

Abstract
Pompe disease is an autosomal recessive glycogen storage disorder caused by deficiency of the lysosomal enzyme acid alpha-glucosidase (GAA). GAA deficiency results in systemic lysosomal glycogen accumulation and cellular disruption in muscle and the central nervous system (CNS). Adeno-associated virus (AAV) gene therapy is ideal for Pompe disease, since a single systemic injection may correct both muscle and CNS pathologies. Using the Pompe mouse (B6;129-GaaTm1Rabn/J), this study sought to explore if AAVB1, a newly engineered vector with a high affinity for muscle and CNS, reduces systemic weakness and improves survival in adult mice. Three-month-old Gaa–/– animals were injected with either AAVB1 or AAV9 vectors expressing GAA and tissues were harvested 6 months later. Both AAV vectors prolonged survival. AAVB1-treated animals had a robust weight gain compared to the AAV9-treated group. Vector genome levels, GAA enzyme activity, and histological analysis indicated that both vectors transduced the heart efficiently, leading to glycogen clearance, and transduced the diaphragm and CNS at comparable levels. AAVB1-treated mice had higher GAA activity and greater glycogen clearance in the tongue. Finally, AAVB1-treated animals showed improved respiratory function comparable to wild-type animals. In conclusion, AAVB1-GAA offers a promising therapeutic option for the treatment of muscle and CNS in Pompe disease.
Keywords: Pompe disease, AAVB1, AAV9, respiratory, glycogen
Introduction
Pompe disease is an autosomal recessive lysosomal storage disorder caused by mutations in the GAA gene encoding acid α-glucosidase (GAA)—a lysosomal enzyme responsible for the hydrolysis of glycogen to glucose. Deficiency in GAA results in accumulation of glycogen in lysosomes and subsequent cellular dysfunction in cardiac, skeletal, and smooth muscles as well as in the central nervous system (CNS).1–6 Pompe disease can present early in life as infantile onset Pompe disease (IOPD) or later in childhood to adulthood as late onset Pompe disease (LOPD). IOPD patients have little to no GAA activity and, if left untreated, typically develop cardiorespiratory failure within the first year of life.7 LOPD patients have some residual GAA activity resulting in a more variable presentation of muscle weakness and respiratory insufficiency.8–11 Respiratory failure is a prominent cause of death in both types of Pompe disease.2,11 The only Food and Drug Administration (FDA)-approved treatment for Pompe disease is enzyme replacement therapy (ERT). However, systemically administered GAA does not cross the blood–brain barrier and therefore cannot treat the CNS pathology and affected respiratory motor neurons. Furthermore, ERT only partially corrects skeletal muscle abnormalities as a result of low uptake into muscle fibers. Consequently, two-thirds of IOPD patients eventually require ventilatory support,12 and respiratory insufficiency persists among LOPD patients.13,14
Gene therapy using adeno-associated virus (AAV) vectors is ideal for Pompe disease, since it is a monogenetic disorder. Contingent on the serotype and promoter, AAV gene therapy has the potential to treat both the muscle and motor neuron pathologies following a single administration. AAV gene therapy for Pompe disease has been extensively studied in the Gaa–/– mouse model.15–25 These studies resulted in a Phase I/II clinical trial using AAV1-GAA intramuscular injections into the diaphragm of Pompe patients.26 This trial was the first to show that AAV1-GAA delivered to the diaphragm was safe and resulted in therapeutic benefits. Patients had increased unassisted tidal volumes and increased tolerance of unassisted breathing, with continued respiratory benefit seen 1 year post injection (p.i.).27 Furthermore, a clinical trial using AAV9-GAA injected intramuscularly into the tibialis anterior is currently underway to test the feasibility of gene therapy re-dosing.28
Systemic delivery of AAV9-GAA in Gaa–/– adult mice has yielded variable results, with some improvements in respiratory outcome measures but with variable glycogen clearance in muscle.17 An ever-evolving area within the gene therapy field is enhancing vector biology for more efficient transduction of disease relevant tissues. Many new capsid variants have emerged over the years by rational design or library screening. Thus, this study sought to explore novel vectors in order to optimize muscle and motor neuron transduction for Pompe disease. One of these new capsids, AAVB1, generated by library selection showed enhanced muscular and neuronal transduction compared to AAV9.29 In addition, this vector is more favorable for clinical application due to a reduced immune profile.29 Since muscle and CNS transduction is more efficient with AAVB1, it was hypothesized that AAVB1-GAA would be a more efficient therapy for overall glycogen clearance in Pompe disease.
Methods
Animals
All experimental procedures were approved by the Institutional Animal Care and Use Committee at the University of Massachusetts Medical School. B6;129-GaaTm1Rabn/J mice (stock number 004154) were obtained from Jackson Laboratory (Bar Harbour, ME) and were fed a standard mouse chow ad libitum. The mice were bred in-house by breeding B6;129-GaaTm1Rabn/J males to B6;129-GaaTm1Rabn/J females; animals heterozygous at the Gaa allele were used to create littermate controls. Prior to enrollment in the study, littermates were genotyped using standard polymerase chain reaction (PCR) methods to confirm the lack of Gaa gene.30,31 Adult male Gaa–/– (n = 36) and wild-type (WT) controls (n = 10) were enrolled in the study at 3 months of age. Gaa–/– mice were randomly and blindly assigned to a one of three treatment groups: AAV9 (n = 10), AAVB1 (n = 10), or phosphate buffered saline (PBS; n = 10). Six additional animals were enrolled in the PBS group because of the high death rate in this group and the increased number of animals needed for the physiological studies. All WT animals were injected with 200 μL of PBS.
AAV vectors
AAV9 and AAVB1 vectors were generated using previously described method.29 Vectors were purified by iodixanol gradient centrifugation followed by buffer exchange, as described previously.29 Final formulations of AAVB1-DES-hGAA and AAV9-DES-hGAA were in PBS. All vectors were generated and titered at the University of Massachusetts in the laboratory of Dr. Sena-Esteves using previously published methods.29 Either AAV9 or AAVB1 (1 × 1012 vector genomes [vg]) were injected in a total volume of 200 μL (or PBS for controls) into the tail vein of the mice.
