable isotopes of all metabolites determined. Chromatographic separation was achieved on an EZ-Fast 250 × 2.0 mm, 4 μm AAA-MS analytical column (Phenomenex, Torrance, CA, USA), maintained at 36°C at a flow of 250 μl/min. The compounds were detected by multiple-reaction monitoring with positive-ESI. Sample injection and separation were performed in a Prominence LC System (Shimadzu, Kyoto, Japan) interfaced with a 5500 QTrap Mass Spectrometer, and the data were collected with Analyst Software v.1.6 (Sciex, Framingham, MA, USA).
The neurotransmitters dopamine and serotonin and related metabolites in various regions of the brain were measured by HPLC with coulometric detection (28, 29). Tissue samples stored at −80°C were deproteinized in 0.1 M perchloric acid containing 0.1 M diethylenetriaminepentaacetic acid and 1 M DTT. The clear extract was injected into the HPLC system with a refrigerated autosampler (Model A1-200; Mettler-Toledo Rainin, Oakland, CA, USA), and metabolites were separated in a reverse-phase Gemini 5μ C18 250 × 3.0 mm column (Phenomenex, Torrance, CA, USA) maintained at 35°C with a flow rate of 0.35 ml/min. Detection of the compounds was performed in a microdialysis cell 5014B and guard cell 5020 (ESA Biosciences, Waltham, MA, USA), with cell potentials set at E1 = −10 mV, E2 = +400 mV, and guard cell = +600 mV, at +600 mV and quantitated with external standards.
Histology and immunohistochemistry
The spinal column was removed and transferred to formalin fixative/decalcifier (95057-810; Avantor, West Chester, PA, USA) and was dissected from the bone after 24 h. The SC was dehydrated in increasing ethanol concentrations of 70–100%, cleared in xylene, and embedded in Paraplast (23-021401; Thermo Fisher Scientific, Waltham, MA, USA). Transverse sections of the SC (7 µm) were made with a Leitz microtome and mounted on silane-coated glass slides (Leica Microsystems, Buffalo Grove, IL, USA). Before histology and immunostaining, tissue sections were cleared in xylene and rehydrated in ethanol (100–70%).
To study myelin in the SC, sections were stained with Luxol Fast Blue-Cresyl Echt Violet Stain (KTLFB; American MasterTech, Lodi, CA, USA), according to the protocol provided by the manufacturer. Sudan black dye was used to stain the myelin in the SN. SNs, initially fixed in 70% ethanol, were transferred to Sudan black (made in 85% propylene glycol) and incubated at 60°C overnight. The nerves were transferred to 85% propylene glycol the next day, incubated at room temperature for 4 h, and placed on a glass slide, and the nerve fibers were separated under a bright-field microscope (Nikon, Tokyo, Japan) with a fine needle.
Immunohistochemistry of brain and SC sections
Tissue sections (7 μm) were washed in PBS and incubated in 3% H2O2 for 5 min, followed by washing in PBS (2 times for 15 min each) and blocking for nonspecific binding by incubation in normal horse serum for 1 h. Primary antibodies, were then added to the sections and incubated for 2 h at room temperature. A pAb to myelin basic protein (MBP) (260746; Abbiotech, San Diego, CA, USA), was used at a 1:10 dilution. For glial fibrillary acidic protein (GFAP), a pAb (Ab7260; Abcam, Cambridge, MA, USA) was used at 1:200 dilution. A pAb to ionized calcium binding adapter molecule (Iba)-1 (Ab107159; Abcam), was used at 1:100 dilution. In addition, expression of TNF-α was also monitored with a polyclonal TNF-α antibody (PA5-19810; Thermo Fisher Scientific) at a 1:100 dilution.
Normal rabbit serum at a similar dilution was used as a negative control. After primary antibody incubation, sections were washed in PBS (2 times for 15 min each) and incubated with anti-rabbit IgG secondary antibody, coupled to a micropolymer of active peroxidase (ImmPress Reagent Kit, MO-7401-15; Vector Laboratories, Burlingame, CA, USA), for 30 min. Subsequently, the sections were washed in PBS (2 times for 15 min each), and the chromogen was developed with diaminobenzidine (Peroxidase Substrate Kit, SK-4100; Vector Laboratories), according to the manufacturer’s instructions. Sections were counterstained with Hematoxylin QS (H-3404; Vector Laboratories), dehydrated in ethanol, cleared in xylene, and prepared for viewing in Permount (SP15-500; Fisher Chemicals, Hampton, NH, USA). Immunostaining was quantified with ImageJ software (National Institutes of Health, Bethesda, MD, USA; https://imagej.nih.gov/ij/index.html).
Thermal response test
To measure thermal nociceptive response in the mouse (30), we used a Plexiglas box (20 × 12 × 12 cm) with a copper base. After the mouse was placed in the box, the box was placed on a hot plate set at 56°C. As the copper bottom of the box heated up, the mouse leapt out of the box when uncomfortable, and the latency to the leaping response was measured in seconds for each mouse. Three trials were performed each day over 3 d for each mouse.
Compound action potential measurements in the SN
Mice were placed in an induction (anesthetizing) chamber, then deeply anesthetized with vaporized isoflurane (5% in 100% O2) for 3 min, and euthanized by decapitation. Both SNs were dissected and transferred to ice-cold artificial cerebrospinal dissection fluid (aCSF; in mM: 119 NaCl, 4 KCl, 7 MgSO4, 0.1 CaCl2, 26.2 NaHCO3, 1 NaH2PO4, and 11 glucose, saturated with 95% O2 and 5% CO2). After collection, both SNs were equilibrated at room temperature for 10 min in recording aCSF (same composition as dissection aCSF, but containing 1.5 mM MgSO4 and 2.5 mM CaCl2) and then transferred into an interface recording chamber with flowing recording aCSF (4 ml/min) saturated with 95% O2 and 5% CO2 at 37°C (23). Both SNs were laid out straight across the extension of the recording chamber and separated from each other. The compound action potential (CAP) response was elicited by stimulating one end of an SN with a bipolar electrode (MX211ESJA1; FHC & Co., Bowdoin, ME, USA) and recording the evoked response at the other end with a glass borosilicate micropipette, 5–8 MΩ filled with recording aCSF (BF150-86-10; Sutter Instrument Co., Novato, CA, USA). CAP responses were evoked by a set of increasing voltage stimuli (0–40 V, at 5-V intervals). The distance between electrodes was set at 4 mm. Measures of CAP latency and amplitude were determined in both Cd320+/+ and Cd320−/− SNs for the responses (peaks) observed, to determine any changes in conduction efficacy. Further, latency and distance between the stimulating and recording electrodes were used to calculate conduction velocity (m/s).
