

Abstract
Introduction
Bone marrow-derived multipotent adult progenitor cells (MAPCs) are adult allogeneic adherent stem cells currently investigated clinically for use in acute respiratory distress syndrome (ARDS). To date, there is no agreement on which is the best method for stem cells delivery in ARDS. Here, we compared the efficacy of two different methods of administration and biodistribution of MAPC for the treatment of ARDS in a sheep model.
Methods
MAPC were labelled with [18F] fluoro-29-deoxy-D-glucose and delivered by endobronchial (EB) or intravenous route 1 hour after lipopolysaccharide infusion in sheep mechanically ventilated. PET/CT images were acquired to determine the biodistribution and retention of the cells at 1 and 5 hours of administration.
Results
The distribution and retention of the MAPC was dependent on the method of cell administration. By EB route, PET images showed that MAPC remained at the site of administration and no changes were observed after 5 hours, whereas with intravenous route, the cells had broad biodistribution to different organs, being the lung the main organ of retention at 1 and 5 hours. MAPC demonstrated an equal effect on arterial oxygenation recovery by either route of administration.
Conclusion
The EB or intravenous routes of administration of MAPC are both effective for the treatment of ARDS in an acute sheep model, and the effect of MAPC therapy is not dependent of parenchymal integration or systemic biodistribution.
Keywords: Multipotent Adult Progenitor Cells (MAPC), Mesenchymal stem cell (MSC), lung injury, Acute Respiratory Distress Syndrome (ARDS)
Key messages.
Does the route of administration of stem cells affect their efficacy and biodistribution?
While the biodistribution of the cells differs depending on the administration route, both routes of administration are equally effective for the treatment of acute respiratory distress syndrome (ARDS).
This study proves two alternatives for the administration of cells for therapy in ARDS that are safe and have similar efficacy; the choice for the route of administration might be of crucial interest depending on the technical limitations presented at the time of treatment.
Introduction
Acute respiratory distress syndrome (ARDS) is a devastating lung condition. Currently, it is the leading cause of death and disability in critically ill patients, with a mortality rate that ranges between 26% and 50%.1–3 ARDS is an inflammatory state; characterised by infiltration of mixed inflammatory cells, diffuse destruction of the alveolar–capillary barrier, severe oedema, consequent hypoxaemia and increase in lung density.4 5 To date, ARDS is only managed with supportive care such as lung protective ventilation, prone positioning and conservative fluid management; and no definitive treatment is available.4 6–9 Cell therapy with multipotent adult progenitor cells (MAPCs) and mesenchymal stem cells (MSCs), two predominant adult stem cell types of the bone marrow stroma, are promising therapeutic options for patients with ARDS.10–16 These cells hold potent immunomodulatory and repair effects. Unlike most adult somatic stem cells, MAPCs appear to proliferate without senescence, have pluripotent differentiation ability in vitro and in vivo and do not express major histocompatibility (MHC) class II antigens.17 18 Previous experimental studies published by our group and others have demonstrated that bone marrow-derived stem cells administered by either intra-tracheal and intravenous route, are able to decrease lung inflammation, enhance lung repair, restore alveolar fluid clearance and improve arterial oxygenation.12 15 19 20 Although the efficacy of cell therapy in ARDS has been demonstrated in animal models, including our sheep lipopolysaccharide (LPS)-induced lung injury model, it is unknown how different routes of administration can affect homing and retention of those cells in ARDS.
Positron emission tomography (PET) has been successfully used in several studies as a quantitative method of cell tracking and localisation in vivo.21–23 The [18F] fluoro-29 -deoxy-D-glucose ([18F] FDG), a glucose analogue, is a positron-emitting radionuclide that is metabolically trapped inside the cells after phosphorylation by hexokinase, and has been successfully used to label and track stem cells.21–24
Objective
Although the endobronchial (EB) and intravenous routes of administration have been recommended for cell therapy in ARDS, the differences of homing and retention of the cells after delivery are currently unknown. Therefore, we aimed to compare the biodistribution of MAPC by EB or intravenous administration and their therapeutic effect in a sheep model of LPS-induced lung injury.
Material and methods
Animal model
All animals received human care in compliance with the ‘Principles of Laboratory Animal Care’ formulated by the National Society for Medical Research and the ‘Guide for the Care and Use of Laboratory Animals’ prepared by the Institute of Laboratory Animal Resources and published by the National Institutes of Health (NIH) (NIH no. 86–23). Eleven adult Dorsett Cross sheep weighing 30–40 kg were used in the present study. Both females and males were included in a relationship of 1:1. Each sheep was fasted overnight and pre-medicated with intramuscular atropine (0.05 mg/kg), and after anaesthesia induction with intravenous ketamine, general anaesthesia was maintained with isoflurane (1.5%–2%) for 6 hours. Animals were placed in prone position on the PET scanner table and mechanically ventilated with PEEP of 5 cmH2O, FiO2 of 0.9, I:E ratio 1:2, tidal volume of 8–10 mL/kg and respiratory rate adjusted to maintain the arterial carbon dioxide PaO2 between 35 and 45 mm Hg. The right carotid artery and a peripheral vein were cannulated and haemodynamic parameters were continuously measured. To induce acute lung injury, the sheep received 5 µg/kg intravenous LPS from Escherichia coli 055:B5 (Sigma, St. Louis, Missouri, USA) in normal saline (Baxter, Deerfield, Illinois, USA), over 21 min at a rate of 1 mL/min. Blood samples and arterial blood gases (ABGs) were collected at baseline and at 1, 2 and 6 hours after LPS or saline infusion. A CT was taken during baseline and PET/CT scans were acquired 1 and 5 hours after cells or free tracer delivery (figure 1A). The degree of lung damage was evaluated by oxygenation and variation of lung density in CT scans measured by hounsefield (HU) units.
