

ABSTRACT
Rab35 and the Rab35 network of GAPs, GEFs, and effectors are important regulators of membrane trafficking for a variety of cellular processes, from cytokinesis and phagocytosis to neurite outgrowth. In the past five years, components of this signaling network have also been implicated as critical mediators of synaptic vesicle (SV) recycling and protein homeostasis. Recent studies by several groups, including our own, have demonstrated that Rab35-mediated endosomal sorting is required for the degradation of SV proteins via the ESCRT pathway, thereby eliminating old or damaged proteins from the SV pool. This sorting process is regulated by Rab35 activation in response to neuronal activity, and potentially by an antagonistic signaling relationship between Rab35 and the small GTPase Arf6 that directs SVs into distinct recycling pathways depending on neuronal activity levels. Furthermore, mutations in genes encoding Rab35 regulatory proteins are emerging as causative factors in human neurologic and neurodegenerative diseases, consistent with their important roles in synaptic and neuronal health. Here, we review these recent findings and offer our perspective on how the Rab35 signaling network functions to maintain neurotransmission and synaptic fitness.
KEYWORDS: Arf6, ESCRT, Hrs, Rab35, Skywalker/TBC1D24, synaptic vesicle
Introduction
The fundamental sites of information transfer in the nervous system are synapses, asymmetric cellular junctions between neurons that mediate electrochemical signaling. The ‘chemical’ component of this signaling occurs through the regulated release of neurotransmitters from synaptic vesicles (SVs) within the presynaptic compartment. SVs are specialized secretory organelles that release neurotransmitter by fusing with the neuronal plasma membrane (exocytosis). SV fusion is followed by the re-uptake of SV proteins and membranes (endocytosis), and finally by SV re-formation to enable subsequent rounds of exocytosis and neurotransmitter release. Although SVs can be directly reformed at the plasma membrane, increasing evidence indicates that SV proteins and membranes undergo endosomal sorting before their re-emergence on functional SVs (see Fig. 1; for reviews, see refs. 1, 2). Such sorting appears necessary for maintaining the proper number and repertoire of SV proteins to support neurotransmission, and for removing damaged or dysfunctional proteins for degradation.3-5 Although the molecular composition of presynaptic endosomal sorting compartments is unclear and somewhat controversial due to their transient nature, essential components include SNARE proteins (e.g. VAMPs, Syntaxins, Vti1a), clathrin adaptor proteins (e.g., AP-2, AP-3), and specific lipids and lipid metabolizing enzymes (e.g., phosphoinositide-3-phosphates) (For a review, see ref.6). Another class of molecules that appear to play key roles in SV sorting and recycling are Rab GTPases, critical regulators of membrane trafficking. These small GTPases act as “molecular switches” that cycle between ‘active’ (GTP-bound) and ‘inactive’ (GDP-bound) states. Rab activation is catalyzed by guanine nucleotide exchange factors (GEFs), which release GDP and allow Rabs to bind cytosolic GTP. Active Rabs in turn recruit effectors to catalyze downstream events such as membrane fusion (i.e. SNARE proteins) and vesicle trafficking (i.e., motor proteins) (For a review, see ref.7). Conversely, Rab inactivation is aided by GTPase activating proteins (GAPs) that hydrolyze GTP to GDP. Thus, regulation of Rab activation/inactivation by GEFs and GAPs allows for the spatiotemporal coordination of critical membrane trafficking events. Remarkably, at least 30 of the ∼60 mammalian Rab GTPases are associated with SV pools,8,9 and several of these have been implicated in SV recycling. For instance, Rabs 3 and 27 are important regulators of SV exocytosis, while Rab5 has a role in clathrin-mediated endocytosis and SV recycling.10-16 Recently emerging studies, including our own, have implicated Rab35 and the Rab35 network of GEFs, GAPs, and effectors in SV protein homeostasis, critical for SV protein sorting, degradation, and the maintenance of functional SV pools. These roles are essential for long-term neuronal health, as evidenced by the identification of human patients with neurologic and neurodegenerative conditions who have mutations in these genes.17-23 In this review, we will discuss how the antagonistic and synergistic functions of molecules within the Rab35 signaling network are necessary for regulating SV protein trafficking, degradation, and neurotransmitter release, and conclude with a discussion of how dysfunction of this signaling network may induce neurologic and neurodegenerative diseases.
Figure 1.
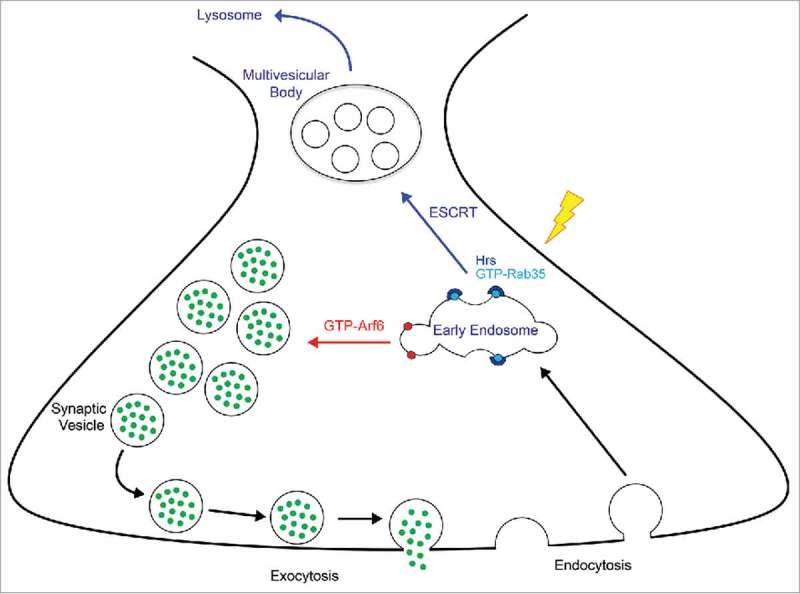
Schematic diagram of Rab35/Arf6 control of SV cycling. After synaptic vesicle (SV) exocytosis, SV proteins and membranes are endocytosed into early endosomes. Under conditions of Arf6 activation (GTP-Arf6; red arrow), SVs are directly reformed and recycled to the SV pool for use in subsequent rounds of exocytosis. Under conditions of Rab35 activation (GTP-Rab35; blue arrow), such as periods of high neuronal activity (lightning bolt), the Rab35 effector and initial ESCRT component Hrs is recruited to presynaptic endosomes. This recruitment initiates ESCRT-mediated formation of multivesicular bodies, leading to the delivery of old or damaged SV proteins to lysosomes for degradation.