Statistical analysis
A 2-way ANOVA with the Tukey honest significant difference (HSD) post hoc test was used for the blood profile, metabolite analysis, neurotransmitter analysis, thermal response test, and CAP measurements. A 2-way ANOVA was used to compare multiple groups simultaneously to determine any significant changes. The Tukey HSD test was further used to separately compare each of the groups to determine specific differences that amounted to values of P < 0.05. In the thermal response test, a 2-way repeated-measures ANOVA with Bonferroni correction was applied to determine whether there were differences of P < 0.05. The nonparametric test was performed before the Tukey HSD because the small sample size (n < 20) could not be tested for normality distribution. Similarly, for immunohistochemistry, the Mann-Whitney U test, a nonparametric test was used to determine P values, because the sample size was small.
RESULTS
Cbl profile
Cd320−/− mice fed a normal diet containing Cbl for 5 mo were severely depleted of Cbl in the brain (Fig. 1A) and SC (Fig. 1B), amounting to a decrease of >90%. The liver (Fig. 1C) and the spleen (Fig. 1D) also showed a significant decrease in Cbl, but this decrease amounted to <35% of the total Cbl. On the other hand, kidneys showed a significantly higher accumulation of Cbl (>6-fold) in the Cd320−/− mice vs. the wild type (Fig. 1E). In groups fed a Cbl-deficient diet, Cd320−/−-Cbl− mice had even lower Cbl levels in the brain vs. the wild type (Fig. 1A). However, other tissues had similar but significantly depleted levels of Cbl in both groups. Groups fed a Cbl-deficient diet supplemented with folic acid had Cbl levels similar to those of groups fed Cbl-deficient diets. In summary, the levels of Cbl in the CNS (brain and SC) were significantly lower in Cd320−/− mice and were similar to levels observed in Cd320+/+-Cbl− mice. However, severe systemic Cbl deficiency was observed only by dietary withholding of Cbl.
Figure 1 .

Cbl levels in the Cd320+/+ and the Cd320−/− groups. A–D) Brain tissue showed significantly lower levels of Cbl in all groups fed a Cbl-deficient diet vs. their respective groups fed a normal diet. Mice fed a Cbl-deficient diet also showed significantly reduced Cbl levels in the SC (B), liver (C), and spleen (D). No significant differences were observed in Cd320−/− and Cd320−/−-F+ groups in Cbl levels compared with their respective wild-type groups fed a Cbl-deficient diet. E) On the other hand, kidneys showed significantly increased levels of Cbl accumulation in Cd320−/− mice fed a normal diet. Values are expressed as picograms per milligram wet weight. For all box plots, the vertical line indicates range, the box includes the second and third quartiles, and the horizontal line within the box indicates median (n = 5 for each group). *P < 0.05, ***P < 0.001.
CBC
The CBC did not show any significant changes in hematologic parameters between Cd320+/+ and Cd320−/− mice fed the normal diet. However, changes consistent with anemia were seen in Cd320−/−-Cbl− mice, with a decrease in white and red blood cell counts and hematocrit (HCT), a significant decrease in hemiglobin (HGB) and mean corpuscular hemoglobin (MCH), and a significant increase in red cell distribution width (RDW) and platelet count. Folate supplementation of Cd320−/−Cbl− (Cd320−/−-Cbl−F+) mice appeared to partially correct some of these abnormalities (Table 1).
TABLE 1.
Complete blood count in Cd320+/+ and Cd320−/− groups
| Group | Cd320+/+ (n = 8) | Cd320−/− (n = 7) | Cd320+/+-Cbl− (n = 13) | CD320−/−-Cbl− (n = 4) | CD320−/−-Cbl−F+ (n = 7) |
|---|---|---|---|---|---|
| WBC (K/μl) | 18.67 ± 0.57 | 14.06 ± 1.65 | 13.06 ± 1.08 | 14.12 ± 2.0 | 13.56 ± 1.97 |
| RBC (M/μl) | 13.0 ± 0.66 | 12.95 ± 0.48 | 12.01 ± 0.36 | 11.19 ± 0.72 | 12.55 ± 0.24 |
| HGB (g/dl) | 19.55 ± 0.91 | 18.29 ± 0.74 | 18.55 ± 0.58 | 15 ± 0.62 | 18.74 ± 0.63 |
| HCT (%) | 63.80 ± 3.31 | 61.37 ± 2.44 | 62.22 ± 1.90 | 53.10 ± 2.24 | 64.29 ± 1.58 |
| MCV (fl) | 49.08 ± 0.54 | 47.36 ± 0.32 | 51.89 ± 0.32 | 47.68 ± 1.56 | 51.20 ± 0.63 |
| MCH (pg) | 15.09 ± 0.21 | 14.10 ± 0.12 | 15.45 ± 0.09 | 13.50 ± 0.75 | 14.96 ± 0.29 |
| MCHC (g/dl) | 30.74 ± 0.46 | 29.80 ± 0.13 | 29.82 ± 0.19 | 28.28 ± 0.67 | 29.17 ± 0.43 |
| RDW (%) | 11.84 ± 0.09 | 12.27 ± 0.14 | 12.22 ± 0.10 | 15.33 ± 1.56 | 12.46 ± 0.25 |
| PLAT (K/μl) | 922.85 ± 65.18 | 643.43 ± 59.96 | 1098.77 ± 88.96 | 1738 ± 826.49 | 1078.86 ± 148.93 |
| MPV (fl) | 7.33 ± 0.22 | 8.16 ± 0.10 | 6.58 ± 0.18 | 7.30 ± 0.62 | 7.37 ± 0.23 |
No significant differences were observed in most parameters between the various groups, except for a significant decrease in HGB and MCH (P < 0.01). The RDW and platelet counts in Cd320+/+ mice fed a normal diet and Cd320−/− mice fed a B12 (Cbl)-deficient diet were compared (P < 0.002). MCV, mean corpuscular volume; MCHC, MCH concentration; MPV, mean platelet volume; PLAT, platelet count. Data are means ± sem.