Figure 1.

Experimental protocol. (A) Timeline: after anaesthesia, a CT scan, blood samples and arterial blood gases (ABGs) were collected before lipopolysaccharide (LPS)/saline and 1, 2 and 6 hours after infusion. Labelled multipotent adultprogenitorcells (MAPCs) or free tracer was delivered 1 hour after LPS/saline infusion. PET/CT scans were acquired 1 and 5 hours after cells or free tracer delivery. (B). Seven groups were studied. EB, endobronchial; [18F] FDG, [18F] fluoro-29-deoxy-D-glucose.
Cell lines
MAPCs were isolated from a human donor through bone marrow aspiration. Cell isolation was processed according to previously described methods.25 Briefly, MAPCs were cultured in fibronectin-coated plastic tissue culture flasks. Cell cultures were maintained under low oxygen tension in a humidified atmosphere of 5% CO2. Cells were cultured in a media containing low-glucose (D)MEM (Life Technologies, Grand Island, New York, USA) supplemented with fetal bovine serum (Atlas, Fort Collins, Colorado, USA), ITS liquid media supplement (Sigma), MCDB (Sigma), platelet-derived growth factor (R&D Systems, Minneapolis, Minnesota, USA), epidermal growth factor (R&D Systems), dexamethasone, penicillin/streptomycin (Life Technologies), 2-phospho-L-ascorbic acid and linoleic acid-albumin (Sigma). Cells were passaged every 3–4 days and harvested using trypsin/EDTA (Life Technologies). The cells were positive for CD49c and CD90 and negative for MHC class II and CD45 (all antibodies (Abs) were from BD Biosciences, San Jose, CaliforniaA, USA). Cells were cryopreserved in media and 5% dimethyl sulfoxide. Before administration, the MAPCs were counted with trypan blue exclusion, and the final concentration was adjusted according with the percentage of live cells.
[18F] FDG MAPC labelling
MAPC were labelled following protocol previously described in detail.26 The initial mixing and incubation processes were conducted in a Class 100 laminar airflow hood. In brief, cells were resuspended in [18F] FDG (provided by Zevacor) in total volume <1.0 mL. The cells were gently mixed and then incubated in a warm water bath for 1 hour with gentle agitation every 5 min. The cell labelling reaction was then centrifuged at 2000 RPM (750 × g) for 7 min to pellet the cells. The cells were rinsed to remove any residual [18F] FDG by removing the supernatant and resuspending the cell fraction in 2.0 mL of 0.9% saline for injection. The rinse procedure was repeated two additional times. Following the final rinse, the cells were resuspended in 0.9% saline for injection in the desired volume for administration. The decay corrected cell labelling yield averaged 68%±19% (n=8).
We also evaluated the stability of the labelled MAPCs. In a single experiment, we incubated the MAPC in 0.9% saline for injection at (37°C±1°C) for 1 hour following the labelling procedure. At the end of the hour, the rinse procedure described above was performed (in triplicate) and the amount of radioactivity in the supernatant was determined. The activity in the supernatant accounted for 35% of the total radioactivity in the labelled cell fraction.
PET/CT scan
A baseline CT including the whole thorax was acquired at breathhold. In the case of LPS administration, a second breathhold CT was acquired 1 hour post insult. Either [18F] FDG or FDG-labelled cells were administered by intravenous or EB route. Also, 1 and 5 hours after radio-isotope administration, a whole-body PET/CT scan was obtained using a Biograph mCT Flow PET/CT scanner (Siemens Medical Solutions USA, Malvern, Pennsylvania, USA). A ‘step-and-shoot’ PET acquisition was employed using multiple 3 min bed positions to cover the head through the pelvis. The corresponding CT scans were acquired at intermediate breathhold to match to the average position during respiration as far as possible, though some degree of mismatch at the location of the diaphragm was noted in the resulting images. Respiratory gating was not employed. PET data were reconstructed using the supplied ultraHD-PET iterative algorithm with all quantitative corrections applied and the CT images were used for attenuation correction. To assess quantitatively biodistribution, numerical PET results were presented as standard uptake values (SUVs) normalised to injected dose and animal weight. Two regions of interest (ROIs) were defined for lung field analysis using the HU scale. The ROIs used for analysis were selected using the baseline CT scans to sample large representative regions of well-aerated (−900 to −501 HU) and moderately aerated (−500 to −101 HU) lung. Careful inspection in choosing suitable regions was required as these were ruminant animals with considerable evidence of prior lung infections, and because atelectasis could also be present due to the use of ventilation. The ROIs were trivially transferred to the later time point images as the animal was continually ventilated and did not move between the scans. Evaluations were made by a board-certified radiologist blinded on experimental subgroup assignation.