Rab35 network proteins as regulators of SV cycling
The first evidence for Rab35's role in SV protein trafficking came several years ago from a study by Uytterhoeven and colleagues, who performed a screen to identify genes that caused synaptic transmission defects in Drosophila. This group identified Skywalker/TBC1D24 (Sky), a Rab GAP whose inactivation led to defects in SV recycling and the accumulation of large cisternal structures at the Drosophila neuromuscular junction (NMJ). These structures were identified as endosomal sorting stations for SVs, indicating that SV proteins sorted excessively through endosomes in the absence of Sky GAP activity. The authors subsequently identified Rab35 as the target Rab GTPase for Sky, and showed that dominant-negative Rab35 rescued sky mutant phenotypes while constitutively-active Rab35 replicated them. This study offered the first indication that Rab35 and its GAP, Sky/TBC1D24, were key players in the endosomal sorting/recycling of SV proteins.3
Interestingly, TBC1D24 has also been reported to interact with and negatively regulate Arf6, another small GTPase in the Rab35 signaling network.22,24 Arf6, like Rab35, is an important mediator of vesicle trafficking in a variety of cellular contexts (For a review, see ref.25). In the nervous system, Arf6 appears to play a critical role during development as a regulator of neuronal migration, dendritic outgrowth, and spine formation.24,26,27 It was recently shown that TBC1D24 modulates Arf6-dependent neuronal migration and outgrowth, as phenotypes induced by knockdown of TBC1D24 (i.e., delayed neuron migration and immature dendritic morphology) were rescued by overexpression of dominant-negative Arf6.24 These findings indicate that TBC1D24 prevents Arf6 activation, suggesting that it serves as a GAP for Arf6.
Although Rab35 and Arf6 are both negatively regulated by TBC1D24/Sky in neurons, studies in non-neuronal cells have demonstrated that these GTPases have an antagonistic relationship. For instance, Arf6 activity is negatively regulated by ACAP2, a Rab35 effector and Arf6 GAP, to facilitate neurite outgrowth in PC12 cells, phagocytosis in macrophages, and myelination of neurons via regulation of oligodendrocyte differentiation.28-30 Conversely, Rab35 activity is negatively regulated by EPI64B/TBC1D10B, an Arf6 effector and Rab35 GAP, to promote the endocytic recycling necessary for successful cytokinesis in mammalian HeLa cells.31 This antagonistic Rab35/Arf6 relationship offers an elegant mechanism for opposing trafficking pathways to be co-regulated by the activation of just one of these GTPases.
Not only does the antagonistic Rab35/Arf6 signaling cascade have essential roles in facilitating neurite outgrowth and phagocytosis,29,32,33 but it may also regulate SV trafficking in neurons. A recent study by Tagliatti and colleagues in cultured rodent hippocampal neurons has demonstrated a role for Arf6 in the direct recycling of SVs to the readily releasable pool (RRP) of vesicles following endocytosis.29 By electron microscopy, they noted that presynaptic boutons expressing shRNAs to knockdown Arf6 exhibited both a decrease in the total number of SVs and an increase in large cisternal structures representing early/sorting endosomes. Using an inhibitor of an Arf6 GEF, the authors were able to recapitulate this phenotype and conclude that the lack of active Arf6 was responsible. They hypothesized that active Arf6 functions to sort SVs directly to the RRP, and that inactivation of Arf6 leads to excessive endosomal sorting of SVs.34 The phenotype of Arf6 loss-of-function is strikingly similar to that observed in sky mutant terminals, suggesting that Arf6 inactivation and Rab35 activation induce similar effects and that these molecules may function antagonistically to regulate SV trafficking. Indeed, the findings of Uytterhoeven et al. and Tagliatti et al. suggest a mechanism wherein Arf6 activation leads to the recycling of SV proteins back to the RRP, while Rab35 activation leads to SV recycling via endosomes (Figs. 1 and 2).
Figure 2.
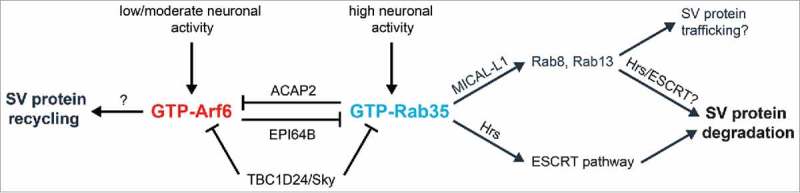
Putative pathway for antagonistic Rab35/Arf6 regulation in presynaptic terminals. Rab35 activation by high neuronal firing rates leads to Hrs binding, ESCRT pathway recruitment, and subsequent SV protein degradation as shown in Figure 1. Rab35 activation may also induce binding with its effector MICAL-L1, leading to the recruitment of additional Rab GTPases (Rabs 8 and 13) to catalyze downstream SV protein degradation and/or trafficking events. Finally, Rab35 activation may simultaneously inactivate Arf6 via ACAP2, a Rab35 effector and Arf6 GAP. On the other hand, Arf6 activation in response to low or moderate neuronal activity is hypothesized to promote SV protein recycling back to the readily-releasable pool, while inactivating Rab35 activity via EPI64B, an Arf6 effector and Rab35 GAP. TBC1D24/Sky may serve as a GAP for both Rab35 and Arf6. Additional work is needed to verify these pathways and determine the cellular signals responsible for Arf6 and Rab35 activation/inactivation at presynaptic terminals.
If this Rab35/Arf6 switch does indeed regulate SV recycling, what are the signals responsible for selectively activating each of these GTPases? Based on our recent work, we hypothesize that neuronal activity is a key signal that activates Rab35 to induce the endosomal sorting of SVs (Figs. 1 and 2). In particular, we find that application of the GABA receptor antagonist bicuculline and the voltage-gated K+ channel blocker 4-amino pyridine (4-AP) to hippocampal neurons for a 12-hour period not only dramatically increases neuronal firing rates, but also significantly increases the levels of GTP-Rab35 and the binding of Rab35 to its downstream effector Hrs/Hgs (Hepatocyte Growth Factor-Regulated Tyrosine Kinase Substrate), an early endosome-associated protein and the first component of the endosomal sorting complex required for transport (ESCRT) pathway.5 Moreover, a series of studies going back to the 1970s have demonstrated a link between intense or prolonged neuronal activity and the appearance of endosomal structures in presynaptic boutons.35-40 Both Tagliatti et al. and Uytterhoeven et al. also observed an increase in cisternal/endosomal structures within presynaptic boutons following neuronal activity, and a decrease upon neuronal silencing.3,34 Together, these findings suggest that high neuronal firing rates activate the Rab35 pathway, thereby increasing the endosomal sorting of SVs. Intriguingly, although the relationship between neuronal activity and presynaptic Arf6 activation is unexplored, a recent study by Kim et al. found that neuronal activity suppresses Arf6 activation in the postsynaptic compartment.26 Specifically, the authors report that enhancing neuronal activity with either the GABA receptor antagonist picrotoxin or by chemical stimulation of postsynaptic glutamate receptors reversed the effects of Arf6 expression on dendritic spine density.26 These findings are consistent with the concept that high levels of neuronal firing lead to Arf6 inactivation (Fig. 2), potentially through an antagonistic Rab35 signaling cascade, although additional studies are needed to investigate this phenomenon in the presynaptic compartment.