Metabolites in Cbl deficiency
Metabolites were measured in the brain (Fig. 2A, B) and the liver (Fig. 2C, D) of wild-type and Cd320−/− groups fed various diets. Both brain and liver SAM and SAH levels were not significantly altered in these groups (Fig. 2A, C), but the SAM:SAH ratio was significantly decreased in the brain in the Cd320−/−-Cbl− and the Cd320−/−-Cbl−F+ groups (Fig. 2A) and increased in the liver (Fig. 2C). Brain methionine levels were increased in Cd320+/+-Cbl− and Cd320−/−-Cbl− groups (Fig. 2B). Cystathionine levels were increased in the Cd320−/− groups only. No significant changes in the levels of betaine and choline were observed in the brains of all the groups (Fig. 2B, D). In the liver, no major changes were observed in the concentration of methionine, cystathionine, betaine, and choline, except for an increase in cystathionine in the Cd320−/−-Cbl−F+ group and increased betaine in the Cd320−/−-Cbl− group; folate supplementation appeared to decrease liver betaine content (Fig. 2D).
Figure 2 .

Metabolites in the brain (A, B) and liver (C, D) of Cd320+/+ and Cd320−/− groups fed a normal diet, a Cbl-deficient diet, or a Cbl-deficient diet supplemented with folic acid. A, B) Significant changes were observed in the concentration of methionine, cystathionine, and SAM:SAH ratio in the brain, but not in the levels of SAM and SAH (A) or betaine and choline (B). C, D) The profile of liver metabolites varied considerably from that in brain tissue. The SAM (C):SAH ratio was increased in Cd320+/+-Cbl− and Cd320−/−-Cbl− mice and decreased in Cd320−/−-Cbl−F+ mice. Cystathionine (D) was increased only in the Cd320−/−-Cbl− group, and betaine was increased only in the Cd320−/−-Cbl− group (n = 5 for each group). *P < 0.05.
Neurotransmitters in the brain
Metabolites of neurotransmitters were measured in Cd320+/+ and Cd320−/− mice fed a normal diet (Fig. 3). Distribution of the dopamine metabolites dopamine, 3,4-dihydroxyphenylacetic acid, and homovanillic acid were similar in specific regions of the brain, but the levels were highest in the cortex and medulla in both groups of mice (Fig. 3A–C). Serotonin metabolites 5-hydroxytryptamine (HT) and 5-hydroxyindoleacetic acid (HIAA) were similar in all regions in both groups (Fig. 3D–F), except for an increase in the 5-HTAA:5-HT ratio in the cerebellum of Cd320−/− mice (Fig. 3F).
Figure 3 .

Neurotransmitter metabolites in the brain. Dopamine (A–C) and serotonin (D–F) metabolites in the brain of Cd320+/+ and Cd320−/− mice fed a normal diet. Dopamine metabolites increased in the cortex and decreased in the medulla of Cd320−/− mice, but the changes were not significant. Dopamine content of the cerebellum was not reported, because it was below the level of detection in the method used. No significant changes were seen in serotonin metabolites, except for a decrease in 5-HIAA and 5-HT and an increase in the 5-HIAA:5-HT ratio in the cerebellum of Cd320−/− mice (n = 5 for each group). *P < 0.05.
Structural and immunohistochemical visualization of the SC and SN
Loss or disruption of myelin was observed in the anterior, posterior, and lateral regions of the SC in Cd320−/− mice, as seen in a cross section from the thoracic region (Fig. 4A). The axonal tracts protruding from gray matter into white matter were also depleted of myelin in the Cd320−/− mice vs. the wild type (Fig. 4B). This effect was accompanied by increased expression of MBP (Fig. 5A, B) and GFAP (Fig. 5C, D). A marker for macrophage/microglia activation, Iba-1, was also upregulated in the SC (Fig. 6A–C). The cytokine ΤΝF-α, an acute-phase reactant and a marker for inflammation, was also increased in the SC (Fig. 6D, E). In the SN, the Cd320−/− mice showed an increased number of short segments of myelin, compared to the wild type (Fig. 7). The internodal distance was short and nonuniform in length. However, the myelination between the nodes appeared to be of the same thickness as that observed in the wild type.
Figure 4 .
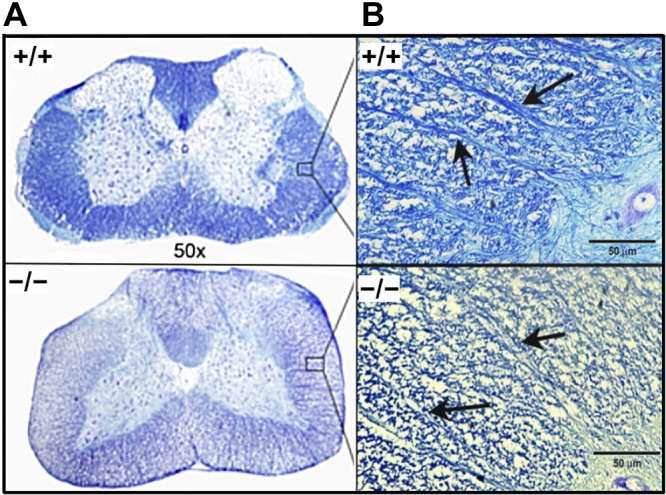
Histopathologic evaluation of the thoracic region of the SC in cross section in Cd320+/+ and Cd320−/− mice. A) Luxol fast blue staining for myelin showed demyelination/dysmyelination in the posterior, lateral and anterior regions of the Cd320−/− SC. B) Examination of the axonal tracts protruding from gray matter to white matter showed disruption of myelin (arrows).
Figure 5 .

Immunohistochemical staining for myelin basic protein in a cross section of the SC. A, B) The overall increase in the expression of MBP can be seen in white matter (anterior funiculus shown in the magnification) with intense staining along the tracts (arrows) in the Cd320−/− mice. C) Increased expression of GFAP in the SC of Cd320−/− mice (arrows; intermediolateral gray column). D) Quantification of immunostaining for GFAP (n = 6). *P < 0.05.
Figure 6 .