Experimental protocol
We studied seven groups (figure 1B).
Experimental groups
Also, 1 hour after LPS infusion two groups received [18F] FDG-labelled MAPC: group 1 (10 million cells/kg by intravenous route, n=3), group 2 (1 million cells/kg by EB route, n=3).
Control groups
After receiving the same dose of LPS, two groups received free [18F] FDG: group 3 (intravenous-free tracer, n=1) and group 4 (EB-free tracer, n=1). Additionally, the biodistribution of EB and intravenous labelled cells in a non-injured animal (saline solution) was also assessed using the same amount of cells in each route: group 5 (intravenous cells, n=1) and group 6 (EB cells, n=1). Finally, the intravenous-free tracer was also evaluated in a non-injured animal (group 7 (IV-free tracer), n=1). The number of cells used during the experiment was calculated in accordance with results from previous preliminary data. Cells delivered by EB route were administered in the lung lobe with higher degree of injury assessed by CT scan.
Statistical analysis
The biodistribution data were analysed using SPSS V.16.0. Comparison between groups was made using the Student’s t-test and two-way analysis of variance as appropriate using GraphPad Prism V.7 (GraphPad Software, San Diego, California, USA). Statistical significance was determined by p value<0.05. We calculate a sample size of at least one subject per group in order to describe the changes after each experiment.
Results
Experimental groups (1 and 2; figure 1) consisted of a mix of female and male sheep averaging 35 and 44 kg, respectively. All groups had normal baseline levels of oxygenation (PaO2/FiO2 levels above 350 mm Hg), a respiration rate of 10 breaths per minute and normal haemodynamics and biochemistries (heart rate averaging between 95 and 99 beats per minute, mean arterial pressure of 61–103 mm Hg and all glucose and protein values within normal levels, indicating no liver, pancreas or kidney damage; see online supplementary figures).
bmjresp-2018-000308supp001.pdf (387.4KB, pdf)
Biodistribution by intravenous route
The PET/CT images acquired 1 and 5 hours after intravenous injection of labelled MAPCs showed a systemic biodistribution of the cells to many organs with special homing and retention in the lung after 5 hours, in both groups, LPS/intravenous cells (n=3) and naive/intravenous cells (n=1) (figure 2A, B). The intensity of labelled cell uptake was heterogeneous in both lungs, with a predominant preference for inferior lobules. Additionally, a decrease of such intensity was seen after 5 hours.
Figure 2.

PET/CT fusion images. Coronal section of whole body and transverse section of the thorax at 1 and 5 hours after intravenous injection of labelled multipotent adultprogenitorcells (MAPCs) in lipopolysaccharide (LPS) (A) and naive (B) sheep and free tracer in LPS (C) and naive (D) sheep. (A) LPS/intravenous cells groups (n=3): 1 hour post-intravenous labelled cells infusion showed a systemic distribution of the cells with an important homing and retention by the lungs, which faded after 5 hours. (B) Naive/IV cells group (n=1). (C) LPS/intravenous-free tracer group (n=1). (D) Naive/IV free tracer (n=1). Dashed lines represent selected transverse images.
The quantitative analysis of the labelled cell uptake showed that at the first hour the majority of the cells were retained in the lungs (table 1). However, the amount of cells detected was reduced after 5 hours, decreasing 42.3% in naive/intravenous cells (figure 3A)(figure 3A) and only 20% in LPS/intravenous cells group (figure 3B). Analysis of other organs (liver, kidney and brain) reported minor cell retention compared with lungs in the LPS group, with no differences among them in the two times points. Interestingly, in naive sheep, a considerable amount of the cells were retained in the brain after 1 hour, with very low percentage of clearance (16%) after 5 hours.
Table 1.
Quantitative analysis of labelled cells uptake in different organs, in lipopolysaccharide (LPS) and naive groups
| Organ | Standard uptake values mean | |||
| 1 hour post intravenous cells injection | 5 hour post intravenous cells injection | |||
| LPS | Naive | LPS | Naive | |
| Lung | 8.0±3.8 | 7.45 | 6.4±2.7 | 4.3 |
| Liver | 2.2±0.8 | 1.7 | 1.9±0.5 | 0.6 |
| Kidney | 2.7±0.6 | 3.6 | 2.8±0.1 | 2 |
| Brain | 3.0±1.3 | 6.6 | 3.2±0.5 | 5.5 |
Labelled cells showed systemic distribution with an important retention in the lungs that persisted after 5 hours. Retention of cells was notably higher in the injured lung, compared with naive lung. Data are expressed as mean±SD.