Also currently missing from this model of Rab35/Arf6 regulation of SV cycling are the identities of the reciprocal negative regulators, as the roles of ACAP2 and EPI64B have not yet been investigated in the context of SV recycling. However, ACAP2 plays an important role in endosomal sorting in PC12 cells,29,41 which are often used a model for studying synaptic vesicle biogenesis and trafficking, and EPI64B is necessary for vesicle secretion from pancreatic acinar cells,42 which share with neurons much of the highly-conserved machinery for regulated exocytosis. These studies suggest that both proteins could have conserved roles in endosomal sorting and/or vesicle exocytosis at nerve terminals. Moreover, a recent study by Fischer and colleagues provides clues as to how Rab35 and Arf6 could be inactivated during SV cycling by TBC1D24/Sky. Specifically, Fischer et al. solved the crystal structure of the TBC domain of Skywalker, responsible for catalyzing GTP hydrolysis and thereby Rab/Arf inactivation. They report that this domain interacts with specific phosphoinositides and is necessary for Sky binding to SVs.43 Thus, it appears likely that Sky association with phosphoinositides on SVs or plasma membrane, potentially in response to neuronal activity or other stimuli, would place Sky in close proximity to Rab35 and Arf6 to mediate their inactivation. Clearly, further studies are needed to test these concepts and determine the precise mechanisms through which Rab35 and Arf6 regulate the differential sorting of SVs and their associated proteins.
Rab35 network proteins as regulators of SV turnover and replenishment
Given the evidence for Rab35-mediated endosomal sorting of SVs, what is its relevance for presynaptic function? Recent studies have demonstrated the importance of endosomal sorting for maintaining a releasable pool of SVs to support neurotransmission. For instance, Uytterhoeven et al. observed that neurotransmitter release was enhanced in sky mutants, indicating that SVs were more efficiently exocytosed upon stimulation. Inhibition of the endosomal sorting of SVs in sky mutants (via heterozygous deletion of the ESCRT protein Hrs) normalized this phenotype, demonstrating that increased endosomal sorting was responsible for creating ‘high-functioning’ SVs capable of more efficient exocytosis.3 Another example of the importance of endosomal sorting for maintaining functional SV pools comes from a study by Watson et al. in teetering mice.44 These mice harbor a single nucleotide mutation in the HRS/HGS gene that leads to the loss of Hrs protein expression. Similar to sky mutant flies, teetering mice have increased numbers of endosomal structures and decreased numbers of SVs at their NMJs,44 consistent with defects in the endosomal sorting of SVs. Unlike sky mutants, teetering mice exhibit significantly decreased spontaneous and evoked neurotransmitter release,44 consistent with the concept that decreased endosomal sorting results in ‘low functioning’ SVs that are less capable of efficient neurotransmitter release. Together, these studies suggest that Hrs-mediated endosomal sorting plays a fundamental role in maintaining healthy, fusion-competent SVs to support neurotransmission.
Mechanistically, how does endosomal sorting maintain fusion-competent SVs? One possibility is that this sorting facilitates the high-fidelity reformation of SVs containing the correct number and repertoire of proteins to mediate exocytosis. Indeed, loss-of-function of endocytic adaptors (e.g., AP180, AP3) that mediate the sorting of specific SV membrane proteins to SVs has been shown to impair SV recycling and neurotransmitter release.45-50 However, Rab35 has not been found to have a role in regulating the localization or function of these adaptors, or the molecular composition of SVs. A second possibility, borne out by the studies discussed in the following paragraphs, is that Rab35/ESCRT-mediated sorting facilitates the removal and degradation of damaged or misfolded SV proteins, thereby maintaining healthy proteins on SV pools. Indeed, the ESCRT pathway is one of the major cellular degradative pathways for removal of ubiquitinated proteins. Ubiquitination is a signal for protein degradation, with specific ubiquitin linkages (i.e., lysine-48 polyubiquitination) targeting cytosolic proteins for degradation by the proteasome, and other linkages (monoubiquitination or lysine-63 polyubiquitination) targeting membrane-associated proteins for degradation by the lysosome. The ESCRT pathway comprises a series of protein complexes that recruit ubiquitinated proteins to the endosomal membrane and internalize these proteins into intraluminal vesicles to create multivesicular bodies (MVBs). These MVBs subsequently deliver cargo proteins to lysosomes for degradation.
Initial evidence that Rab35/ESCRT-mediated endosomal sorting was required for SV protein degradation came from Uytterhoeven et al., who created a degradative substrate by fusing ubiquitin to Synaptobrevin (Ub-Syb), the Drosophila vSNARE protein localized to SVs. They found that while Ub-Syb was efficiently cleared from wild-type boutons, its clearance was markedly faster in sky mutants. Moreover, Hrs loss-of-function led to the accumulation of Ub-Syb within both wild-type and sky mutant boutons, indicating that ESCRT-mediated sorting was required for Ub-Syb degradation.3 In a follow-up study from the same laboratory, Fernandes et al. used fluorescent timer fusion proteins, which change color as they mature, to monitor the age of chimeric Syb-timer proteins in response to stimulation or inhibition of endosomal sorting. They found that Sky mutation or overexpression of active Rab35 to stimulate endosomal sorting resulted in a ‘younger’ pool of Syb-timer on SVs.4 Conversely, inhibition of endosome-to-lysosome trafficking via mutation of the homotypic fusion and vacuole protein sorting (HOPS) complex resulted in the accumulation of ‘older’ Syb-timer at boutons, and also led to reduced neurotransmitter release.4 These studies showed that Rab35/ESCRT/HOPS-mediated endo-lysosomal sorting promoted the degradation of SV proteins, leaving the youngest and presumably healthiest proteins to populate SVs and mediate exocytosis.