A–C) Immunohistochemical staining for Iba-1 in a cross section of the SC showed a significant increase in the expression of this protein in Cd320−/− mice (intermediolateral gray column). D, E) Similarly, the expression of TNF-α was increased in this tissue (anterior funiculus; arrows). ***P < 0.001.
Figure 7 .
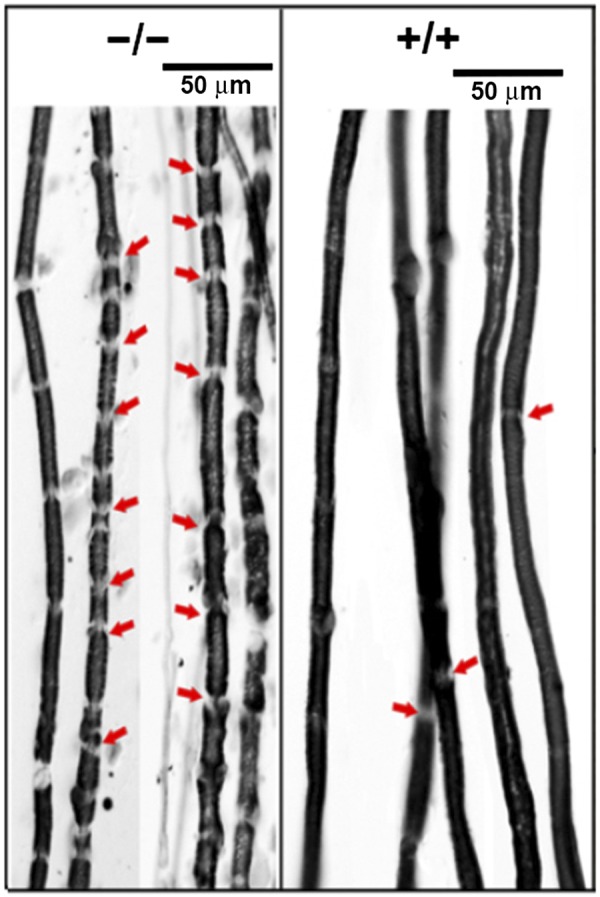
Sudan black staining of myelin sheaths on nerve fibers showed smaller segments of various lengths with increased nodes of Ranvier (arrows) in the Cd320−/− mice.
Thermal response to heat and nerve conduction studies
Loss of peripheral sensation was evident from the significantly increased latency to thermal nociceptive sensitivity in the Cd320−/− vs. the Cd320+/+ mice (Fig. 8A). For nerve conduction studies, we examined the CAP response elicited in the SN of these mice and measured CAP latency and amplitude from the peaks of the CAP responses. We detected 2 peaks (Fig. 8B): the first peak (P1) was observed almost immediately after stimulation and was characteristic of axonal fibers of high conduction velocity, including type-A fibers (31–34). The second peak (P2), observed was characteristic of fibers with a lower conduction velocity, such as type-B fibers. We did not detect a third peak characteristic of type-C fibers, with further lower conduction velocity (31–34). Time to peak was not different between Cd320−/− and Cd320+/+ mice for P1 or P2 (Fig. 8C, D). Therefore, the conduction velocity, calculated over the 10–40 V of stimulation for each peak, was similar. For P1, mean conduction velocity for Cd320+/+ was 55.9 ± 19.2 m/s (range, 22.4–109.1 m/s). P1 mean conduction velocity in Cd320−/− was 55.4 ± 17.1 m/s (range, 21.2–101.3 m/s). For P2, mean conduction velocity for Cd320+/+ was 5.58 ± 0.19 m/s (range, 5–5.8 m/s). P2 mean conduction velocity in Cd320−/− was 5.3 ± 0.18 m/s (range, 4.8–5.5 m/s). Examination of CAP amplitude, however, revealed a significant decrease in amplitude in Cd320−/− compared to Cd320+/+ mice (Fig. 8E, F). CAP amplitudes for P1 were significantly lower at all voltages tested in Cd320−/− vs. Cd320+/+ mice (Fig. 8E), suggesting a massive impact on the amount of type-A fibers in the Cd320−/− mouse. Decreased CAP amplitudes for P2 in the Cd320−/− mouse were revealed only at the highest (40 V) stimulation (Fig. 8F), suggesting a limited impact on type-B fibers.
Figure 8 .

A) Latency to heat response was significantly increased in the Cd320−/− vs. the Cd320+/+ mice. The CAP in the SNs of wild-type (6 nerves from 3 mice) and Cd320−/− (6 nerves from 3 mice) mice was recorded. B) CAP traces showed 2 peaks (P1 and P2) that were characteristic of type-A and -B fibers, respectively. C, D) No significant changes in the latency of CAP was observed for each peak (measured at 10–40 V). E, F) Significant changes were found between Cd320+/+ and Cd320−/− mice in the amplitude of CAP for peak P1 across all stimulation voltages and at 40 V for P2. *P < 0.05, **P < 0.01, ***P < 0.001.
DISCUSSION
Lack of functional TCblR causes Cbl deficiency in humans (35) and in the Cd320−/− mouse (22). As a consequence, HCY and MMA are elevated and serve as biomarkers of Cbl deficiency (13). The Cd320−/− mouse fed a normal diet develops Cbl deficiency within 3 mo of birth, significantly in the nervous system (22). In the current study, we observed that the Cd320−/− mouse, at the age of 5 mo, developed CNS Cbl deficiency when fed a Cbl-replete normal diet, whereas both wild-type and Cd320−/− mice developed systemic and CNS Cbl deficiency when fed a Cbl-deficient diet. Our results show that the Cd320−/− mouse develops severe Cbl deficiency when fed a normal diet and therefore is a suitable model for the long-term effects of Cbl deficiency in the CNS. The study also showed that systemic and CNS Cbl deficiency can be produced in a rodent model by depriving the animal of dietary Cbl for 3–5 mo. Some mice fed a Cbl-deficient diet were given additional folate supplementation to determine whether high folate intake would place additional demand on the available Cbl and worsen the Cbl deficiency and its phenotype. In this mouse model, high folate supplementation of Cd320−/− mice corrected some of the hematologic parameters, such as HGB, HCT, and RDW but did not correct or worsen the neuropathology. Although, the levels of Cbl in the Cd320−/− mouse liver and spleen were lower, they were not as severely depleted as in the brain and SC, suggesting an alternate pathway for Cbl uptake in these organs if additional Cbl is available through diet (23, 24). Furthermore, lack of Cbl uptake in the CNS of the Cd320−/− mouse fed a normal diet contributes to elevated TC-Cbl in the blood and results in increased Cbl in the kidneys via uptake by megalin (3). Higher circulating TC-Cbl may also contribute to the lack of hematologic changes indicative of megaloblastic anemia in the Cd320−/− mouse fed a normal diet. Some hematologic changes consistent with anemia were seen in both Cd320+/+ and Cd320−/− mice fed a Cbl-deficient diet, indicating that dietary restriction of Cbl can produce mild anemia. Nevertheless, the normal fetal and adult development of the Cd320−/− mouse and the severe loss of Cbl in the nervous system provides an ideal model to study the CNS manifestations of Cbl deficiency in the adult mouse (24).