Figure 3.

Quantitative analysis of labelled cells uptake in different organs, in lipopolysaccharide (LPS) and naive groups. Labelled cells showed systemic distribution with an important retention in the lungs that persisted after 5 hours. (A, B) Retention of cells was notably higher in the injured lung (80%), compared with the naive lung (57.7%). Data are expressed as % of change after cells injection.
Due to the heterogeneous uptake of cells in the lung parenchyma, two ROIs were defined: normally aerated lung tissue (−900 to −501 HU) and poorly aerated lung tissue (−500 to −101 HU; figure 4). The retention of labelled cells was higher by poorly aerated regions, especially in the LPS group (p<0.005) and remained highly concentrated in these regions after 5 hours.
Figure 4.

Evaluation of SUVmax in the lung by regions of interest (RIOs). Labelled cells were highly retained in poorly aerated regions, especially in the lipopolysaccharide (LPS) group (*p<0.005), where cells remained highly concentrated in these regions after 5 hours. No significant decrease of cell retention was reported after 5 hours in either the LPS or the naive group. The small amount of free tracer was equally distributed in both regions with no significant differences between them.[18F]FDG, [18F] fluoro-29-deoxy-D-glucose; SUV, standard uptake values.
To determine whether the biodistribution of the labelled cells traced in PET images could correspond to radiotracer efflux, we tracked free [18F]-FDG in a naive sheep and a sheep with LPS-induced acute lung injury at 1 and 5 hours after intravenous injection of the tracer. Contrary to labelled cells, the intravenous-free tracer did not show retention preference to any organ after 1 and 5 hours of administration in both groups (figures 2C and 3D). The lung was the organ with less uptake of the tracer in control groups, both LPS and naive. Kidney and brain were the organs with higher free tracer uptake over time in both groups.
Biodistribution by EB route
The EB-labelled cells or free tracer was delivered in the right lower lobe, with the exception of one sheep in which the cells were administered in the left lower lobe. The PET/CT images in the EB groups showed a small focal area of intense uptake in the lung parenchyma, corresponding to the site where the cells were delivered. The labelled cells did not spread in the lung parenchyma or to other organs, at 1 and 5 hours after administration in LPS (figure 5A) and naive (figure 5B) groups. In the image inspection, no differences were found between the two groups. The distribution of the free tracer, with no cells, was also evaluated in an LPS sheep. In a similar pattern, the free tracer remained focally located in the area where it was administrated and no signal of the tracer was detected in any other organ (figure 5C). No EB-free tracer was evaluated after the intraoperative loss of the subjects assigned to this experiment.
Figure 5.

PET of whole body distribution in coronal section at 1 and 5 hours after labelled multipotent adultprogenitorcells (MAPCs) delivery intravenous or endobronchial (EB) in the acute respiratory distress syndrome (ARDS) model. (A) 1 hour post-intravenous labelled cell infusion showed a systemic distribution of the cells with an important homing and retention in the lungs that faded after 5 hours. (B) EB administration of labelled cells in the naive sheep showed retention in the compartment in which they were delivered with some of them remaining in the lower airway, and no differences after 5 hours were found. Dashed lines represent selected transverse images.
CT images and quantitative analysis
Using CT imaging, the lung tissue density was evaluated. The LPS group presented higher density in ROIs after 1 hour endotoxin infusion compared with naive groups (figure 6). The LPS group that received intravenous cells showed a significant decrease in the radiological attenuation at 1 hour post administration. However, there were no differences in density between groups after 5 hours.
Figure 6.

Per cent change of lung density (HU) evaluated at 1, 2 and 6 hours after lipopolysaccharide (LPS). The cells were injected 1 hour after LPS or saline by intravenous or endobronchial (EB) routes in the lipopolysaccharide (LPS) (A) or naive (B) groups. *p<0.05 between two time points. [18F]FDG, [18F] fluoro-29-deoxy-D-glucose;
ARDS and cell therapy
We have previously reported that a single dose of LPS (3.5 mg/kg) was able to cause progressive hypoxaemia within the first hours after infusion in sheep on right lateral recumbency.12 In this study, we compared the effect of MAPC administered by EB or intravenous route on arterial oxygen (PO2/FiO2) levels. After LPS infusion, a reduction in PO2/FiO2 values was observed in both groups, reaching levels of hypoxaemia. MAPC were administered 1 hour after LPS, and the PO2/FiO2 levels recovered to normal values in both groups within the first hour of cell delivery, remaining constant throughout the study. There were no significant differences in arterial oxygenation between both routes of administration (figure 7). However, the group of sheep that after LPS did not receive MAPC (free tracer groups 3 and 4), significantly worsened PO2/FiO2 ratio at the end of the experiments, concordantly with an ARDS model.
Figure 7.