Behavior testing
Four-limb grip strength was determined by a blinded investigator by measuring peak force using an Almeno® digital grip strength meter (Ahlborn, Holzkirchen, Germany) equipped with a mesh screen. Mice were placed on the metal grid until they gripped it, and then they were gently pulled by the tail until they could no longer hold the grid. The grip force test was repeated twice, with a 15 min rest period in between, and the maximum force was recorded.
Inverted screen testing was performed by an investigator blinded to the treatment group or genotype on an apparatus, as described previously.32 Animals were placed on a square wire mesh of 30 cm2 with 25 mm2 holes, over a cushioned surface, which was rotated 180°. Animals were assessed for latency to falling for up to 2 min. This was repeated twice per session, with a 15 min rest period in between, and the longest hang time was recorded.
Ventilation
These studies determined the impact of systemic vector injection on ventilation. Ventilation was quantified 180 days p.i. using whole-body plethysmography in unrestrained, unanesthetized mice, as previously described.5 An investigator blinded to the treatment group or genotype performed these studies. Awake, unrestrained mice were placed inside a 3.5″ × 5.75″ Plexiglas chamber (SCIREQ, Montreal, Canada), and data were collected in 10 s intervals. The Drorbaugh and Fenn equation33 was used to calculate respiratory volumes, including tidal volume and minute ventilation. During both a 30 min acclimation period and subsequent 60–90 min eupnea period, mice were exposed to normoxic air (FiO2: 0.21; nitrogen balance). Then, the mice underwent a 10 min hypercapnic challenge (FiO2: 0.21; FiCO2: 0.07; nitrogen balance).
Measures of airway resistance
Pulmonary mechanics were performed at study endpoint using forced oscillometry (FlexiVent system, SCIREQ) at baseline and in response to incremental doses of methacholine, as previously described.3 In brief, animals were anesthetized with an intraperitoneal injection (i.p.) injection of a mixture of ketamine (90 mg/kg; Animal Health International) and xylazine (4.5 mg/kg; Propharma), and a tracheotomy was performed followed by insertion of pre-calibrated cannula into the trachea. Spontaneous respiratory effort was prevented using a neuromuscular blocking agent (pancuronium bromide: 2.5 mg/kg; Hospira, Inc., Lake Forest, IL). Respiratory mechanics were obtained and calculated using FlexiWare software (SCIREQ), as previously described.34,35 After the initial mechanical scan protocol, incremental doses (3.125, 6.25, 12.5, and 25 mg/mL) of methacholine were nebulized, as previously described.36 Between each dose, it was confirmed that the mouse was still alive by assessing the heartbeat.
Vector genomes
gDNA was extracted using a DNeasy blood and tissue kit (Qiagen, Valencia, CA) from the tissues harvested from AAV- and PBS-injected animals 180 days after injection. gDNA concentrations were determined using a Nanodrop 2000c (Thermo Fisher Scientific, Carlsbad CA). Recombinant AAV genome copies in the gDNA were quantified by quantitative PCR with an Applied Biosystems Vii7 (Life Technologies, Carlsbad, CA), as previously described.19 Briefly, DNA samples were assayed in triplicate, using primers and probe designed to amplify a sequence from the SV40 poly-A signal in the DES-hGAA vector (forward: AGCAATAGCATCACAAATTTCACAA; reverse: GCAGACATGATAAGATACATT-GATGAGTT; and probe: 6FAM-AGCATTTTTTTCACTGCATTCTAGTTGTGGTTTGTC). A standard curve was generated using plasmid DNA containing the same SV40 poly-A target sequence.
Histology
The following tissues were harvested at 180 days after injection immediately following the final respiratory mechanics test: the medulla and spinal cord, heart, tongue, diaphragm, gastrocnemius muscle, and trachea.
GAA staining
The heart, gastrocnemius, tongue, diaphragm, medulla, and spinal cord tissues were extracted and post fixed by immersion in 4% paraformaldehyde (n = 4 animals per group) for 24 h and then transferred to 30% sucrose for cryoprotection until sinking. The brain stem and spinal cord were then blocked into the brain stem, cervical spinal cord, thoracic spinal cord, and lumber spinal cord. Samples were embedded in optimal cutting temperature (OCT) compound and frozen. Sections (8 μm) were cut for the heart, gastrocnemius, tongue, and diaphragm; 40 μm sections were cut for the medulla and spinal cord. Sections stained by immunohistochemistry (IHC) for GAA, as previously described.19 Briefly, the tissue was incubated overnight in primary antibody against hGAA, 1:2,000 (rabbit polyclonal anti-hGAA antibody; Covance, Emeryville, CA). The following day, the tissue was washed in PBS, incubated in a biotinylated anti-rabbit IgG secondary antibody, 1:200 (Vector Laboratories, Inc., Burlingame, CA) and coupled with a Vectastain ABC Kit and DAB for bright field microscopy.
Periodic acid–Schiff staining
The heart, tongue, diaphragm, trachea, and gastrocnemius muscle were either fixed in glutaraldehyde and then embedded in epoxy resin or immediately frozen in OCT. Tissues were processed, as previously described, for epoxy resin embedding.36 Polymerized blocks were trimmed and cut at 1.5 μm on a Leica Ultracut E ultramicrotome and mounted on charged slides. To evaluate the histological glycogen load, periodic acid–Schiff (PAS) staining was used, as previously described.4,36 Briefly, tissues were incubated in 1% periodic acid for 10 min at 60°C, rinsed with tap water for 1 min, and then immediately stained with Schiff's reagent (Sigma–Aldrich, St. Louis, MO) at room temperature for 10 min, followed by a tap-water rinse until the water ran clear. Once dried completely, slides were coverslipped with Permount mounting medium (Thermo Fisher Scientific, Hampton NH) without dehydration in ethanol and the use of xylene. For the fresh-frozen tissue, 8 μm sections were stained with PAS according to standard techniques and counterstained with hematoxylin and by IHC to GAA, as described above. Images were taken on a Leica DM5500B bright-field microscope.
Enzyme assay
At 180 days p.i., tissue homogenates were assayed for GAA enzyme activity, as previously described.17,25 Briefly, tissues were homogenized using stainless-steel beads (Qiagen, Hilden, Germany) in water with protease inhibitors (Roche, Basel, Switzerland). Lysates were assayed for GAA using 4-methylumbelliferyl-α-D-glucoside (Sigma M9766, Sigma–Aldrich). Protein concentrations were determined by Bradford assay (Bio-Rad, Hercules, CA) using a bovine serum albumin standard (Sigma–Aldrich).