The Cd320−/− mouse brain is hypomethylated (36), a condition attributed to lower levels of SAM, a universal methyl group donor. However, in the current study, SAM level and SAM:SAH ratios were not consistently decreased in the brain and liver. It is likely that other mechanisms are involved in regulating DNA methylation (37). Lack of any significant change in neurotransmitter metabolites as a result of Cbl deficiency in the brain suggests that the CNS manifestations are related to structural changes. Myelin loss in the brain caused by Cbl deficiency is not well documented (38–40). However, Cbl deficiency leads to metabolic stress and alterations in the hippocampus that can cause functional deficits, as observed in Cd320−/− mouse (23). On the other hand, disruption of myelin in the SC and peripheral nerves is seen in human Cbl deficiency, suggesting that Cbl plays an important role in myelin homeostasis (19, 39). Similar to that in humans, we observed myelin disruption in the axonal tracts of the SC in the Cd320−/− mouse. These axons are involved in carrying signals from peripheral nerves to the brain and vice versa (33, 41). Further, the overexpression of MBP suggests attempts at remyelination or repair (42, 43). Myelin disruption can lead to accumulation of myelin debris, which could generate an inflammatory response in clearing this debris (44). Activation of astrocytes is indicated by increased GFAP expression in the SC, as reported in other neuropathies (45, 46) and Cbl-deficient rats (47). Iba-1 is overexpressed in macrophages in response to inflammation. In the brain, this protein is overexpressed in activated microglia and has been observed in neurodegenerative diseases, such as multiple sclerosis, Alzheimer’s disease, Parkinson’s disease, and Huntington’s disease (48, 49). TNF-α, secreted by activated microglia, is also a major component of the neuroinflammatory response in several CNS diseases, including Cbl deficiency (47, 50). These observations are consistent with the pathology of inflammation in the SC of the Cd320−/− mouse.
The SN originates from the L3 to L5 vertebrae in the lumbar region of the spinal column in mice and extends to the hind limb (51). This nerve contains afferent axons from sensory neurons in the dorsal root ganglion and efferent motor neurons extending from the anterior horn cells of the SC (34, 52). These axons are type-A fibers because of their thick myelin insulation and high conduction velocity (33). Besides myelinated motor and sensory axons, SN also contains less myelinated and nonmyelinated sensory and sympathetic axons, known as type-B and -C fibers, respectively (33, 34). Based on myelination, the different fibers present in the SN have different conduction velocities and produce different action potentials based on time and amplitude (31, 32). In humans, myelin loss, in the form of segmental demyelination in peripheral nerves, reduces conduction velocity and amplitude of CAPs and, as a consequence, causes loss of sensation in the hands and feet (53).
The Cd320−/− mouse SN stained for myelin showed short nonuniform internodal segments. A similar phenotype has been reported in Charcot-Marie-Tooth disease type 1A (54). The myelin sheath between the internodal segments of the SN appeared to be similar in thickness to the wild type. The Cd320−/− mouse showed no difference in conduction velocity for type-A and -B fibers. However, the amplitude of CAP was significantly lower in the Cd320−/− mouse than in the wild type. This conduction block was more pronounced in type-A fibers than in type-B. A decrease in internodal distance could contribute to this conduction block (55). In concordance, the Cd320−/− mouse showed increased latency to thermal nociceptive sensitivity. These studies show that Cbl deficiency in the Cd320−/− mouse affects the SC and the SN and produces a phenotype with loss of peripheral sensation. This functional deficit is similar to the clinical phenotype of neurologic manifestations observed in humans. Only 2 enzymatic pathways are dependent on Cbl in mammalian cells (Fig. 9). These pathways can be affected by lack of Cbl uptake via the receptor TCblR or lack of TC, the transporter that carries the Cbl to the receptor (9). Many genetic defects in the intracellular processing of Cbl and the synthesis of Cbl cofactors can also lead to the clinical phenotype (56). The cause of megaloblastic anemia is linked to a block in the processing of folate via the Cbl-dependent MS enzyme, but the precise causes of the myelin pathology and the CNS manifestations in Cbl deficiency have not been identified. The MMCoA mutase–dependent production of succinyl-CoA appears to be the link that connects to the production of glycolipids and sphingomyelin, the 2 major components of myelin (57). Therefore, disruption of the TCA cycle and mitochondrial function may play a role in the pathology. This mouse model can enable identification of changes in the lipidome and methylome in the CNS in Cbl deficiency, to identify pathways and genes affected. The mouse model can be further explored to elucidate the underlying mechanisms. Although early intervention has been beneficial in correcting the CNS manifestations of Cbl deficiency, a deeper understanding of the underlying mechanisms could help in the treatment of advanced clinical disorder, which appears to be irreversible by Cbl treatment.
Figure 9 .

Cobalamin uptake and utilization in the cell. The TC protein carries absorbed Cbl to the TCblR on the cell surface. The TC-Cbl complex is internalized, TC is degraded in the lysosome, and free Cbl is released in the cytoplasm. Some of the Cbl is converted to methyl Cbl and participates as a cofactor for the enzyme. Additional Cbl is transported into the mitochondria and converted to 5′-deoxyadenosyl Cbl (AdoCbl) for utilization in the MMCoA mutase dependent conversion of MMCoA to succinyl CoA. Production of succinyl-CoA to feed the TCA cycle and optimum mitochondrial function appear to be critical determinants for the production of glycolipids and sphingomyelin.