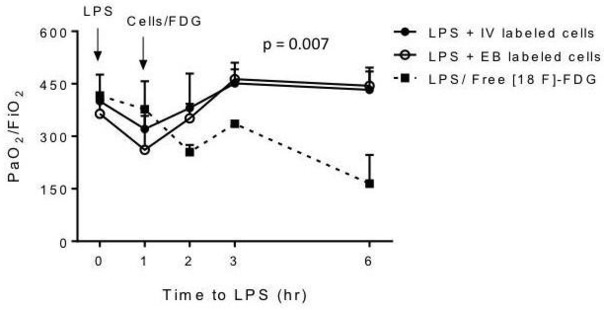
Effect of lipopolysaccharide (LPS) and multipotent adultprogenitorcells (MAPCs) on the PaO2/FiO2 ratio. After the administration of MAPCs, the PaO2/FiO2 ratio recovered and remained in normal ranges until the study was completed. No differences were observed between both routes of administration. The LPS group that did not received MAPCs (groups 3 and 4) significantly worsened PaO2/FiO2 ratio after LPS infusion. EB, endobronchial.
Haemodynamic and metabolic variables
In the LPS groups, the heart rate remained stable throughout the study and there were no changes after either EB or intravenous MACP administration. As expected in an endotoxin model of ARDS,12 27 28 MAP decreased in both groups showing lower readings in LPS/EB cells group after cell administration. Serum glucose, BUN, creatinine, alkaline phosphatase and gamma-glutamyl transferase were measured before and after cell administration, revealing no differences between both groups. Alanine aminotransferase concentration decreased in both groups but remained within normal range after cell administration (table 2).
Table 2.
Routine chemistry in the multipotent adultprogenitorcells groups for both routes of administration
| Haemodynamics and chemical variables | LPS/before cells | LPS/after cells | ||||
| Intravenou | EB | P value | Intravenous | EB | P value | |
| HR, bmp | 98.6±9.7 | 103.6±7.8 | 0.24 | 94.7±6.8 | 91.6±7 | 0.11 |
| MAP, mm Hg | 55.8±5.8 | 75.6±26.6 | 0.05 | 44±4.3 | 39±3.6 | 0.01 |
| Glucose | 60.2±16 | 65.6±8.3 | 0.59 | 20±6 | 36.6±12 | 0.32 |
| BUN, mg/dL | 12.5±0.5 | 14.6±2.7 | 0.45 | 13.9±0.05 | 19.6±2.3 | 0.13 |
| Creatinine, mg/dL | 0.85±0.15 | 0.6±0.03 | 0.5 | 1.1±0.2 | 1.8±0.06 | 0.14 |
| ALT, IU/L | <10 ± 0 | 17.3±0.8 | 0.12 | 15.6±1 | 16.5±0.5 | 0.04 |
| ALK P, IU/L | 92.6±28.5 | 178.6±68 | 0.42 | 126.5±45 | 257.6±82 | 0.26 |
| GGT, U/L | 29.5±0.5 | 33.6±2.3 | 0.54 | 36.5±0.5 | 36.3±3.1 | 0.96 |
Kidney and liver panels are within normal range. The EB group had a higher increase in MAP after infusion of LPS compared with the IT group. After the infusion of the cells both groups maintained similar values throughout the experiment. HR was initially high in both groups and was maintained stable during the entire experiment.
ALK, alkaline phosphatase; ALT, alanine aminotransferase; EB, endobronchial; GGT, gamma-glutamyl transferase; HR, heart rate; IT, Intratracheal; LPS, lipopolysaccharide; MAP, mean arterial pressure.
Discussion
Our study compares the efficacy of two methods of administration of MAPC for the treatment of ARDS in a sheep model. We evaluated the early distribution and retention of MAPCs delivered by EB and intravenous route, using PET/CT to track the cells in vivo. Despite showing different cell biodistribution, the therapeutic benefit of MAPC was similar by either EB or intravenous route of administration.
This study evaluates for the first time the feasibility of using endobronchial [18F]-FDG as a way to assess MAPC biodistribution. By this route of administration, we observed a trapping of labelled cells inside the lung with no systemic distribution. In ARDS, the equilibrium between the lung interstitial tissue and the capillary vessels is disrupted in early stages of the disease29 and the possible migration of the cells would hypothetically occur in the early stages. The permanence of the cells inside the lower airways after EB delivery observed in this study may be due to multiple factors: the integrity of the alveolar capillary membrane, the absence of signalling factors from other organs to promote the cell migration and the lack of mechanisms of cell diffusion from inside the alveoli to the systemic circulation. It is possible that the biodistribution of MAPC could be more extensive in advanced stages.
The intravenous route, as we expected, resulted in a systemic distribution of the cells, with a predominant retention in the lung. This finding is consistent with the results presented on other studies using PET in models different to ARDS, where the stem cells were trapped in the lung after intravenous injection.21 24 30 Although there was a slight clearance of MAPC after 5 hours, the majority of cells remained trapped in the lungs, predominantly in injured areas from lungs that received LPS. These findings suggest that the injury could be a mechanism to retain cells, concordantly with previous studies, that recommend intravenous route for cellular therapy in ARDS.16 31–34 Cell retention in the brain, liver and kidney was lower in the LPS group.