Glycogen assay
Homogenates from GAA enzyme assay were assayed for glycogen accumulation utilizing a glycogen assay kit (ab65620; Abcam, Cambridge, MA) following the manufacturer's instructions. In brief, samples were boiled for 10 min and run in duplicate, with duplicate background controls for each sample run in parallel to correct for free glucose within the sample. The glycogen assay kit was read at Ex535/Em587.
Statistics
All statistical analyses were performed using GraphPad Prism v7 (GraphPad, La Jolla, CA). A survival log-rank (Mantel–Cox) test was performed for survival analysis between all four treatment groups. A two-way analysis of variance (ANOVA) was performed for weights, strength testing, and measures of respiratory mechanics. Multiple comparisons between groups and comparisons to baselines were performed using Fisher's Least Significant Difference (LSD). Vector genomes analysis was performed using Student's t-test. Significance for the enzyme assay and ventilation data was determined by one-way ANOVA. Fisher's LSD was used to evaluate multiple comparisons between groups. Significance was considered at a p-value of <0.05.
Results
Strength and survival
To compare the therapeutic benefits of novel capsid AAVB1 to AAV9, 3-month-old male Gaa–/– mice were treated by systemic injection of 1 × 1012 vg of either AAVB1-DES-hGAA or AAV9-DES-hGAA, further referred to as “AAVB1” and “AAV9,” respectively. The desmin promoter (DES) was used because it has high activity in cardiac, skeletal, and smooth muscle as well as in motor neurons, but limited activity in the liver.37 Gaa–/– mice and WT mice were injected with PBS as controls. Several parameters were assayed at 30, 90, and 180 days p.i. The experimental endpoint was at 180 days when the animals were approximately 9 months old. By 180 days p.i., 50% of PBS-treated Gaa–/– controls had survived, with the rest of the animals spontaneously dying between 76 and 173 days (Fig. 1A). Survival significantly improved in both the AAVB1- and AAV9-treated animals (p = 0.0037; Fig. 1A). However, only the AAVB1-treated animals displayed robust weight gain over time in comparison to the AAV9- and PBS-treated Gaa–/– mice (Fig. 1B). WT animals were significantly heavier than all three Gaa–/– groups at 30 days (p < 0.02). However, by 90 and 180 days, the difference in weight was only significant when comparing the WT group to both AAV9- and PBS-treated groups (p < 0.04). At 90 days, the AAVB1-treated Gaa–/– animal weights were not significantly different from WT (p = 0.16), and by 180 days, their weights had significantly increased (p < 0.05), reaching near WT weights (p = 0.92).
Figure 1.

Improved survival and behavior in Gaa–/– animals treated with AAVB1 and AAV9 gene therapy. (A) Kaplan–Meier curve representing the percentage of animals surviving to the experimental endpoint at 180 days post injection. All wild type (WT) animals and 91% of both AAV9- and AAVB1-treated Gaa–/– mice survived to 180 days, whereas only 50% of phosphate-buffered saline (PBS)-treated Gaa–/– mice survived for the duration of the study. *p < 0.05 compared to PBS-treated Gaa–/– animals. (B) Body weights (g) after injection until experimental endpoint. *p < 0.05 compared to 30 days. #p < 0.05 compared to WT. (C) Four-limb grip strength test measured in centiNewtons (cN) at 30, 90, and 180 days. *p < 0.05. (D) Inverted screen test time at 30, 90, and 180 days. *p < 0.05. Data are represented as mean ± standard error of the mean (SEM).
Next, the study looked at the muscle strengths of treated and control groups. At 180 days p.i., AAV9- and PBS-treated groups had significantly lower four-limb strength (Fig. 1C) and inverted screen hang time (Fig. 1D) compared to WT, whereas AAVB1-treated animals were comparable to WT animals (p = 0.24) in four-limb strength (Fig. 1C). At all time points, Gaa–/–-untreated and AAV9-treated mice had significantly lower inverted screen hang times (Fig. 1D) than WT controls, whereas AAVB1-treated animals were able to maintain strength at near WT levels at 30 and 90 days p.i.
Vector genome copies
In order to compare the transduction of AAVB1 to AAV9, vector genome copies were measured in respiratory-specific tissues, peripheral tissues, and the spinal cord. In the respiratory tissues (Fig. 2A), a trend of increased transduction of AAVB1 compared to AAV9 was observed in the diaphragm and lung, neither of which was statistically significant. Significantly greater levels were, however, measured in the tongue base, which includes the genioglossus muscle—a muscle necessary for maintaining airway patency. In contrast, there was lower transduction of AAVB1 compared to AAV9 in the trachea (p = 0.3 trachea). In the medulla, cervical, and lumbar spinal cord (Fig. 2B), there was similar transduction between the two groups, but a significantly greater transduction was observed with AAVB1 in the thoracic spinal cord (p = 0.001). Genome copy numbers were identical for both AAV vectors in the heart, gastrocnemius muscle, and liver (Fig. 2C).
Figure 2.
