
Figure 7. E. coli DNA Pol IV, Human DNMT1 Promote DNA Damage via Binding the Replisome Clamp.
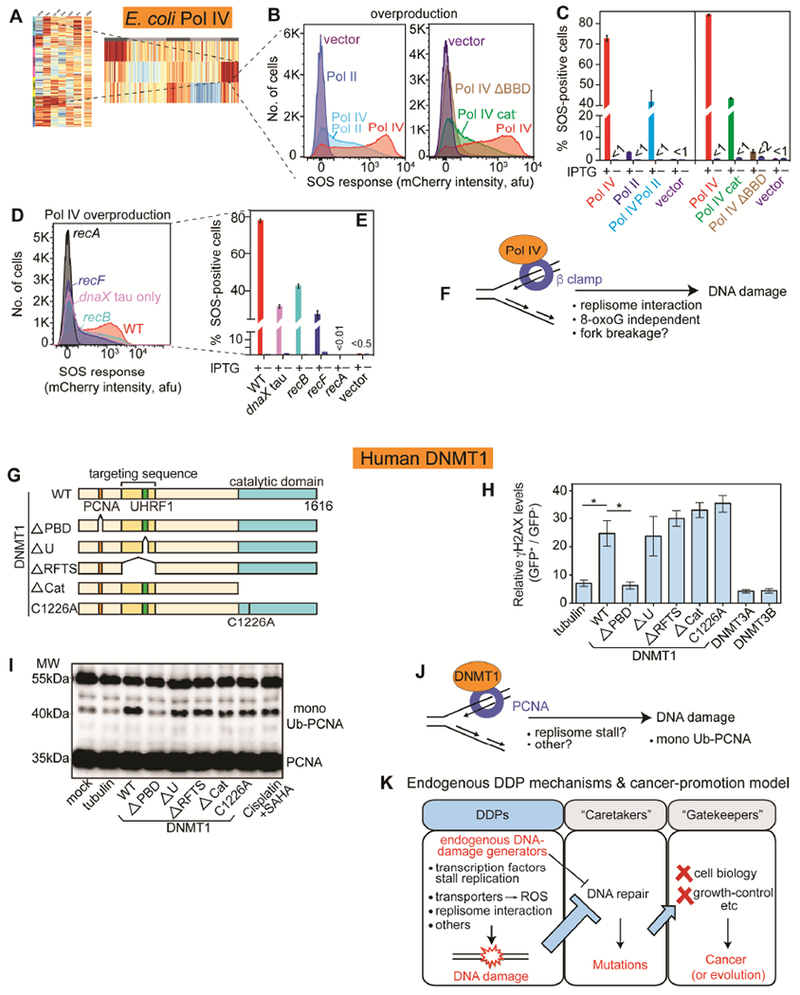
(A) E. coli Pol IV in cluster with high DNA loss.
(B) Left: Pol IV promotion of DNA damage is reduced by overproducing its competitor Pol II. Right: Pol IV promotion of DNA damage requires interaction with beta (β) replisome sliding clamp—ΔBBD, deleted β-binding domain—and is partly independent of Pol IV catalytic activity (cat− mutant).
(C) Mean ± SEM of ≥3 experiments.
(D) DNA damage reduced by reduction of Pol IV-β interaction with tau-only mutant, which favors Pol III. RecB- and RecF-dependence of Pol IV-induced DNA damage implicate DSBs and single-strand gaps.
(E) Mean ± SEM of ≥3 experiments. Pol IV is induced by IPTG.
(F) Model: Pol IV induces DNA damage by excess binding the β clamp. Excess β interaction might slow the replisome causing fork breakage/collapse, or displace β-binding DNA-repair proteins, among other possibilities. 8-oxo-dG-independence, Figure S7F,G.
(G) Mutant derivatives of human DNA methyltransferase DNMT1 (WT, wild-type). PBD, PCNA-binding domain; U, UHRF1 (ubiquitin-like PHD and RING-finger 1 interacting domain; RFTS, (recruits DNMT1 to DNA-methylation sites); Cat and C1226A, catalytically inactivate mutants, all N-terminally GFP tagged.
(H) Human DNMT1 overproduction in human cells promotes γH2AX accumulation methylase-independently and replisome-clamp-interaction dependently.
(I) Elevated DNMT1 promotes PCNA monoubiquitination (replication-stress) replisome-interaction dependently. Western blot with anti-PCNA antibody.
(J) Model/hypotheses for how excess DNMT1 promotes DNA damage.
(K) Hypothesis: DDPs, a cancer-protein function class upstream of DNA repair. Excessive endogenous DNA damage could titrate (thick blue -|) or inhibit (thin black -|) DNA repair causing DNA-repair-protein deficiency without a DNA-repair-gene mutation. Repair deficiency increases mutation rate, and cancer- (or evolution-) driving mutations in cell-biology-altering “gatekeeper” genes that cause the cancer phenotypes.