Independently of cell biodistribution, we observed similar effects on arterial oxygenation either in EB or intravenous routes. This finding suggests that the beneficial effect of MAPC is not dependent on parenchymal integration or systemic biodistribution of the cells. Instead, a strong paracrine capacity might be the principal mechanism that contributes to immunomodulation and tissue repair.,
Both routes of administration have potential limitations. Intravenous administration requires large quantities of cells to guarantee the delivery of an effective therapeutic cell number to the target organ due to the fact that cells are trapped in the lung capillaries in addition to the spleen, liver and kidney.35 The trapping effect is the major determinant of vascular obstruction and complications emerging thereof.35 36 Lung and cerebral microembolism are another important adverse event described by intravenous cell delivery and it might be a matter for concern when high cell doses are considered due to risk for clotting formation.37 However, different doses have been tested in the past for efficacy and toxicity in both routes of administration. No adverse effects have been reported with a dose of 10×106 cells/kg but a higher efficacy was observed than with a 5×106 cells/kg dose.38 In the present study, we did not observe any of the abovementioned adverse events and no animal died during or after cell delivery by intravenous and no haemodynamic changes such as tachycardia or severe hypotension were reported.
On the other hand, EB administration of the cells requires a flexible bronchoscopy, a procedure usually safe if performed by a trained specialist.39 However, for critically ill patients with acute hypoxaemic respiratory failure, this procedure may lead to deterioration of their condition. Complications such as severe hypoxaemia, cardiac arrhythmia, hypercapnia, haemorrhage, pneumothorax, laryngospasm and bronchospasm have been reported in these types of patients.40 Notably, as previously shown, we required 10-fold less stem cell number in the EB route compared with the intravenous route, ensuring the safety and efficacy of cells when administered intrabronchially.12
The use of non-invasive image modalities provides real-time information of the cells in vivo. Previous studies in small animals demonstrated the labelling of MAPC or MSCs with [18F] FDG do not affect their biological properties or their cell proliferative activity.23 26 The PET/CT image fusion allows obtaining anatomical references for regions with increased [18F] FDG uptake.41 SUV is the parameter most widely used for molecular image quantification. However, image inspection by SUV parameter in the EB route is limited by the fact that there is no distribution of cells over time.
In summary, labelled MAPCs showed different biodistribution patterns and retention in the lungs after EB and intravenous administration in our ARDS sheep model. Despite these findings and the lower dose used by EB route, we observed similar therapeutic benefits of MAPC delivered by both routes of administration. We also found that this method of cell labelling is safe and applicable in a large animal model. Therefore, this combined technique could also be useful for the conduction of human clinical trials.
Acknowledgments
The authors thank Dr Tomas Drabek for his clinical assistance with the animal work and Chandler for technical support.
Footnotes
NC and PA-V contributed equally.
Contributors: NC designed and analysed, coordinated the experiments and wrote the manuscript; PAV and JS analysed data and wrote the manuscript; JPC, DA, ZY, ES and BJL analysed data; EK, LL and SM conducted experiments; JAWS and AET provided reagents; MR conceived and designed this study, analysed data and wrote the manuscript.
Funding: This work was supported by National Institute of Health grant RO1 BMJ Open Respiratory Research HL123766-01A1.
Competing interests: None declared.
Patient consent for publication: Not required.
Ethics approval: The Institutional Animal Care and Use Committee (IACUC) for Animal Research of the University of Pittsburgh approved all experimental procedures in advance. Bone marrow aspirates were acquired with consent and in accordance with 21 CFR Part 1271 Human Cells, Tissues, and Cellular and Tissue Based Products and approved by the Institutional Review Board.
Provenance and peer review: Not commissioned; externally peer reviewed.
Data sharing statement: No additional data are available.