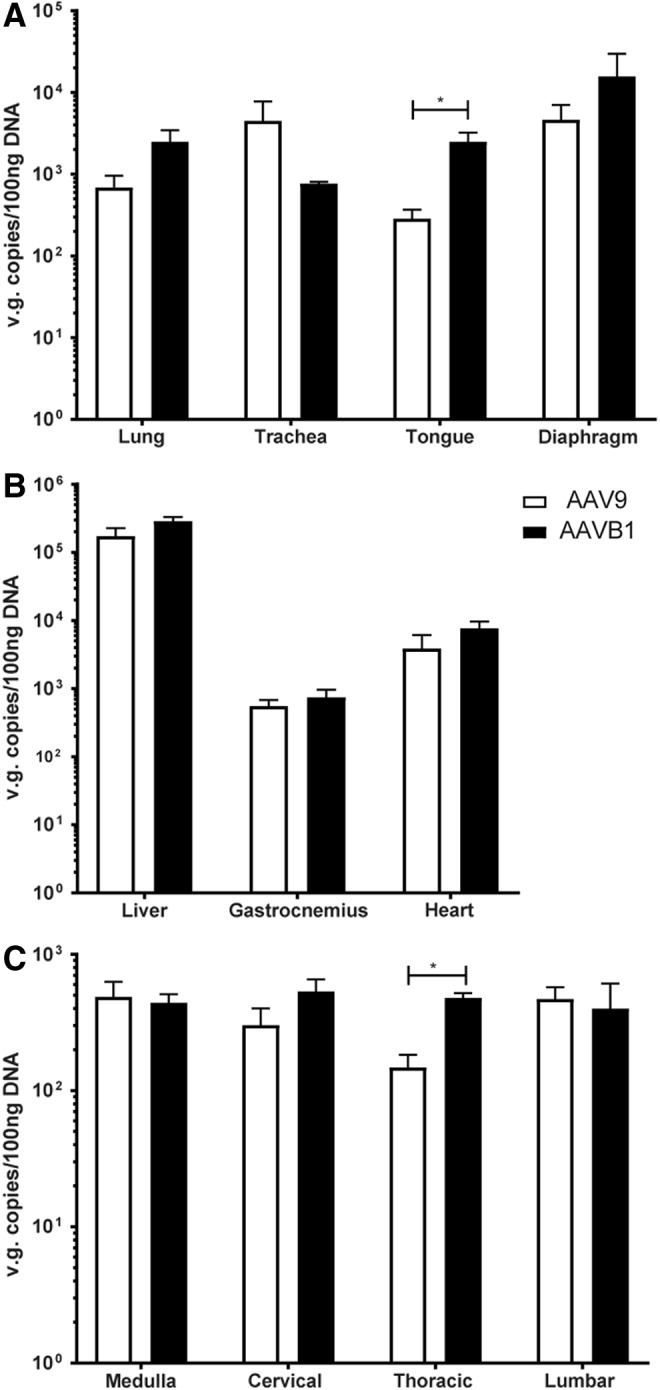
Biodistribution of vector genomes in AAV9- and AAVB1-treated Gaa–/– animals. Vector genomes (vg) contents in (A) respiratory related tissues—the lung, trachea, tongue, and diaphragm, (B) the liver, gastrocnemius muscle, and heart, and (C) the medulla and cervical, thoracic, and lumbar spinal cord 180 days after AAVB1 or AAV9 injection. AAVB1-injected animals had higher vg content in the tongue and in the thoracic cord compared to AAV9. Data are represented as mean ± SEM. *p < 0.05.
GAA immunohistochemistry
IHC for hGAA protein was performed in order to assess vector expression in AAVB1- and AAV9-treated animals 180 days p.i. Cross-sections of cardiac, respiratory, and hind-limb muscles (Fig. 3A), as well as sections of the spinal cord (Fig. 3B), were stained with an anti-hGAA antibody, specific for human GAA produced from the vector. Robust GAA expression was evident in the cardiac muscle of both the AAVB1- and AAV9-treated animals. Expression in the diaphragm and gastrocnemius muscle occurred, but this was weaker than in the heart. Superior expression in the tongue was observed in AAVB1-treated animals compared to AAV9. In sections of the spinal cord from the medulla, cervical, and thoracic regions, motor neurons were transduced in both AAVB1- and AAV9-treated animals.
Figure 3.

Robust acid alpha-glucosidase (GAA) expression was noted by immunohistochemistry in both AAV9 and AAVB1 groups. Tissues were harvested 180 days following systemic AAV9 and AAVB1 injections, and the presence of GAA was detected by immunohistochemistry (brown). (A) GAA expression was seen in the heart, diaphragm, tongue, and gastrocnemius muscle. (B) In the medulla and spinal cord, motor neurons in both AAV9- and AAVB1-injected animals stained positive for GAA. Scale bar = 100 μm. Color images available online at www.liebertpub.com/hum
GAA activity assay
To assess the amount of functional GAA enzyme present, GAA enzyme activity assays were performed 180 days p.i. Supraphysiological levels of GAA enzyme activity were observed in the heart, diaphragm, tongue, gastrocnemius muscle, and lung but not in liver from both AAV-treated groups (Fig. 4A–F). GAA activity was comparable between AAVB1- and AAV9-treated animals in all muscles, but only in the heart was it significantly elevated compared to that in WT animals (p = 0.01 for both groups; Fig. 4A). Lung GAA activity in AAVB1-treated animals was significantly higher than in WT controls (p = 0.03; Fig. 4E). In contrast, the liver GAA levels in AAV-treated animals were considerably below WT, most likely due to the use of the tissue-restrictive desmin promoter (Fig. 4F)37. GAA activity was below the detection limit of the enzyme assay in the trachea, medulla, cervical, thoracic, and lumbar spinal cord of both AAV treatment groups (data not shown).
Figure 4.

GAA enzymatic activity evident 6 months following AAV injection. (A) GAA activity in both AAV9- and AAVB1-injected Gaa–/– mice was robust in the heart, with levels well above WT levels. (B–D) GAA activity was not significantly different between AAV9 and AAVB1 animals in the diaphragm (B), tongue (C), and gastrocnemius muscle (D). (E) Lung GAA activity was significantly higher in the AAVB1-treated group compared to the AAV9 group. (F) Liver GAA activity levels were significantly lower in both AAV groups compared to WT. Data are represented as mean ± SEM. N.D., not detected. *p < 0.05.
Glycogen clearance
Next, the study examined the impact of AAV treatment on lysosomal glycogen accumulation by histological analysis of glycogen abundance in sections of respiratory, cardiac, and hind-limb muscles stained with PAS. Reduced glycogen accumulation was observed in each of the muscles studied from both AAVB1- and AAV9-treated mice. A nearly equal, robust clearance of glycogen storage was observed in both the heart (Fig. 5A) and diaphragm (Fig. 5B) in animals injected with either AAV. However, in AAVB1-treated animals, glycogen clearance was greater in the tongue (Fig. 5C), whereas clearance in the gastrocnemius appeared greater in AAV9-treated animals (Fig. 5D). Quantification of clearance of glycogen was determined by biochemical glycogen assay (Fig. 5E–H). Near-complete clearance of glycogen was observed in the heart, mirroring the histology (Fig. 5A), with significant differences when comparing PBS-treated Gaa–/– mice groups to WT, AAV9, and AAVB1, and both treatment groups were not statistically significant from WT (Fig. 5E). Similarly, within the gastrocnemius and diaphragm, significant reduction of glycogen was observed in all groups when compared to PBS-treated Gaa–/– mice (Fig. 5F and 5H). In the tongue, glycogen clearance was pronounced in AAVB1-treated animals, similar to the observation of PAS staining, and glycogen accumulation was significantly decreased from PBS-treated Gaa–/– mice and not significantly different from WT (Fig. 5G). In contrast, in the tongues of AAV9-treated animals, glycogen accumulation was significantly increased over WT and not significantly different from PBS-treated Gaa–/– mice.