ACKNOWLEDGMENTS
This work was supported, in part, by U.S. National Institutes of Health (NIH), National Institute of Diabetes and Digestive and Kidney Diseases Grant DK064732 (to E.V.Q.), and NIH National Institute of Neurological Disorders and Stroke Grant NS091830 (to J.M.A.) The authors declare no conflicts of interest.
Glossary
- aCSF
artificial cerebrospinal fluid
- CAP
compound action potential
- CBC
complete blood count
- Cbl
cobalamin
- ESI
electrospray ionization
- GFAP
glial fibrillary acidic protein
- HCT
hematocrit
- HCY
homocysteine
- HGB
hemoglobin
- HIAA
5-hydroxyindoleacetic acid
- HSD
honest significant difference
- HT
5-hydroxytryptamine
- Iba
ionized calcium binding protein
- MBP
myelin basic protein
- MCH
mean corpuscular hemoglobin
- MMA
methylmalonic acid
- MMCoA
methylmalonyl-coenzyme A
- MS
methionine synthase
- RDW
red cell distribution width
- SAH
S-adenosylhomocysteine
- SAM
S-adenosyl methionine
- SC
spinal cord
- SN
sciatic nerve
- TC
transcobalamin
- TCA
tricarboxylic acid
- TCblR
transcobalamin receptor
AUTHOR CONTRIBUTIONS
K. Arora, J. M. Sequeira, J. M. Alarcon, and E. V. Quadros designed the study; K. Arora and J. M. Sequeira performed the experiments; T. Bottiglieri, B. Wasek, and E. Arning analyzed metabolites in tissues; and all authors contributed to the interpretation of data and writing the manuscript.
REFERENCES
- 1.Institute of Medicine ; Food and Nutrition Board ; A Report of the Standing Committee on the Scientific Evaluation of Dietary Reference Intakes and its Panel on Folate, Other B Vitamins, and Choline and Subcommittee on Upper Reference Levels of Nutrients . (1998) Dietary Reference Intakes for Thiamin, Riboflavin, Niacin, Vitamin B6, Folate, Vitamin B12, Pantothenic Acid, Biotin, and Choline. National Academies Press, Washington, DC, USA: [PubMed] [Google Scholar]
- 2.Ståhlberg, K. G., Radner, S., Nordén, A. (1967) Liver B12 in subjects with and without vitamin B12 deficiency; a quantitative and qualitative study. Scand. J. Haematol. 4, 312–330 10.1111/j.1600-0609.1967.tb01632.x [DOI] [PubMed] [Google Scholar]
- 3.Birn, H., Willnow, T. E., Nielsen, R., Norden, A. G., Bönsch, C., Moestrup, S. K., Nexø, E., Christensen, E. I. (2002) Megalin is essential for renal proximal tubule reabsorption and accumulation of transcobalamin-B(12). Am. J. Physiol. Renal Physiol. 282, F408–F416 10.1152/ajprenal.00206.2000 [DOI] [PubMed] [Google Scholar]
- 4.Harte, R. A., Chow, B. F., Barrows, L. (1953) Storage and elimination of vitamin B12 in the rat. J. Nutr. 49, 669–678 10.1093/jn/49.4.669 [DOI] [PubMed] [Google Scholar]
- 5.Scott, J. S., Treston, A. M., Bowman, E. P., Owens, J. A., Cooksley, W. G. (1984) The regulatory roles of liver and kidney in cobalamin (vitamin B12) metabolism in the rat: the uptake and intracellular binding of cobalamin and the activity of the cobalamin-dependent enzymes in response to varying cobalamin supply. Clin. Sci. (Lond.) 67, 299–306 10.1042/cs0670299 [DOI] [PubMed] [Google Scholar]
- 6.Quadros, E. V., Rothenberg, S. P., Jaffe, E. A. (1989) Endothelial cells from human umbilical vein secrete functional transcobalamin II. Am. J. Physiol. 256, C296–C303 10.1152/ajpcell.1989.256.2.C296 [DOI] [PubMed] [Google Scholar]
- 7.Quadros, E. V., Nakayama, Y., Sequeira, J. M. (2005) The binding properties of the human receptor for the cellular uptake of vitamin B12. Biochem. Biophys. Res. Commun. 327, 1006–1010 10.1016/j.bbrc.2004.12.103 [DOI] [PubMed] [Google Scholar]
- 8.Quadros, E. V., Nakayama, Y., Sequeira, J. M. (2009) The protein and the gene encoding the receptor for the cellular uptake of transcobalamin-bound cobalamin. Blood 113, 186–192 10.1182/blood-2008-05-158949 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Quadros, E. V. (2010) Advances in the understanding of cobalamin assimilation and metabolism. Br. J. Haematol. 148, 195–204 10.1111/j.1365-2141.2009.07937.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Taylor, R. T., Weissbach, H. (1967) N5-methyltetrahydrofolate-homocysteine transmethylase. Propylation characteristics with the use of a chemical reducing system and purified enzyme. J. Biol. Chem. 242, 1509–1516 [PubMed] [Google Scholar]
- 11.Cannata, J. J., Focesi, A., Jr., Mazumder, R., Warner, R. C., Ochoa, S. (1965) Metabolism of propionic acid in animal tissues, XII: properties of mammalian methylmalonyl coenzyme a mutase. J. Biol. Chem. 240, 3249–3257 [PubMed] [Google Scholar]
- 12.Agamanolis, D. P., Victor, M., Harris, J. W., Hines, J. D., Chester, E. M., Kark, J. A. (1978) An ultrastructural study of subacute combined degeneration of the spinal cord in vitamin B12-deficient rhesus monkeys. J. Neuropathol. Exp. Neurol. 37, 273–299 10.1097/00005072-197805000-00006 [DOI] [PubMed] [Google Scholar]
- 13.Hannibal, L., Lysne, V., Bjørke-Monsen, A. L., Behringer, S., Grünert, S. C., Spiekerkoetter, U., Jacobsen, D. W., Blom, H. J. (2016) Biomarkers and algorithms for the diagnosis of vitamin B12 deficiency. Front. Mol. Biosci. 3, 27; erratum: 4, 53 10.3389/fmolb.2016.00027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chanarin, I., Deacon, R., Lumb, M., Muir, M., Perry, J. (1985) Cobalamin-folate interrelations: a critical review. Blood 66, 479–489 [PubMed] [Google Scholar]
- 15.Healton, E. B., Savage, D. G., Brust, J. C., Garrett, T. J., Lindenbaum, J. (1991) Neurologic aspects of cobalamin deficiency. Medicine (Baltimore) 70, 229–245 10.1097/00005792-199107000-00001 [DOI] [PubMed] [Google Scholar]