Our recent study provides further mechanistic insight into the degradation of SV proteins by Rab35 and the ESCRT pathway. In accordance with previous studies implicating Rab35 in the endosomal sorting of SVs, we observed that overexpression of Rab35 stimulates the degradation of SV membrane proteins (but notably not peripheral SV proteins or active zone proteins) in mammalian hippocampal neurons. Additionally, we found that knockdown of Rab35 or ESCRT pathway components inhibited SV protein turnover and led to their accumulation over time, suggesting that the Rab35/ESCRT pathway is the major pathway for SV protein degradation in neurons. Given the putative link between high neuronal firing rates and endosomal sorting, we also examined the effect of neuronal activity on SV protein degradation. Here, we found that neuronal activity, induced by application of bicuculline and 4-AP, significantly increased the degradation of the SV proteins VAMP2/Synaptobrevin2 and SV2. Moreover, this activity-dependent degradation required Rab35 and the ESCRT pathway, indicating that endosomal sorting is necessary for the effective clearance of SV proteins during periods of high neuronal firing. Remarkably, we also observed that Rab35 was activated in response to bicuculline/4-AP treatment, and that this activation promoted binding to Hrs, which we identified as a novel Rab35 effector. As the first component of the ESCRT pathway, Hrs not only concentrates ubiquitinated cargo on the endosome membrane, but also initiates the recruitment of downstream ESCRT machinery, thereby mediating MVB formation and the delivery of SV proteins to lysosomes for their degradation. Indeed, we also observed higher levels of CHMP2B, a downstream ESCRT component and MVB marker, in axons and presynaptic boutons following bicuculline/4-AP treatment. Overall, our findings demonstrate a direct mechanistic link between Rab35 and the ESCRT pathway, and show that activation of Rab35 by neuronal activity catalyzes the recruitment of ESCRT machinery to SV pools to mediate the degradation of SV proteins.5
Our study did not examine the effects of Rab35/ESCRT pathway loss-of-function on neurotransmission, but work from multiple groups suggests that impaired protein clearance leads to presynaptic dysfunction and degeneration in mouse models. For instance, Watson et al. observed an increase in ubiquitinated proteins within the synaptosomal fraction of cortical neurons from teetering mice,44 suggesting that the accumulation of old/damaged proteins contributes to the impaired neurotransmitter release observed in these animals, potentially by compromising SV fusion. Notably, teetering mice also exhibit growth retardation, motor and sensory defects, dysregulated myelination, and early perinatal lethality,44 indicating widespread consequences of ESCRT pathway dysfunction. Similar phenotypes were reported in another study in which the HRS gene was specifically deleted from the mouse central nervous system.51 Here, mice lacking neuronal Hrs also exhibited growth retardation, accumulation of ubiquitinated proteins, and motor defects, as well as learning and memory defects and prominent neurodegeneration in the hippocampus. Together, these studies demonstrate the critical role of Hrs and the ESCRT pathway in maintaining synaptic transmission and neuronal health. Interestingly, phenotypes reminiscent of those seen in teetering and Hrs knockout mice were also reported in transgenic mice expressing VAMP2/Synaptobrevin2 fused to non-cleavable ubiquitin. These phenotypes included impaired neurotransmitter release at the NMJ, as well as decreased number of SVs, increased endosomal structures, and progressive NMJ degeneration.52 We hypothesize that fusion of non-cleavable ubiquitin to VAMP2 likely prevents its proper clearance and degradation, leading to the accumulation of non-functional VAMP2 on SVs, impaired neurotransmitter release, and ultimately degeneration of presynaptic terminals. Further evidence for the link between impaired protein degradation and presynaptic dysfunction comes from studies of mice deficient in the deubiquitinating enzymes Usp14 or UCHL1. These animals have lower levels of free ubiquitin, leading to reduced polyubiquitination and degradation of protein substrates,53,54 as well as prominent NMJ phenotypes, including decreased neurotransmitter release, loss of SVs, and NMJ degeneration.54,55 Together, these studies indicate that presynaptic boutons are particularly sensitive to defects in protein clearance and/or buildup of damaged and ubiquitinated SV proteins, and suggest that Rab35/ESCRT-mediated degradation is crucial for the function and long-term maintenance of mammalian synapses.
Rab35 as a master Rab?
Rab35 has important roles in many cellular processes, including neurite outgrowth, cytokinesis, and phagocytosis. Intriguingly, studies of these processes in non-neuronal cells indicate that Rab35 functions as a master regulator to recruit and coordinate downstream Rab GTPases. For example, Rab35 has been shown to undergo nerve growth factor-dependent recruitment to Arf6-positive recycling endosomes in PC12 cells, in a process necessary for neurite outgrowth.41 Active Rab35 subsequently recruits its effectors ACAP2 and MICAL-L1 onto these recycling endosomes. ACAP2 inactivates Arf6 as part of the antagonistic Rab35/Arf6 signaling cascade discussed earlier in this review, while MICAL-L1 mediates the recruitment of downstream Rabs, including Rabs 8, 13, and 36, to Arf6-positive endosomes.56 Using siRNA depletion, Kobayashi and colleagues demonstrated that both Rab35 and MICAL-L1 were necessary for the localization of these Rabs to recycling endosomes, and they proposed that Rab35 functions as a “master Rab” to regulate the localization of MICAL-L1, which in turn acts as a scaffold for Rab clustering on recycling endosomes. Each of these downstream Rabs (8, 13, 36) was shown to have an independent role in neurite outgrowth that did not depend upon the localization of the other two. Intriguingly, another recent study has implicated a similar Rab cascade in a completely different cellular process, phagocytosis in macrophages.57 Specifically, Yeo et al. find that a sequential cascade of Rab 35, 13, 8, 27a, 10, and 31 is necessary for the proper closure of the phagocytic cup. This study again implicates Rab35 as the major initiator of subsequent Rab GTPase recruitment to mediate downstream trafficking events. An interesting possibility is that Rab35 acts in a similar manner at presynaptic terminals to initiate a cascade of Rab recruitment through MICAL-L1 or another effector to coordinate SV protein endosomal trafficking and/or degradation (Fig. 2). Intriguingly, two of the Rabs that function downstream of Rab35 and MICAL-L1, Rabs 8 and 13, are implicated in the clearance/degradation of α-synuclein, an SV-associated protein whose misfolding and aggregation is linked to Parkinson disease and dementia with Lewy bodies.58 These findings, although obtained in non-neuronal cells, suggest that Rabs 8 and 13 also have roles in degradative protein sorting. However, it remains to be determined whether these Rabs function in the Rab35/ESCRT pathway, in a parallel pathway to mediate the clearance of α-synuclein and other aggregation-prone proteins, and/or in additional SV protein trafficking or recycling steps (Fig. 2).
Rab35 network in neurological and neurodegenerative disease
Given that dysregulation of membrane trafficking is emerging as a central cause of neurodegenerative diseases (For reviews, see refs. 59, 60), it will be critically important to unravel the roles of Rab GTPases in neuronal protein sorting events. Indeed, mutations in proteins of the Rab35 signaling network have been implicated in several neurologic and neurodegenerative disorders. For instance, loss-of-function mutations in TBC1D24 are linked to epilepsy, non-syndromic deafness, neurodegeneration, and DOORS (deafness, onychodystrophy, osteodystrophy, mental retardation, and seizures) syndrome. Indeed, phenotypes of patients carrying TBC1D24 mutations are extremely variable, ranging from isolated deafness to mild seizures to drug-resistant epilepsy with severe developmental delay and early death.17-23 This heterogeneity likely reflects the location of mutations within the TBC1D24 gene, as some are reported to result in the loss of protein expression,23 and others in the loss of phosphoinositide binding and SV association.43 Interestingly, Fischer et al. were able to rescue behavioral defects (seizures and temperature sensitivity) associated with mutant Sky/TBC1D24 expression in Drosophila by genetically increasing levels of the phosphoinositide PI(4,5)P2 within presynaptic boutons. This remarkable finding suggests that the major phenotypes associated with some TBC1D24 mutations, such as epilepsy, may result from presynaptic defects in SV trafficking/turnover, and could be mitigated with pharmacological agents that boost PI(4,5)P2 production. However, given that TBC1D24 negatively regulates Arf6 and Rab35, its loss-of-function presumably also causes defects in other Arf6 and Rab35-dependent processes that impact neuronal excitability, such as neuronal migration, neurite outgrowth, and dendritic spine formation. These processes may prove more difficult to reverse with pharmacological treatments.
As mentioned previously, mutation or deletion of Hrs, a Rab35 effector and ESCRT pathway component, causes synaptic dysfunction, accumulation of ubiquitinated proteins, and neurodegeneration in animal models.3,44 In humans, mutations in another ESCRT component, CHMP2B, are linked to frontotemporal dementia with or without parkinsonism and to amyotrophic lateral sclerosis.61-67 Not only do such mutations demonstrate the importance of the ESCRT pathway for neuronal health, they also illustrate the selective vulnerability of specific populations of neurons (i.e., motor neurons and cortical neurons) to dysfunction of endo-lysosomal sorting and the accumulation of damaged proteins.