Figure 5.

Systemic administration of AAV9 and AAVB1 reduced glycogen storage in Gaa–/– mice. Periodic acid–Schiff staining of 1.5 μm sections of (A) the heart, (B) the diaphragm, (C) the tongue, and (D) the gastrocnemius muscle of Gaa–/– animals injected with PBS, AAV9, or AAVB1 and WT animals. Dark staining corresponds to glycogen accumulation in cells. Note the robust accumulation of glycogen in the heart, diaphragm, tongue, and gastrocnemius muscle of the PBS-treated Gaa–/– mice. In contrast, the cardiac muscle was almost completely cleared in animals treated with either AAV9 or AAVB1. The Gaa−/− animals treated with AAVB1 had almost complete clearance of glycogen in the tongue, unlike the AAV9 group where considerable amounts of glycogen remained. The diaphragm and gastrocnemius muscle had partial clearance, with complete clearance seen in many muscle fibers. Scale bar = 100 μm. Biochemical assessment of glycogen accumulation in (E) the heart, (F) the diaphragm, (G) the tongue, and (H) the gastrocnemius muscle of Gaa–/– animals injected with PBS, AAV9, or AAVB1 and WT animals. Data are represented as mean ± SEM. *p < 0.05.
Respiratory outcome measures
The impact of gene therapy on breathing and lung mechanics was examined over time. Using whole-body plethysmography, lung volumes and ventilation parameters were tested during eupnea (normoxic conditions: FiO2: 0.21; nitrogen balance) and during a respiratory challenge (hypercapnic conditions: FiO2: 0.21; FiCO2: 0.07; nitrogen balance). No significant differences occurred between groups at baseline, 30 days p.i., or 90 days p.i. (data not shown). Thus, the impact on breathing was evaluated at 180 days p.i. as a percent change during respiratory challenge over baseline breathing (Fig. 6A–G). Statistically significant differences between WT and Gaa–/– mice were observed at 180 days under hypercapnic conditions, specifically in tidal volume (TV; Fig. 6B; p = 0.02), minute ventilation (MV; Fig. 5C; p < 0.05), peak inspiratory flow (PIF; Fig. 6D; p = 0.001), and inspiratory time (Ti; Fig. 6F; p = 0.01). PIF is a reflection of diaphragm and inspiratory muscle strength,38 and a higher Ti is a reflection of increased upper airway resistance.19 In addition, AAV9-treated animals showed reduced TV (Fig. 6B) and PIF (Fig. 6D) in response to a respiratory challenge compared to WT animals (p < 0.05 and p = 0.01, respectively), but AAVB1 animals did not. No significant differences in MV or Ti were observed between AAV9 and WT (p > 0.09) or AAVB1 and WT animals (p > 0.44). In addition, AAVB1-treated animals had a more robust decline in Ti during a respiratory challenge than PBS-treated Gaa–/– animals (Fig. 6E; p = 0.04).
Figure 6.

Respiratory phenotype was partially corrected by AAVB1 gene therapy in Gaa–/– mice. Spontaneous breathing in Gaa–/– animals 180 days following injections with PBS, AAV9, or AAVB1, or WT mice treated with PBS. These measurements were taken during a respiratory challenge with hypercapnia, and are presented as percent above baseline breathing. (A) No significant differences in breathing frequency were observed between groups. (B–D) AAVB1-treated Gaa–/– animals responded to hypercapnic challenge with similar tidal volume (B), minute ventilation (C), and peak inspiratory flow rate (D) compared to WT animals. (E) No differences were observed in peak expiratory flow between groups. (F) AAVB1-treated mice had significantly lower inspiratory time compared to PBS-treated Gaa–/– mice. (H) Airway and tissue resistance was measured in ventilated and sedated animals in response to incremental doses of methacholine. Data are represented as mean ± SEM. #p < 0.05 compared to WT. (I) PAS staining of plastic embedded tracheal sections of AAV9- and AAVB1-treated animals, respectively, revealed persistent glycogen accumulation in smooth muscle fibers. Scale bar = 100 μm. Color images available online at www.liebertpub.com/hum
As has been shown, robust accumulation of glycogen in the airway smooth muscle occurs in the Gaa–/– mouse model.3 This results in disruption of pulmonary mechanics, namely an inability of the airways to react to a bronchoconstrictive agent (i.e., methacholine).36 Consistent with these findings, when a methacholine challenge was performed on sedated, intubated, and ventilated WT and Gaa–/– mice, significant differences in tissue and small airway resistance was seen between WT and Gaa–/– mice at a methacholine dose of 12.5 mg/mL and 25 mg/mL (Fig. 6H). However, no difference was observed in resistance between PBS-treated Gaa–/– animals and either AAV9- or AAVB1-treated groups (p = 0.55 and p = 0.82, respectively). Moreover, both AAV- and PBS-treated Gaa–/– animals had significant airway hyporesponsiveness to methacholine when compared to WT animals (Fig. 6H). This finding was consistent with histological evidence showing continued glycogen accumulation in trachea of AAV9- and AAVB1-treated animals (Fig. 6I and J, respectively).
Discussion
The most important finding in this study is that a single systemic injection of either AAVB1-DES-hGAA or AAV9-DES-hGAA significantly prolongs survival of Pompe ((B6;129-GaaTm1Rabn/J Gaa–/–) mice. Both vectors robustly transduced the heart, increased enzyme activity, and cleared glycogen in the myocardium. This cardiac transduction is the most likely explanation for the increase in survival. Furthermore, based on these studies, it appears that the novel AAVB1 vector is superior to AAV9 in targeting the upper airway and respiratory system, as evidenced by the enhanced ventilatory parameters during a respiratory challenge. In addition, the improved gene delivery by AAVB1 to the tongue enhanced glycogen clearance and resulted in significant weight gain in comparison to AAV9- and PBS-treated Gaa–/– animals.