- 16.McCombe, P. A., McLeod, J. G. (1984) The peripheral neuropathy of vitamin B12 deficiency. J. Neurol. Sci. 66, 117–126 10.1016/0022-510X(84)90147-3 [DOI] [PubMed] [Google Scholar]
- 17.Savage, D. G., Lindenbaum, J. (1995) Neurological complications of acquired cobalamin deficiency: clinical aspects. Baillieres Clin. Haematol. 8, 657–678 10.1016/S0950-3536(05)80225-2 [DOI] [PubMed] [Google Scholar]
- 18.Fenton, J., Rajakulendran, S., Chinn, R., Janssen, J. C. (2011) Subacute combined degeneration of the spinal cord due to vitamin B12 deficiency. BMJ Case Rep. 2011, bcr0320114030 10.1136/bcr.03.2011.4030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kalita, J., Chandra, S., Bhoi, S. K., Agarwal, R., Misra, U. K., Shankar, S. K., Mahadevan, A. (2014) Clinical, nerve conduction and nerve biopsy study in vitamin B12 deficiency neurological syndrome with a short-term follow-up. Nutr. Neurosci. 17, 156–163 10.1179/1476830513Y.0000000073 [DOI] [PubMed] [Google Scholar]
- 20.Lachner, C., Steinle, N. I., Regenold, W. T. (2012) The neuropsychiatry of vitamin B12 deficiency in elderly patients. J. Neuropsychiatry Clin. Neurosci. 24, 5–15 10.1176/appi.neuropsych.11020052 [DOI] [PubMed] [Google Scholar]
- 21.Shipton, M. J., Thachil, J. (2015) Vitamin B12 deficiency: a 21st century perspective. Clin. Med. (Lond.) 15, 145–150 10.7861/clinmedicine.15-2-145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lai, S. C., Nakayama, Y., Sequeira, J. M., Wlodarczyk, B. J., Cabrera, R. M., Finnell, R. H., Bottiglieri, T., Quadros, E. V. (2013) The transcobalamin receptor knockout mouse: a model for vitamin B12 deficiency in the central nervous system. FASEB J. 27, 2468–2475 10.1096/fj.12-219055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Arora, K., Sequeira, J. M., Hernández, A. I., Alarcon, J. M., Quadros, E. V. (2017) Behavioral alterations are associated with vitamin B12 deficiency in the transcobalamin receptor/CD320 KO mouse. PLoS One 12, e0177156 10.1371/journal.pone.0177156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Arora, K., Sequeira, J. M., Quadros, E. V. (2017) Maternofetal transport of vitamin B12: role of TCblR/CD320 and megalin. FASEB J. 31, 3098–3106 10.1096/fj.201700025R [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Paul, L., Selhub, J. (2017) Interaction between excess folate and low vitamin B12 status. Mol. Aspects Med. 53, 43–47 10.1016/j.mam.2016.11.004 [DOI] [PubMed] [Google Scholar]
- 26. Quadros, E., Song, W., Beecher, G., Eitenmiller, R. (2000) Vitamin B12. In Modern Analytical Methodologies in Fat-and Water-Soluble Vitamins, pp. 313–328, Wiley, Hoboken, New Jersey, USA.
- 27.Inoue-Choi, M., Nelson, H. H., Robien, K., Arning, E., Bottiglieri, T., Koh, W. P., Yuan, J. M. (2012) One-carbon metabolism nutrient status and plasma S-adenosylmethionine concentrations in middle-aged and older Chinese in Singapore. Int. J. Mol. Epidemiol. Genet. 3, 160–173 [PMC free article] [PubMed] [Google Scholar]
- 28.Ogburn, K. D., Bottiglieri, T., Wang, Z., Figueiredo-Pereira, M. E. (2006) Prostaglandin J2 reduces catechol-O-methyltransferase activity and enhances dopamine toxicity in neuronal cells. Neurobiol. Dis. 22, 294–301 10.1016/j.nbd.2005.11.006 [DOI] [PubMed] [Google Scholar]
- 29.Wasek, B., Arning, E., Bottiglieri, T. (2018) The use of microwave irradiation for quantitative analysis of neurotransmitters in the mouse brain. J. Neurosci. Methods 307, 188–193 10.1016/j.jneumeth.2018.05.016 [DOI] [PubMed] [Google Scholar]
- 30.Jacob, J. J., Ramabadran, K. (1978) Enhancement of a nociceptive reaction by opioid antagonists in mice. Br. J. Pharmacol. 64, 91–98 10.1111/j.1476-5381.1978.tb08645.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ahmed, Z. (2014) Trans-spinal direct current stimulation modifies spinal cord excitability through synaptic and axonal mechanisms. Physiol. Rep. 2, e12157 10.14814/phy2.12157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cain, D. M., Khasabov, S. G., Simone, D. A. (2001) Response properties of mechanoreceptors and nociceptors in mouse glabrous skin: an in vivo study. J. Neurophysiol. 85, 1561–1574 10.1152/jn.2001.85.4.1561 [DOI] [PubMed] [Google Scholar]
- 33.Gartner, L. P., Hiatt, J. L. (2001) Colour Textbook of Histology. WB Saunders, London, United Kingdom [Google Scholar]
- 34.Schmalbruch, H. (1986) Fiber composition of the rat sciatic nerve. Anat. Rec. 215, 71–81 10.1002/ar.1092150111 [DOI] [PubMed] [Google Scholar]
- 35.Quadros, E. V., Lai, S. C., Nakayama, Y., Sequeira, J. M., Hannibal, L., Wang, S., Jacobsen, D. W., Fedosov, S., Wright, E., Gallagher, R. C., Anastasio, N., Watkins, D., Rosenblatt, D. S. (2010) Positive newborn screen for methylmalonic aciduria identifies the first mutation in TCblR/CD320, the gene for cellular uptake of transcobalamin-bound vitamin B(12). Hum. Mutat. 31, 924–929 10.1002/humu.21297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fernàndez-Roig, S., Lai, S. C., Murphy, M. M., Fernandez-Ballart, J., Quadros, E. V. (2012) Vitamin B12 deficiency in the brain leads to DNA hypomethylation in the TCblR/CD320 knockout mouse. Nutr. Metab. (Lond.) 9, 41 10.1186/1743-7075-9-41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chiang, P. K., Gordon, R. K., Tal, J., Zeng, G. C., Doctor, B. P., Pardhasaradhi, K., McCann, P. P. (1996) S-Adenosylmethionine and methylation. FASEB J. 10, 471–480 10.1096/fasebj.10.4.8647346 [DOI] [PubMed] [Google Scholar]
- 38. Kunze, K., Leitenmaier, K. (1976) Vitamin B12 deficiency and subacute combined degeneration of the spinal cord. In Handbook of Clinical Neurology, pp. 141–198, North Holland Publishing Co, Amsterdam, The Netherlands.