References
- 1.Bellani G, Laffey JG, Pham T, et al. . Epidemiology, patterns of care, and mortality for patients with acute respiratory distress syndrome in intensive care units in 50 countries. JAMA 2016;315:788–800. 10.1001/jama.2016.0291 [DOI] [PubMed] [Google Scholar]
- 2.Chiumello D, Coppola S, Froio S, et al. . What's next after ARDS: long-term outcomes. Respir Care 2016;61:689–99. 10.4187/respcare.04644 [DOI] [PubMed] [Google Scholar]
- 3.MacCallum NS, Evans TW. Epidemiology of acute lung injury. Curr Opin Crit Care 2005;11:43–9. 10.1097/00075198-200502000-00007 [DOI] [PubMed] [Google Scholar]
- 4.Fanelli V, Vlachou A, Ghannadian S, et al. . Acute respiratory distress syndrome: new definition, current and future therapeutic options. J Thorac Dis 2013;5:326–34. 10.3978/j.issn.2072-1439.2013.04.05 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.McCaffree DR, Rogers RM. The adult respiratory distress syndrome. Disease-a-month : DM. 27, 1981. [DOI] [PubMed] [Google Scholar]
- 6.Bersten AD, Cooper DJ. Better supportive care, less ARDS: just do it? Am J Respir Crit Care Med 2011;183:6–7. 10.1164/rccm.201007-1139ED [DOI] [PubMed] [Google Scholar]
- 7.Kallet RH. A comprehensive review of prone position in ARDS. Respir Care 2015;60:1660–87. 10.4187/respcare.04271 [DOI] [PubMed] [Google Scholar]
- 8.Bein T, Grasso S, Moerer O, et al. . The standard of care of patients with ARDS: ventilatory settings and rescue therapies for refractory hypoxemia. Intensive Care Med 2016;42:699–711. 10.1007/s00134-016-4325-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.National Heart, Lung, and Blood Institute Acute Respiratory Distress Syndrome (ARDS) Clinical Trials Network, Wiedemann HP, Wheeler AP, et al. . Comparison of two fluid-management strategies in acute lung injury. N Engl J Med 2006;354:909–75. 10.1016/j.jvs.2006.08.053 [DOI] [Google Scholar]
- 10.Huleihel L, Levine M, Rojas M. The potential of cell-based therapy in lung diseases. Expert Opin Biol Ther 2013;13:1429–40. 10.1517/14712598.2013.833603 [DOI] [PubMed] [Google Scholar]
- 11.Prockop DJ, Oh JY. Medical therapies with adult stem/progenitor cells (MSCs): a backward journey from dramatic results in vivo to the cellular and molecular explanations. J Cell Biochem 2012;113:n/a–9. 10.1002/jcb.24046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rojas M, Cárdenes N, Kocyildirim E, et al. . Human adult bone marrow-derived stem cells decrease severity of lipopolysaccharide-induced acute respiratory distress syndrome in sheep. Stem Cell Res Ther 2014;5:42. 10.1186/scrt430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rojas M, Parker RE, Thorn N, et al. . Infusion of freshly isolated autologous bone marrow derived mononuclear cells prevents endotoxin-induced lung injury in an ex-vivo perfused swine model. Stem Cell Res Ther 2013;4:26. 10.1186/scrt174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Devaney J, Horie S, Masterson C, et al. . Human mesenchymal stromal cells decrease the severity of acute lung injury induced by E. coli in the rat. Thorax 2015;70:625–35. 10.1136/thoraxjnl-2015-206813 [DOI] [PubMed] [Google Scholar]
- 15.La Francesca S, Ting AE, Sakamoto J, et al. . Multipotent adult progenitor cells decrease cold ischemic injury in ex vivo perfused human lungs: an initial pilot and feasibility study. Transplant Res 2014;3:19. 10.1186/2047-1440-3-19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rojas M, Xu J, Woods CR, et al. . Bone marrow-derived mesenchymal stem cells in repair of the injured lung. Am J Respir Cell Mol Biol 2005;33:145–52. 10.1165/rcmb.2004-0330OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jiang Y, Jahagirdar BN, Reinhardt RL, et al. . Pluripotency of mesenchymal stem cells derived from adult marrow. Nature 2002;418:41–9. 10.1038/nature00870 [DOI] [PubMed] [Google Scholar]
- 18.Jacobs SA, Roobrouck VD, Verfaillie CM, et al. . Immunological characteristics of human mesenchymal stem cells and multipotent adult progenitor cells. Immunol Cell Biol 2013;91:32–9. 10.1038/icb.2012.64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Danchuk S, Ylostalo JH, Hossain F, et al. . Human multipotent stromal cells attenuate lipopolysaccharide-induced acute lung injury in mice via secretion of tumor necrosis factor-α-induced protein 6. Stem Cell Res Ther 2011;2:27. 10.1186/scrt68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Matthay MA, Anversa P, Bhattacharya J. Cell therapy for lung diseases. Report from an NIH-NHLBI workshop. Am J Respir Crit Care Med 2012;188:370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Elhami E, Dietz B, Xiang B, et al. . Assessment of three techniques for delivering stem cells to the heart using PET and MR imaging. EJNMMI Res 2013;3:72. 10.1186/2191-219X-3-72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gao LR, Pei XT, Ding QA, et al. . A critical challenge: dosage-related efficacy and acute complication intracoronary injection of autologous bone marrow mesenchymal stem cells in acute myocardial infarction. Int J Cardiol 2013;168:3191–9. 10.1016/j.ijcard.2013.04.112 [DOI] [PubMed] [Google Scholar]