AAV gene therapy
AAV gene delivery offers a promising therapeutic option for monogenetic disorders such as Pompe disease, since a single intravenous administration has the potential to deliver the absent or deficient gene to the affected cells and results in an endogenous production of the missing GAA enzyme. The current FDA-approved therapy, ERT, requires weekly or biweekly infusions of rhGAA for the lifetime of the patient, as well as prolonged immune suppression.1,39,40 Although ERT leads to prolonged survival, many patients continue to have respiratory insufficiency secondary to untreated CNS involvement and ineffective enzyme uptake into skeletal muscle fibers.5,41 Systemic delivery of AAV9 vectors is an effective approach to treat both the muscle and CNS pathology, as these vectors can cross the blood–brain barrier. In several animal studies, AAV9 has successfully transduced muscle and motor neurons when injected directly into the muscle or intra-pleural cavity.19,42 In addition, systemic administration of AAV1-hGAA in neonatal Gaa–/– mice resulted in significant clearance of glycogen and enhanced respiratory measures.30 Furthermore, a previous study of AAV9-hGAA in adult Gaa–/– mice was found to be superior to ERT in respiratory outcome measures, which was attributed to effective motor neuron transduction.17 Although glycogen accumulation remained in the myocardium and diaphragm of AAV9-hGAA-injected animals, that study underscored the importance of treating the CNS. In this study, 3-month-old adult Gaa–/– mice were chosen to assess if gene therapy can clear the glycogen accumulation that is already present.43,44 To date, AAV gene therapy studies in adult mice with Pompe disease have had a limited transduction of respiratory and cardiac muscles, and motor neurons. Here, robust expression, enzyme activity, and clearance of glycogen in the myocardium and diaphragm are shown with both AAVB1 and AAV9.
Strength and survival
Higher mortality rates of Gaa–/– mice have been observed by others.17,45 Both AAVB1 and AAV9 resulted in significant cardiac transduction and increased survival. The desmin promotor has robust cardiac activity, which may account for the almost complete clearance of glycogen seen in both treatment groups. Transduction in the gastrocnemius muscle was similar to WT animals, and glycogen clearance occurred in many fibers. Nonetheless, the animals continued to have abnormal grip strength and a decreased ability to hang on to the wire net in the inverted screen test. Recently, Lee et al. used AAVN to target CNS disease and found that intracerebroventricular injections of AAVN-GAA resulted in endogenous GAA production in brain and spinal cord neurons.46 However, glycogen accumulation persisted in some of the motor neurons, and their muscle strength did not improve significantly. Similarly, moderate transduction efficiency in motor neurons and muscle was found in the present study, but this does not seem to be enough to reverse weakness and improve strength. In the future, exploration of a promoter that has greater expression in skeletal muscle, as well as cardiac and motor neuron activity, may enhance strength as well as prolong survival in Pompe disease.
Tongue and respiratory pathology
AAVB1 transduced the tongue and the hypoglossal motor neurons innervating the genioglossal muscle. The tongue plays a vital role in adequate nutrition and in maintaining upper airway patency. Contraction of the extrinsic tongue muscles dilates and stiffens the pharyngeal lumen, thereby minimizing airway narrowing and/or collapse in the face of negative inspiratory pressures.47,48 Tongue pathology is a significant cause of morbidity in IOPD and is an important therapeutic target. Tongue weakness leads to a weak suck, feeding problems early in life, and an increased risk for aspiration pneumonia.49 In LOPD, tongue weakness has also been documented, even in asymptomatic patients with a family history of Pompe disease.50 These clinical findings underscore the importance of ensuring that novel therapies adequately treat the tongue pathology. In the Pompe mouse, a direct intra-lingual injection of AAV9-hGAA can treat the tongue and the hypoglossal motor neurons, resulting in enhanced weight gain and sustained correction.19 Similar improvements in weight gain occurred with systemic AAVB1 therapy, presumably due to efficient vector transduction of the tongue, with subsequent clearance of glycogen in the tongue, which led to increased muscle function, allowing a greater food intake. In contrast, systemic AAV9 did not transduce the tongue as well as AAVB1. Interestingly, the Ti in AAVB1-treated animals was significantly lower than in PBS-treated mice, and it was comparable to WT animals. The decrease in Ti is a reflection of decreased upper airway resistance and improved airflow past the tongue.
Pompe disease often results in respiratory insufficiency in both IOPD and LOPD patients.51 Respiratory insufficiency occurs secondary to upper airway and respiratory muscle and CNS pathology.5,52 Several studies exploring respiratory directed therapy with AAV were very encouraging20,42,53 and resulted in a clinical trial of diaphragmatic injections of AAV1-hGAA administered into the diaphragms of children with Pompe disease. This clinical trial proved that AAV gene therapy was safe and resulted in a significant increase in the duration of time these children tolerated unassisted ventilation.54 In the current study, both AAVB1 and AAV9 transduced the diaphragm and the cervical ventral motor neurons where the phrenic motor neurons are located. However, during a respiratory challenge, the AAVB1 treatment group outperformed the AAV9 group, with a tidal volume and peak inspiratory flow comparable to WT animals. Both of these respiratory parameters are a reflection of inspiratory muscle strength as well as increased upper airway patency, which allows for more efficient air entry, resulting in increased tidal volume and inspiratory flow.55
Conclusions
In conclusion, AAVB1-hGAA is a novel vector with muscle and CNS transduction that is superior to AAV9-hGAA in an adult Pompe disease mouse model. In this model, AAVB1-hGAA robustly decreased glycogen in the tongue and resulted in improved weight gain. In addition, Gaa–/– animals treated with AAVB1-hGAA had improved inspiratory muscle strength 6 months after treatment. Finally, although the immunological impact of both vectors in this model were not studied, when Choudhury et al. assessed the immunogenic potential of AAVB1, they found that AAVB1 was less prone to neutralization by a pooled human immunoglobulin G.29 This is important to consider when adult, late onset patients with Pompe disease are being targeting where pre-existing immunity is a concern. Thus, based on all these factors, AAVB1 offers a potential therapeutic option for Pompe disease.