Finally, a recent study has reported that Rab35 levels are elevated in the cerebrospinal fluid of patients with Parkinson disease (PD) compared with age matched controls.68 The authors link this elevation in Rab35 levels to the increased aggregation and secretion of mutant α-synuclein from dopaminergic neurons. Several studies report that α-synuclein is secreted by exosomes, small secretory vesicles released via fusion of MVBs with the plasma membrane.69-72 Intriguingly, Rab35 was identified as a regulator of exosome secretion in glial cells,73 and its appearance in PD patients may indicate that it can serve as a biomarker for these vesicles. An alternative explanation for this finding is that Rab35 levels are upregulated by neurons in response to α-synuclein misfolding/aggregation, as part of an intracellular stress response to stimulate α-synuclein clearance and degradation. Consistent with this concept, two recent studies by Spencer and colleagues show that α-synuclein is degraded via the ESCRT pathway, and that lentiviral expression of CHMP2B can rescue neurodegeneration in transgenic mice overexpressing α-synuclein.71,72 Further work to identify the molecules and pathways involved in α-synuclein-mediated toxicity, and the role of Rab35 in its secretion and/or degradation, will help to clarify the relationship between Rab35 and PD pathogenesis.
In summary, Rab35 and the Rab35 signaling network have important roles in many aspects of nervous system development and function, from the regulation of early neurite outgrowth to the maintenance of synaptic transmission via SV protein recycling and degradation. Indeed, recent findings from Drosophila and human genetic studies suggest that Rab35-mediated membrane trafficking has a highly conserved role in the nervous system, and that disruption of these trafficking events triggers synaptic dysfunction and neurodegeneration in both invertebrate and vertebrate organisms. Further work to elucidate Rab35 trafficking pathways and identify the intracellular and extracellular signals responsible for their regulation will be vital for understanding how the brain functions under normal and pathological conditions.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
References
- [1].Jahne S, Rizzoli SO, Helm MS. The structure and function of presynaptic endosomes. Exp Cell Res 2015; 335(2):172-9; PMID:25939282; http://dx.doi.org/ 10.1016/j.yexcr.2015.04.017 [DOI] [PubMed] [Google Scholar]
- [2].Morgan JR, Comstra HS, Cohen M, Faundez V. Presynaptic membrane retrieval and endosome biology: defining molecularly heterogeneous synaptic vesicles. Cold Spring Harb Perspect Biol 2013; 5(10):a016915; PMID:24086045; http://dx.doi.org/ 10.1101/cshperspect.a016915 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Uytterhoeven V, Kuenen S, Kasprowicz J, Miskiewicz K, Verstreken P. Loss of skywalker reveals synaptic endosomes as sorting stations for synaptic vesicle proteins. Cell 2011; 145(1):117-32; PMID:21458671; http://dx.doi.org/ 10.1016/j.cell.2011.02.039 [DOI] [PubMed] [Google Scholar]
- [4].Fernandes AC, Uytterhoeven V, Kuenen S, Wang YC, Slabbaert JR, Swerts J, Kasprowicz J, Aerts S, Verstreken P. Reduced synaptic vesicle protein degradation at lysosomes curbs TBC1D24/sky-induced neurodegeneration. J Cell Biol 2014; 207(4):453-62; PMID:25422373; http://dx.doi.org/ 10.1083/jcb.201406026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Sheehan P, Zhu M, Beskow A, Vollmer C, Waites CL. Activity-Dependent Degradation of Synaptic Vesicle Proteins Requires Rab35 and the ESCRT Pathway. J Neurosci 2016; 36(33):8668-86; PMID:27535913; http://dx.doi.org/ 10.1523/JNEUROSCI.0725-16.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Jovic M, Sharma M, Rahajeng J, Caplan S. The early endosome: a busy sorting station for proteins at the crossroads. Histol Histopathol 2010; 25(1):99-112; PMID:19924646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Stenmark H. Rab GTPases as coordinators of vesicle traffic. Nat Rev Mol Cell Biol 2009; 10(8):513-25. Epub 2009/07/16; PMID:19603039; http://dx.doi.org/ 10.1038/nrm2728 [DOI] [PubMed] [Google Scholar]
- [8].Takamori S, Holt M, Stenius K, Lemke EA, Gronborg M, Riedel D, Urlaub H, Schenck S, Brügger B, Ringler P, et al.. Molecular anatomy of a trafficking organelle. Cell 2006; 127(4):831-46; PMID:17110340; http://dx.doi.org/ 10.1016/j.cell.2006.10.030 [DOI] [PubMed] [Google Scholar]
- [9].Pavlos NJ, Jahn R. Distinct yet overlapping roles of Rab GTPases on synaptic vesicles. Small GTPases 2011; 2(2):77-81; PMID:21776405; http://dx.doi.org/ 10.4161/sgtp.2.2.15201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Mahoney TR, Liu Q, Itoh T, Luo S, Hadwiger G, Vincent R, Wang ZW, Fukuda M, Nonet ML. Regulation of synaptic transmission by RAB-3 and RAB-27 in Caenorhabditis elegans. Mol Biol Cell 2006; 17(6):2617-25; PMID:16571673; http://dx.doi.org/ 10.1091/mbc.E05-12-1170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Schluter OM, Schmitz F, Jahn R, Rosenmund C, Sudhof TC. A complete genetic analysis of neuronal Rab3 function. J Neurosci 2004; 24(29):6629-37; PMID:15269275; http://dx.doi.org/ 10.1523/JNEUROSCI.1610-04.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Schluter OM, Schnell E, Verhage M, Tzonopoulos T, Nicoll RA, Janz R, Malenka RC, Geppert M, Südhof TC. Rabphilin knock-out mice reveal that rabphilin is not required for rab3 function in regulating neurotransmitter release. J Neurosci 1999; 19(14):5834-46; PMID: 10407024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Yu E, Kanno E, Choi S, Sugimori M, Moreira JE, Llinas RR, Fukuda M. Role of Rab27 in synaptic transmission at the squid giant synapse. Proc Natl Acad Sci U S A 2008; 105(41):16003-8; PMID:18840683; http://dx.doi.org/ 10.1073/pnas.0804825105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Hoopmann P, Punge A, Barysch SV, Westphal V, Buckers J, Opazo F, Bethani I, Lauterbach MA, Hell SW, Rizzoli SO. Endosomal sorting of readily releasable synaptic vesicles. Proc Natl Acad Sci U S A 2010; 107(44):19055-60; PMID:20956291; http://dx.doi.org/ 10.1073/pnas.1007037107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Shimizu H, Kawamura S, Ozaki K. An essential role of Rab5 in uniformity of synaptic vesicle size. J Cell Sci 2003; 116(Pt 17):3583-90; PMID:12876219; http://dx.doi.org/ 10.1242/jcs.00676 [DOI] [PubMed] [Google Scholar]