- 23.Elhami E, Goertzen AL, Xiang B, et al. . Viability and proliferation potential of adipose-derived stem cells following labeling with a positron-emitting radiotracer. Eur J Nucl Med Mol Imaging 2011;38:1323–34. 10.1007/s00259-011-1753-9 [DOI] [PubMed] [Google Scholar]
- 24.Sood V, Mittal BR, Bhansali A, et al. . Biodistribution of 18F-FDG-labeled autologous bone marrow-derived stem cells in patients with type 2 diabetes mellitus: exploring targeted and intravenous routes of delivery. Clin Nucl Med 2015;40:697–700. 10.1097/RLU.0000000000000850 [DOI] [PubMed] [Google Scholar]
- 25.Boozer S, Lehman N, Lakshmipathy U, et al. . Global characterization and genomic stability of human multistem, a multipotent adult progenitor cell. J Stem Cells 2009;4:17–28. doi:jsc.2009.4.1.17 [PubMed] [Google Scholar]
- 26.Wolfs E, Struys T, Notelaers T, et al. . 18F-FDG labeling of mesenchymal stem cells and multipotent adult progenitor cells for PET imaging: effects on ultrastructure and differentiation capacity. J Nucl Med 2013;54:447–54. 10.2967/jnumed.112.108316 [DOI] [PubMed] [Google Scholar]
- 27.Ballard-Croft C, Wang D, Sumpter LR, et al. . Large-animal models of acute respiratory distress syndrome. Ann Thorac Surg 2012;93:1331–9. 10.1016/j.athoracsur.2011.06.107 [DOI] [PubMed] [Google Scholar]
- 28.Squara P, Dhainaut JF, Artigas A, et al. . Hemodynamic profile in severe ARDS: results of the European collaborative ARDS Study. Intensive Care Med 1998;24:1018–28. 10.1007/s001340050710 [DOI] [PubMed] [Google Scholar]
- 29.Bellingan GJ. The pulmonary physician in critical care * 6: The pathogenesis of ALI/ARDS. Thorax 2002;57:540–6. 10.1136/thorax.57.6.540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Stojanov K, de Vries EF, Hoekstra D, et al. . [18F]FDG labeling of neural stem cells for in vivo cell tracking with positron emission tomography: inhibition of tracer release by phloretin. Mol Imaging 2012;11:1–12. 10.2310/7290.2011.00021 [DOI] [PubMed] [Google Scholar]
- 31.Ortiz LA, Gambelli F, McBride C, et al. . Mesenchymal stem cell engraftment in lung is enhanced in response to bleomycin exposure and ameliorates its fibrotic effects. Proc Natl Acad Sci U S A 2003;100:8407–11. 10.1073/pnas.1432929100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mei SH, McCarter SD, Deng Y, et al. . Prevention of LPS-induced acute lung injury in mice by mesenchymal stem cells overexpressing angiopoietin 1. PLoS Med 2007;4:e269. 10.1371/journal.pmed.0040269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mei SH, Haitsma JJ, Dos Santos CC, et al. . Mesenchymal stem cells reduce inflammation while enhancing bacterial clearance and improving survival in sepsis. Am J Respir Crit Care Med 2010;182:1047–57. 10.1164/rccm.201001-0010OC [DOI] [PubMed] [Google Scholar]
- 34.Xu YL, Liu YL, Wang Q. Intravenous transplantation of mesenchymal stem cells attenuates oleic acid induced acute lung injury in rats. Chin Med J 20122012;125:2012-8–8. [PubMed] [Google Scholar]
- 35.Fischer UM, Harting MT, Jimenez F, et al. . Pulmonary passage is a major obstacle for intravenous stem cell delivery: the pulmonary first-pass effect. Stem Cells Dev 2009;18:683–92. 10.1089/scd.2008.0253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Schrepfer S, Deuse T, Reichenspurner H, et al. . Stem cell transplantation: the lung barrier. Transplant Proc 2007;39:573–6. 10.1016/j.transproceed.2006.12.019 [DOI] [PubMed] [Google Scholar]
- 37.Lee RH, Pulin AA, Seo MJ, et al. . Intravenous hMSCs improve myocardial infarction in mice because cells embolized in lung are activated to secrete the anti-inflammatory protein TSG-6. Cell Stem Cell 2009;5:54–63. 10.1016/j.stem.2009.05.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Matthay MA. Therapeutic potential of mesenchymal stromal cells for acute respiratory distress syndrome. Ann Am Thorac Soc 2015;12S54–57. 10.1513/AnnalsATS.201406-254MG [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Beaudoin EL, Chee A, Stather DR. Interventional pulmonology: an update for internal medicine physicians. Minerva Med 2014;105:197–209. [PubMed] [Google Scholar]
- 40.Guerreiro da Cunha Fragoso E, Gonçalves JM. Role of fiberoptic bronchoscopy in intensive care unit: current practice. J Bronchology Interv Pulmonol 2011;18:69–83. 10.1097/LBR.0b013e31820700f6 [DOI] [PubMed] [Google Scholar]
- 41.Townsend DW, Beyer T. A combined PET/CT scanner: the path to true image fusion. Br J Radiol 2002;75 Spec No:S24–S30. 10.1259/bjr.75.suppl_9.750024 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
bmjresp-2018-000308supp001.pdf (387.4KB, pdf)