- 39.Pant, S. S., Asbury, A. K., Richardson, E. P., Jr. (1968) The myelopathy of pernicious anemia: a neuropathological reappraisal. Acta Neurol. Scand. 44(S35 Suppl 5), 7–36 [PubMed] [Google Scholar]
- 40.Roos, D. (1978) Neurological complications in patients with impaired vitamin B12 absorption following partial gastrectomy. Acta Neurol. Scand. Suppl. 69, 1–77 [PubMed] [Google Scholar]
- 41.Hemmer, B., Glocker, F. X., Schumacher, M., Deuschl, G., Lücking, C. H. (1998) Subacute combined degeneration: clinical, electrophysiological, and magnetic resonance imaging findings. J. Neurol. Neurosurg. Psychiatry 65, 822–827 10.1136/jnnp.65.6.822 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kristensson, K., Holmes, K. V., Duchala, C. S., Zeller, N. K., Lazzarini, R. A., Dubois-Dalcq, M. (1986) Increased levels of myelin basic protein transcripts in virus-induced demyelination. Nature 322, 544–547; erratum: 323, 91 10.1038/322544a0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Loughlin, A. J., Copelman, C. A., Hall, A., Armer, T., Young, B. C., Landon, D. N., Cuzner, M. L. (1997) Myelination and remyelination of aggregate rat brain cell cultures enriched with macrophages. J. Neurosci. Res. 47, 384–392 [DOI] [PubMed] [Google Scholar]
- 44.Wang, X., Cao, K., Sun, X., Chen, Y., Duan, Z., Sun, L., Guo, L., Bai, P., Sun, D., Fan, J., He, X., Young, W., Ren, Y. (2015) Macrophages in spinal cord injury: phenotypic and functional change from exposure to myelin debris. Glia 63, 635–651 10.1002/glia.22774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Garrison, C. J., Dougherty, P. M., Carlton, S. M. (1994) GFAP expression in lumbar spinal cord of naive and neuropathic rats treated with MK-801. Exp. Neurol. 129, 237–243 10.1006/exnr.1994.1165 [DOI] [PubMed] [Google Scholar]
- 46.Garrison, C. J., Dougherty, P. M., Kajander, K. C., Carlton, S. M. (1991) Staining of glial fibrillary acidic protein (GFAP) in lumbar spinal cord increases following a sciatic nerve constriction injury. Brain Res. 565, 1–7 10.1016/0006-8993(91)91729-K [DOI] [PubMed] [Google Scholar]
- 47.Scalabrino, G. (2009) The multi-faceted basis of vitamin B12 (cobalamin) neurotrophism in adult central nervous system: lessons learned from its deficiency. Prog. Neurobiol. 88, 203–220 10.1016/j.pneurobio.2009.04.004 [DOI] [PubMed] [Google Scholar]
- 48.Friedlander, R. M. (2003) Apoptosis and caspases in neurodegenerative diseases. N. Engl. J. Med. 348, 1365–1375 10.1056/NEJMra022366 [DOI] [PubMed] [Google Scholar]
- 49.Vila, M., Przedborski, S. (2003) Targeting programmed cell death in neurodegenerative diseases. Nat. Rev. Neurosci. 4, 365–375 10.1038/nrn1100 [DOI] [PubMed] [Google Scholar]
- 50.Olmos, G., Lladó, J. (2014) Tumor necrosis factor alpha: a link between neuroinflammation and excitotoxicity. Mediators Inflamm. 2014, 861231 10.1155/2014/861231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Rigaud, M., Gemes, G., Barabas, M. E., Chernoff, D. I., Abram, S. E., Stucky, C. L., Hogan, Q. H. (2008) Species and strain differences in rodent sciatic nerve anatomy: implications for studies of neuropathic pain. Pain 136, 188–201 10.1016/j.pain.2008.01.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Swett, J. E., Torigoe, Y., Elie, V. R., Bourassa, C. M., Miller, P. G. (1991) Sensory neurons of the rat sciatic nerve. Exp. Neurol. 114, 82–103 10.1016/0014-4886(91)90087-S [DOI] [PubMed] [Google Scholar]
- 53.Chung, T., Prasad, K., Lloyd, T. E. (2014) Peripheral neuropathy: clinical and electrophysiological considerations. Neuroimaging Clin. N. Am. 24, 49–65 10.1016/j.nic.2013.03.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Saporta, M. A., Katona, I., Lewis, R. A., Masse, S., Shy, M. E., Li, J. (2009) Shortened internodal length of dermal myelinated nerve fibres in Charcot-Marie-Tooth disease type 1A. Brain 132, 3263–3273 10.1093/brain/awp274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sumner, A. J., Saida, K., Saida, T., Silberberg, D. H., Asbury, A. K. (1982) Acute conduction block associated with experimental antiserum-mediated demyelination of peripheral nerve. Ann. Neurol. 11, 469–477 10.1002/ana.410110506 [DOI] [PubMed] [Google Scholar]
- 56.Watkins, D., Rosenblatt, D. S. (2011) Inborn errors of cobalamin absorption and metabolism. Am. J. Med. Genet. C. Semin. Med. Genet. 157C, 33–44 10.1002/ajmg.c.30288 [DOI] [PubMed] [Google Scholar]
- 57.Baron, W., Hoekstra, D. (2010) On the biogenesis of myelin membranes: sorting, trafficking and cell polarity. FEBS Lett. 584, 1760–1770 10.1016/j.febslet.2009.10.085 [DOI] [PubMed] [Google Scholar]