- [16].Wucherpfennig T, Wilsch-Brauninger M, Gonzalez-Gaitan M. Role of Drosophila Rab5 during endosomal trafficking at the synapse and evoked neurotransmitter release. J Cell Biol 2003; 161(3):609-24; PMID:12743108; http://dx.doi.org/ 10.1083/jcb.200211087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Guven A, Tolun A. TBC1D24 truncating mutation resulting in severe neurodegeneration. J Med Genet 2013; 50(3):199-202; PMID:23343562; http://dx.doi.org/ 10.1136/jmedgenet-2012-101313 [DOI] [PubMed] [Google Scholar]
- [18].Milh M, Falace A, Villeneuve N, Vanni N, Cacciagli P, Assereto S, Nabbout R, Benfenati F, Zara F, Chabrol B, et al.. Novel compound heterozygous mutations in TBC1D24 cause familial malignant migrating partial seizures of infancy. Hum Mutat 2013; 34(6):869-72; PMID:23526554; http://dx.doi.org/ 10.1002/humu.22318 [DOI] [PubMed] [Google Scholar]
- [19].Balestrini S, Milh M, Castiglioni C, Luthy K, Finelli MJ, Verstreken P, Cardon A, Stražišar BG, Holder JL Jr, Lesca G, et al.. TBC1D24 genotype-phenotype correlation: Epilepsies and other neurologic features. Neurology 2016; 87(1):77-85; PMID:27281533; http://dx.doi.org/ 10.1212/WNL.0000000000002807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Corbett MA, Bahlo M, Jolly L, Afawi Z, Gardner AE, Oliver KL, Tan S, Coffey A, Mulley JC, Dibbens LM, et al.. A focal epilepsy and intellectual disability syndrome is due to a mutation in TBC1D24. Am J Hum Genet 2010; 87(3):371-5; PMID:20797691; http://dx.doi.org/ 10.1016/j.ajhg.2010.08.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Campeau PM, Kasperaviciute D, Lu JT, Burrage LC, Kim C, Hori M, Powell BR, Stewart F, Félix TM, van den Ende J, et al.. The genetic basis of DOORS syndrome: an exome-sequencing study. Lancet Neurol 2014; 13(1):44-58; PMID:24291220; http://dx.doi.org/ 10.1016/S1474-4422(13)70265-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Falace A, Filipello F, La Padula V, Vanni N, Madia F, De Pietri Tonelli D, de Falco FA, Striano P, Dagna Bricarelli F, Minetti C, et al.. TBC1D24, an ARF6-interacting protein, is mutated in familial infantile myoclonic epilepsy. Am J Hum Genet 2010; 87(3):365-70; PMID:20727515; http://dx.doi.org/ 10.1016/j.ajhg.2010.07.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Lozano R, Herman K, Rothfuss M, Rieger H, Bayrak-Toydemir P, Aprile D, Fruscione F, Zara F, Fassio A. Clinical intrafamilial variability in lethal familial neonatal seizure disorder caused by TBC1D24 mutations. Am J Med Genet A 2016; 170(12):3207-14; PMID:27541164; http://dx.doi.org/ 10.1002/ajmg.a.37933 [DOI] [PubMed] [Google Scholar]
- [24].Falace A, Buhler E, Fadda M, Watrin F, Lippiello P, Pallesi-Pocachard E, Baldelli P, Benfenati F, Zara F, Represa A, et al.. TBC1D24 regulates neuronal migration and maturation through modulation of the ARF6-dependent pathway. Proc Natl Acad Sci U S A 2014; 111(6):2337-42; PMID:24469796; http://dx.doi.org/ 10.1073/pnas.1316294111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].D'Souza-Schorey C, Chavrier P. ARF proteins: roles in membrane traffic and beyond. Nat Rev Mol Cell Biol 2006; 7(5):347-58; PMID:16633337; http://dx.doi.org/ 10.1038/nrm1910 [DOI] [PubMed] [Google Scholar]
- [26].Kim Y, Lee SE, Park J, Kim M, Lee B, Hwang D, Chang S. ADP-ribosylation factor 6 (ARF6) bidirectionally regulates dendritic spine formation depending on neuronal maturation and activity. J Biol Chem 2015; 290(12):7323-35; PMID:25605715; http://dx.doi.org/ 10.1074/jbc.M114.634527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Choi S, Ko J, Lee JR, Lee HW, Kim K, Chung HS, Kim H, Kim E. ARF6 and EFA6A regulate the development and maintenance of dendritic spines. J Neurosci 2006; 26(18):4811-9; PMID:16672654; http://dx.doi.org/ 10.1523/JNEUROSCI.4182-05.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Kanno E, Ishibashi K, Kobayashi H, Matsui T, Ohbayashi N, Fukuda M. Comprehensive screening for novel rab-binding proteins by GST pull-down assay using 60 different mammalian Rabs. Traffic 2010; 11(4):491-507; PMID:20070612; http://dx.doi.org/ 10.1111/j.1600-0854.2010.01038.x [DOI] [PubMed] [Google Scholar]
- [29].Kobayashi H, Fukuda M. Rab35 regulates Arf6 activity through centaurin-beta2 (ACAP2) during neurite outgrowth. J Cell Sci 2012; 125(Pt 9):2235-43; PMID:22344257; http://dx.doi.org/ 10.1242/jcs.098657 [DOI] [PubMed] [Google Scholar]
- [30].Miyamoto Y, Yamamori N, Torii T, Tanoue A, Yamauchi J. Rab35, acting through ACAP2 switching off Arf6, negatively regulates oligodendrocyte differentiation and myelination. Mol Biol Cell 2014; 25(9):1532-42; PMID:24600047; http://dx.doi.org/ 10.1091/mbc.E13-10-0600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Chesneau L, Dambournet D, Machicoane M, Kouranti I, Fukuda M, Goud B, Echard A. An ARF6/Rab35 GTPase cascade for endocytic recycling and successful cytokinesis. Curr Biol 2012; 22(2):147-53; PMID:22226746; http://dx.doi.org/ 10.1016/j.cub.2011.11.058 [DOI] [PubMed] [Google Scholar]
- [32].Egami Y, Fujii M, Kawai K, Ishikawa Y, Fukuda M, Araki N. Activation-inactivation cycling of Rab35 and ARF6 is required for phagocytosis of Zymosan in RAW264 macrophages. J Immunol Res 2015; 2015:429439; PMID:26229970; http://dx.doi.org/ 10.1155/2015/429439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Egami Y, Fukuda M, Araki N. Rab35 regulates phagosome formation through recruitment of ACAP2 in macrophages during FcgammaR-mediated phagocytosis. J Cell Sci 2011; 124(Pt 21):3557-67; PMID:22045739; http://dx.doi.org/ 10.1242/jcs.083881 [DOI] [PubMed] [Google Scholar]