Acknowledgments
This study was funded by NIH NICHD K08 HD077040-01A1, Parker B. Francis Fellowship, NIH NINDS 1R21NS098131-01 (M.K.E.), and NIH 2R01HD052682-06A1 (B.J.B.). We would like to thank the UMass Electron Microscopy Core for their assistance with this project.
Author Disclosure
No competing financial interests exist.
References
- 1. Byrne BJ, Falk DJ, Pacak CA, et al. . Pompe disease gene therapy. Hum Mol Genet 2011;20:R61–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Fuller DD, ElMallah MK, Smith BK, et al. . The respiratory neuromuscular system in Pompe disease. Respir Physiol Neurobiol 2013;189:241–249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Keeler AM, Liu D, Zieger M, et al. . Airway smooth muscle dysfunction in Pompe (Gaa–/–) mice. Am J Physiol Lung Cell Mol Physiol 2017;312:L873–L881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Lee KZ, Qiu K, Sandhu MS, et al. . Hypoglossal neuropathology and respiratory activity in pompe mice. Front Physiol 2011;2:31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. DeRuisseau LR, Fuller DD, Qiu K, et al. . Neural deficits contribute to respiratory insufficiency in Pompe disease. Proc Natl Acad Sci U S A 2009;106:9419–9424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Falk DJ, Todd AG, Lee S, et al. . Peripheral nerve and neuromuscular junction pathology in Pompe disease. Hum Mol Genet 2015;24:625–636 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Byrne BJ, Kishnani PS, Case LE, et al. . Pompe disease: design, methodology, and early findings from the Pompe Registry. Mol Genet Metab 2011;103:1–11 [DOI] [PubMed] [Google Scholar]
- 8. Mellies U, Lofaso F. Pompe disease: a neuromuscular disease with respiratory muscle involvement. Respir Med 2009;103:477–484 [DOI] [PubMed] [Google Scholar]
- 9. Hagemans ML, Winkel LP, Van Doorn PA, et al. . Clinical manifestation and natural course of late-onset Pompe's disease in 54 Dutch patients. Brain 2005;128:671–677 [DOI] [PubMed] [Google Scholar]
- 10. Mellies U, Ragette R, Schwake C, et al. . Sleep-disordered breathing and respiratory failure in acid maltase deficiency. Neurology 2001;57:1290–1295 [DOI] [PubMed] [Google Scholar]
- 11. Winkel LP, Hagemans ML, van Doorn PA, et al. . The natural course of non-classic Pompe's disease; a review of 225 published cases. J Neurol 2005;252:875–884 [DOI] [PubMed] [Google Scholar]
- 12. Van den Hout JM, Kamphoven JH, Winkel LP, et al. . Long-term intravenous treatment of Pompe disease with recombinant human alpha-glucosidase from milk. Pediatrics 2004;113:e448–457 [DOI] [PubMed] [Google Scholar]
- 13. van der Ploeg AT, Clemens PR, Corzo D, et al. . A randomized study of alglucosidase alfa in late-onset Pompe's disease. N Engl J Med 2010;362:1396–1406 [DOI] [PubMed] [Google Scholar]
- 14. Schneider I, Hanisch F, Muller T, et al. . Respiratory function in late-onset Pompe disease patients receiving long-term enzyme replacement therapy for more than 48 months. Wien Med Wochenschr 2013;163:40–44 [DOI] [PubMed] [Google Scholar]
- 15. Han SO, Ronzitti G, Arnson B, et al. . Low-dose liver-targeted gene therapy for Pompe disease enhances therapeutic efficacy of ERT via immune tolerance induction. Mol Ther Methods Clin Dev 2017;4:126–136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Doerfler PA, Todd AG, Clement N, et al. . Copackaged AAV9 vectors promote simultaneous immune tolerance and phenotypic correction of Pompe disease. Hum Gene Ther 2016;27:43–59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Falk DJ, Soustek MS, Todd AG, et al. . Comparative impact of AAV and enzyme replacement therapy on respiratory and cardiac function in adult Pompe mice. Mol Ther Methods Clin Dev 2015;2:15007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Todd AG, McElroy JA, Grange RW, et al. . Correcting neuromuscular deficits with gene therapy in Pompe disease. Ann Neurol 2015;78:222–234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Elmallah MK, Falk DJ, Nayak S, et al. . Sustained correction of motoneuron histopathology following intramuscular delivery of AAV in pompe mice. Mol Ther 2014;22:702–712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Qiu K, Falk DJ, Reier PJ, et al. . Spinal delivery of AAV vector restores enzyme activity and increases ventilation in Pompe mice. Mol Ther 2012;20:21–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Sun B, Li S, Bird A, et al. . Hydrostatic isolated limb perfusion with adeno-associated virus vectors enhances correction of skeletal muscle in Pompe disease. Gene Ther 2010;17:1500–1505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Sun B, Zhang H, Benjamin DK Jr, et al. . Enhanced efficacy of an AAV vector encoding chimeric, highly secreted acid alpha-glucosidase in glycogen storage disease type II. Mol Ther 2006;14:822–830 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Sun B, Zhang H, Franco LM, et al. . Correction of glycogen storage disease type II by an adeno-associated virus vector containing a muscle-specific promoter. Mol Ther 2005;11:889–898 [DOI] [PubMed] [Google Scholar]
- 24. Sun B, Zhang H, Franco LM, et al. . Efficacy of an adeno-associated virus 8-pseudotyped vector in glycogen storage disease type II. Mol Ther 2005;11:57–65 [DOI] [PubMed] [Google Scholar]
- 25. Fraites TJ, Jr, Schleissing MR, Shanely RA, et al. . Correction of the enzymatic and functional deficits in a model of Pompe disease using adeno-associated virus vectors. Mol Ther 2002;5:571–578 [DOI] [PubMed] [Google Scholar]