- [34].Tagliatti E, Fadda M, Falace A, Benfenati F, Fassio A. Arf6 regulates the cycling and the readily releasable pool of synaptic vesicles at hippocampal synapse. Elife 2016; 5:e10116; PMID:26731518; http://dx.doi.org/ 10.7554/eLife.10116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Heuser JE, Reese TS. Evidence for recycling of synaptic vesicle membrane during transmitter release at the frog neuromuscular junction. J Cell Biol 1973; 57(2):315-44; PMID:4348786; http://dx.doi.org/ 10.1083/jcb.57.2.315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Holt M, Cooke A, Wu MM, Lagnado L. Bulk membrane retrieval in the synaptic terminal of retinal bipolar cells. J Neurosci 2003; 23(4):1329-39; PMID:12598621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].de Lange RP, de Roos AD, Borst JG. Two modes of vesicle recycling in the rat calyx of Held. J Neurosci 2003; 23(31):10164-73; PMID:14602833 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Clayton EL, Cousin MA. The molecular physiology of activity-dependent bulk endocytosis of synaptic vesicles. J Neurochem 2009; 111(4):901-14; PMID:19765184; http://dx.doi.org/ 10.1111/j.1471-4159.2009.06384.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Hayashi M, Raimondi A, O'Toole E, Paradise S, Collesi C, Cremona O, Ferguson SM, De Camilli P. Cell- and stimulus-dependent heterogeneity of synaptic vesicle endocytic recycling mechanisms revealed by studies of dynamin 1-null neurons. Proc Natl Acad Sci U S A 2008; 105(6):2175-80; PMID:18250322; http://dx.doi.org/ 10.1073/pnas.0712171105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Paillart C, Li J, Matthews G, Sterling P. Endocytosis and vesicle recycling at a ribbon synapse. J Neurosci 2003; 23(10):4092-9; PMID:12764096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Kobayashi H, Fukuda M. Rab35 establishes the EHD1-association site by coordinating two distinct effectors during neurite outgrowth. J Cell Sci 2013; 126(Pt 11):2424-35; PMID:23572513; http://dx.doi.org/ 10.1242/jcs.117846 [DOI] [PubMed] [Google Scholar]
- [42].Hou Y, Chen X, Tolmachova T, Ernst SA, Williams JA. EPI64B acts as a GTPase-activating protein for Rab27B in pancreatic acinar cells. J Biol Chem 2013; 288(27):19548-57; PMID:23671284; http://dx.doi.org/ 10.1074/jbc.M113.472134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Fischer B, Luthy K, Paesmans J, De Koninck C, Maes I, Swerts J, Kuenen S, Uytterhoeven V, Verstreken P, Versées W. Skywalker-TBC1D24 has a lipid-binding pocket mutated in epilepsy and required for synaptic function. Nat Struct Mol Biol 2016; 23(11):965-73; PMID:27669036; http://dx.doi.org/ 10.1038/nsmb.3297 [DOI] [PubMed] [Google Scholar]
- [44].Watson JA, Bhattacharyya BJ, Vaden JH, Wilson JA, Icyuz M, Howard AD, Phillips E, DeSilva TM, Siegal GP, Bean AJ, et al.. Motor and Sensory Deficits in the teetering Mice Result from Mutation of the ESCRT Component HGS. PLoS Genet 2015; 11(6):e1005290; PMID:26115514; http://dx.doi.org/ 10.1371/journal.pgen.1005290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Kaempf N, Kochlamazashvili G, Puchkov D, Maritzen T, Bajjalieh SM, Kononenko NL, Haucke V. Overlapping functions of stonin 2 and SV2 in sorting of the calcium sensor synaptotagmin 1 to synaptic vesicles. Proc Natl Acad Sci U S A 2015; 112(23):7297-302; PMID:26015569; http://dx.doi.org/ 10.1073/pnas.1501627112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Koo SJ, Kochlamazashvili G, Rost B, Puchkov D, Gimber N, Lehmann M, Tadeus G, Schmoranzer J, Rosenmund C, Haucke V, et al.. Vesicular Synaptobrevin/VAMP2 Levels Guarded by AP180 Control Efficient Neurotransmission. Neuron 2015; 88(2):330-44; PMID:26412491; http://dx.doi.org/ 10.1016/j.neuron.2015.08.034 [DOI] [PubMed] [Google Scholar]
- [47].Koo SJ, Markovic S, Puchkov D, Mahrenholz CC, Beceren-Braun F, Maritzen T, Dernedde J, Volkmer R, Oschkinat H, Haucke V. SNARE motif-mediated sorting of synaptobrevin by the endocytic adaptors clathrin assembly lymphoid myeloid leukemia (CALM) and AP180 at synapses. Proc Natl Acad Sci U S A 2011; 108(33):13540-5; PMID:21808019; http://dx.doi.org/ 10.1073/pnas.1107067108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Kantheti P, Qiao X, Diaz ME, Peden AA, Meyer GE, Carskadon SL, Kapfhamer D, Sufalko D, Robinson MS, Noebels JL, et al.. Mutation in AP-3 delta in the mocha mouse links endosomal transport to storage deficiency in platelets, melanosomes, and synaptic vesicles. Neuron 1998; 21(1):111-22; PMID:9697856; http://dx.doi.org/ 10.1016/S0896-6273(00)80519-X [DOI] [PubMed] [Google Scholar]
- [49].Salazar G, Love R, Werner E, Doucette MM, Cheng S, Levey A, Faundez V. The zinc transporter ZnT3 interacts with AP-3 and it is preferentially targeted to a distinct synaptic vesicle subpopulation. Mol Biol Cell 2004; 15(2):575-87; PMID:14657250; http://dx.doi.org/ 10.1091/mbc.E03-06-0401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Scheuber A, Rudge R, Danglot L, Raposo G, Binz T, Poncer JC, Galli T. Loss of AP-3 function affects spontaneous and evoked release at hippocampal mossy fiber synapses. Proc Natl Acad Sci U S A 2006; 103(44):16562-7; PMID:17056716; http://dx.doi.org/ 10.1073/pnas.0603511103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Tamai K, Toyoshima M, Tanaka N, Yamamoto N, Owada Y, Kiyonari H, Murata K, Ueno Y, Ono M, Shimosegawa T, et al.. Loss of hrs in the central nervous system causes accumulation of ubiquitinated proteins and neurodegeneration. Am J Pathol 2008; 173(6):1806-17; PMID:19008375; http://dx.doi.org/ 10.2353/ajpath.2008.080684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Liu Y, Li H, Sugiura Y, Han W, Gallardo G, Khvotchev M, Zhang Y, Kavalali ET, Südhof TC, Lin W. Ubiquitin-Synaptobrevin Fusion Protein Causes Degeneration of Presynaptic Motor Terminals in Mice. J Neurosci 2015; 35(33):11514-31; PMID:26290230; http://dx.doi.org/ 10.1523/JNEUROSCI.5288-14.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Chen PC, Qin LN, Li XM, Walters BJ, Wilson JA, Mei L, Wilson SM. The proteasome-associated deubiquitinating enzyme Usp14 is essential for the maintenance of synaptic ubiquitin levels and the development of neuromuscular junctions. J Neurosci 2009; 29(35):10909-19; PMID:19726649; http://dx.doi.org/ 10.1523/JNEUROSCI.2635-09.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Walters BJ, Campbell SL, Chen PC, Taylor AP, Schroeder DG, Dobrunz LE, Artavanis-Tsakonas K, Ploegh HL, Wilson JA, Cox GA, et al.. Differential effects of Usp14 and Uch-L1 on the ubiquitin proteasome system and synaptic activity. Mol Cell Neurosci 2008; 39(4):539-48; PMID:18771733; http://dx.doi.org/ 10.1016/j.mcn.2008.07.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Chen F, Sugiura Y, Myers KG, Liu Y, Lin W. Ubiquitin carboxyl-terminal hydrolase L1 is required for maintaining the structure and function of the neuromuscular junction. Proc Natl Acad Sci U S A 2010; 107(4):1636-41; PMID:20080621; http://dx.doi.org/ 10.1073/pnas.0911516107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Kobayashi H, Etoh K, Ohbayashi N, Fukuda M. Rab35 promotes the recruitment of Rab8, Rab13 and Rab36 to recycling endosomes through MICAL-L1 during neurite outgrowth. Biol Open 2014; 3(9):803-14; PMID:25086062; http://dx.doi.org/ 10.1242/bio.20148771 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Yeo JC, Wall AA, Luo L, Stow JL. Sequential recruitment of Rab GTPases during early stages of phagocytosis. Cell Logist 2016; 6(1):e1140615; PMID:27217977; http://dx.doi.org/ 10.1080/21592799.2016.1140615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Goncalves SA, Macedo D, Raquel H, Simoes PD, Giorgini F, Ramalho JS, Barral DC, Ferreira Moita L, Outeiro TF. shRNA-Based Screen Identifies Endocytic Recycling Pathway Components That Act as Genetic Modifiers of Alpha-Synuclein Aggregation, Secretion and Toxicity. PLoS Genet 2016; 12(4):e1005995; PMID:27123591; http://dx.doi.org/ 10.1371/journal.pgen.1005995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Wang D, Chan CC, Cherry S, Hiesinger PR. Membrane trafficking in neuronal maintenance and degeneration. Cell Mol Life Sci 2013; 70(16):2919-34; PMID:23132096; http://dx.doi.org/ 10.1007/s00018-012-1201-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Schreij AM, Fon EA, McPherson PS. Endocytic membrane trafficking and neurodegenerative disease. Cell Mol Life Sci 2016; 73(8):1529-45; PMID:26721251; http://dx.doi.org/ 10.1007/s00018-015-2105-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Parkinson N, Ince PG, Smith MO, Highley R, Skibinski G, Andersen PM, Morrison KE, Pall HS, Hardiman O, Collinge J, et al.. ALS phenotypes with mutations in CHMP2B (charged multivesicular body protein 2B). Neurology 2006; 67(6):1074-7; PMID:16807408; http://dx.doi.org/ 10.1212/01.wnl.0000231510.89311.8b [DOI] [PubMed] [Google Scholar]
- [62].Skibinski G, Parkinson NJ, Brown JM, Chakrabarti L, Lloyd SL, Hummerich H, Nielsen JE, Hodges JR, Spillantini MG, Thusgaard T, et al.. Mutations in the endosomal ESCRTIII-complex subunit CHMP2B in frontotemporal dementia. Nat Genet 2005; 37(8):806-8; PMID:16041373; http://dx.doi.org/ 10.1038/ng1609 [DOI] [PubMed] [Google Scholar]
- [63].Cox LE, Ferraiuolo L, Goodall EF, Heath PR, Higginbottom A, Mortiboys H, Hollinger HC, Hartley JA, Brockington A, Burness CE, et al.. Mutations in CHMP2B in lower motor neuron predominant amyotrophic lateral sclerosis (ALS). PLoS One 2010; 5(3):e9872; PMID:20352044; http://dx.doi.org/ 10.1371/journal.pone.0009872 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].van Blitterswijk M, Vlam L, van Es MA, van der Pol WL, Hennekam EA, Dooijes D, Schelhaas HJ, van der Kooi AJ, de Visser M, Veldink JH, et al.. Genetic overlap between apparently sporadic motor neuron diseases. PLoS One 2012; 7(11):e48983; PMID:23155438; http://dx.doi.org/ 10.1371/journal.pone.0048983 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].West RJ, Lu Y, Marie B, Gao FB, Sweeney ST. Rab8, POSH, and TAK1 regulate synaptic growth in a Drosophila model of frontotemporal dementia. J Cell Biol 2015; 208(7):931-47; PMID:25800055; http://dx.doi.org/ 10.1083/jcb.201404066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Oshima R, Hasegawa T, Tamai K, Sugeno N, Yoshida S, Kobayashi J, Kikuchi A, Baba T, Futatsugi A, Sato I, et al.. ESCRT-0 dysfunction compromises autophagic degradation of protein aggregates and facilitates ER stress-mediated neurodegeneration via apoptotic and necroptotic pathways. Sci Rep 2016; 6:24997; PMID:27112194; http://dx.doi.org/ 10.1038/srep24997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Hu YB, Dammer EB, Ren RJ, Wang G. The endosomal-lysosomal system: from acidification and cargo sorting to neurodegeneration. Transl Neurodegener 2015; 4:18; PMID:26448863; http://dx.doi.org/ 10.1186/s40035-015-0041-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Chiu CC, Yeh TH, Lai SC, Weng YH, Huang YC, Cheng YC, Chen RS, Huang YZ, Hung J, Chen CC, et al.. Increased Rab35 expression is a potential biomarker and implicated in the pathogenesis of Parkinson's disease. Oncotarget 2016; 7(34):54215-54227; PMID:27509057; http://dx.doi.org/20484626 10.18632/oncotarget.11090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Emmanouilidou E, Melachroinou K, Roumeliotis T, Garbis SD, Ntzouni M, Margaritis LH, Stefanis L, Vekrellis K. Cell-produced alpha-synuclein is secreted in a calcium-dependent manner by exosomes and impacts neuronal survival. J Neurosci 2010; 30(20):6838-51; PMID:20484626; http://dx.doi.org/ 10.1523/JNEUROSCI.5699-09.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Alvarez-Erviti L, Seow Y, Schapira AH, Gardiner C, Sargent IL, Wood MJ, Cooper JM. Lysosomal dysfunction increases exosome-mediated alpha-synuclein release and transmission. Neurobiol Dis 2011; 42(3):360-7; PMID:21303699; http://dx.doi.org/ 10.1016/j.nbd.2011.01.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Spencer B, Emadi S, Desplats P, Eleuteri S, Michael S, Kosberg K, Shen J, Rockenstein E, Patrick C, Adame A, et al.. ESCRT-mediated uptake and degradation of brain-targeted alpha-synuclein single chain antibody attenuates neuronal degeneration in vivo. Mol Ther 2014; 22(10):1753-67; PMID:25008355; http://dx.doi.org/ 10.1038/mt.2014.129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Spencer B, Kim C, Gonzalez T, Bisquertt A, Patrick C, Rockenstein E, Adame A, Lee SJ, Desplats P, Masliah E. alpha-Synuclein interferes with the ESCRT-III complex contributing to the pathogenesis of Lewy body disease. Hum Mol Genet 2016; 25(6):1100-15; PMID:26740557; http://dx.doi.org/ 10.1093/hmg/ddv633 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Hsu C, Morohashi Y, Yoshimura S, Manrique-Hoyos N, Jung S, Lauterbach MA, Bakhti M, Grønborg M, Möbius W, Rhee J, et al.. Regulation of exosome secretion by Rab35 and its GTPase-activating proteins TBC1D10A-C. J Cell Biol 2010; 189(2):223-32; PMID:20404108; http://dx.doi.org/ 10.1083/jcb.200911018 [DOI] [PMC free article] [PubMed] [Google Scholar]