- 26. Smith BK, Collins SW, Conlon TJ, et al. . Phase I/II trial of adeno-associated virus-mediated alpha-glucosidase gene therapy to the diaphragm for chronic respiratory failure in Pompe disease: initial safety and ventilatory outcomes. Hum Gene Ther 2013;24:630–640 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Smith BK, Martin AD, Lawson LA, et al. . Inspiratory muscle conditioning exercise and diaphragm gene therapy in Pompe disease: clinical evidence of respiratory plasticity. Exp Neurol 2017;287:216–224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Corti M, Cleaver B, Clement N, et al. . Evaluation of readministration of a recombinant adeno-associated virus vector expressing acid alpha-glucosidase in Pompe disease: preclinical to clinical planning. Hum Gene Ther Clin Dev 2015;26:185–193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Choudhury SR, Fitzpatrick Z, Harris AF, et al. . In vivo selection yields AAV-B1 capsid for central nervous system and muscle gene therapy. Mol Ther 2016;24:1247–1257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Mah C, Pacak CA, Cresawn KO, et al. . Physiological correction of Pompe disease by systemic delivery of adeno-associated virus serotype 1 vectors. Mol Ther 2007;15:501–507 [DOI] [PubMed] [Google Scholar]
- 31. Raben N, Nagaraju K, Lee E, et al. . Targeted disruption of the acid alpha-glucosidase gene in mice causes an illness with critical features of both infantile and adult human glycogen storage disease type II. J Biol Chem 1998;273:19086–19092 [DOI] [PubMed] [Google Scholar]
- 32. Jeyakumar M, Butters TD, Cortina-Borja M, et al. . Delayed symptom onset and increased life expectancy in Sandhoff disease mice treated with N-butyldeoxynojirimycin. Proc Natl Acad Sci U S A 1999;96:6388–6393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Drorbaugh JE, Fenn WO. A barometric method for measuring ventilation in newborn infants. Pediatrics 1955;16:81–87 [PubMed] [Google Scholar]
- 34. Robichaud A, Fereydoonzad L, Schuessler TF. Delivered dose estimate to standardize airway hyperresponsiveness assessment in mice. Am J Physiol Lung Cell Mol Physiol 2015;308:L837–846 [DOI] [PubMed] [Google Scholar]
- 35. McGovern TK, Robichaud A, Fereydoonzad L, et al. . Evaluation of respiratory system mechanics in mice using the forced oscillation technique. J Vis Exp 2013;e50172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Keeler AM, Liu D, Zieger M, et al. . Airway smooth muscle dysfunction in Pompe (Gaa–/–) mice. Am J Physiol Lung Cell Mol Physiol 2017;312:L873–L881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Pacak CA, Sakai Y, Thattaliyath BD, et al. . Tissue specific promoters improve specificity of AAV9 mediated transgene expression following intra-vascular gene delivery in neonatal mice. Genet Vaccines Ther 2008;6:13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Stoica L, Keeler AM, Xiong L, et al. . Restrictive lung disease in the Cu/Zn superoxide-dismutase 1 G93A amyotrophic lateral sclerosis mouse model. Am J Respir Cell Mol Biol 2017;56:405–408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Berrier KL, Kazi ZB, Prater SN, et al. . CRIM-negative infantile Pompe disease: characterization of immune responses in patients treated with ERT monotherapy. Genet Med 2015;17:912–918 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Kazi ZB, Desai AK, Berrier KL, et al. . Sustained immune tolerance induction in enzyme replacement therapy-treated CRIM-negative patients with infantile Pompe disease. JCI insight 2017;2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Fuller DD, ElMallah MK, Smith BK, et al. . Pompe disease and the respiratory system. Res Physiol Neurobiol 2013;189:241–249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Falk DJ, Mah CS, Soustek MS, et al. . Intrapleural administration of AAV9 improves neural and cardiorespiratory function in Pompe disease. Mol Ther 2013;21:1661–1667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Sidman RL, Taksir T, Fidler J, et al. . Temporal neuropathologic and behavioral phenotype of 6neo/6neo Pompe disease mice. J Neuropathol Exp Neurol 2008;67:803–818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Turner SM, Hoyt AK, ElMallah MK, et al. . Neuropathology in respiratory-related motoneurons in young Pompe (Gaa(–/–)) mice. Respir Physiol Neurobiol 2016;227:48–55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Puzzo F, Colella P, Biferi MG, et al. . Rescue of Pompe disease in mice by AAV-mediated liver delivery of secretable acid alpha-glucosidase. Sci Transl Med 2017;9:eaam6375 DOI: 10.1126/scitranslmed.aam6375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Lee NC, Hwu WL, Muramatsu SI, et al. . A neuron-specific gene therapy relieves motor deficits in Pompe disease mice. Mol Neurobiol 2018;55:5299–5309 [DOI] [PubMed] [Google Scholar]
- 47. Fuller D, Mateika JH, Fregosi RF. Co-activation of tongue protrudor and retractor muscles during chemoreceptor stimulation in the rat. J Physiol 1998;507:265–276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Fuller DD, Williams JS, Janssen PL, et al. . Effect of co-activation of tongue protrudor and retractor muscles on tongue movements and pharyngeal airflow mechanics in the rat. J Physiol 1999;519:601–613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Jones HN, Muller CW, Lin M, et al. . Oropharyngeal dysphagia in infants and children with infantile Pompe disease. Dysphagia 2010;25:277–283 [DOI] [PubMed] [Google Scholar]
- 50. Dubrovsky A, Corderi J, Lin M, et al. . Expanding the phenotype of late-onset Pompe disease: tongue weakness: a new clinical observation. Muscle Nerve 2011;44:897–901 [DOI] [PubMed] [Google Scholar]
- 51. Kishnani PS, Steiner RD, Bali D, et al. . Pompe disease diagnosis and management guideline. Genet Med 2006;8:267–288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Burrow TA, Bailey LA, Kinnett DG, et al. . Acute progression of neuromuscular findings in infantile Pompe disease. Pediatr Neurol 2010;42:455–458 [DOI] [PubMed] [Google Scholar]
- 53. Mah CS, Falk DJ, Germain SA, Kelley JS, et al. . Gel-mediated delivery of AAV1 vectors corrects ventilatory function in Pompe mice with established disease. Mol Ther 2010;18:502–510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Smith BK, Collins S, Conlon T, et al. . Phase I/II trial of AAV1-GAA gene therapy to the diaphragm for chronic respiratory failure in Pompe disease: initial safety and ventilatory outcomes. Hum Gene Ther 2013;24:630–640 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Huang P, Cheng G, Lu H, et al. . Impaired respiratory function in mdx and mdx/utrn(+/–) mice. Muscle Nerve 2011;43:263–267 [DOI] [PMC free article] [PubMed] [Google Scholar]