
Figure 7.
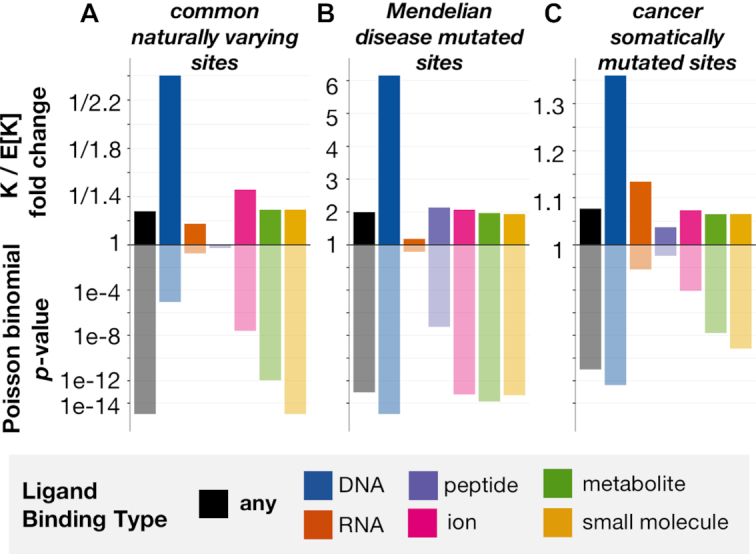
Natural variants show opposite trends from disease mutations with respect to ligand-binding sites. Putative ligand-binding sites correspond to protein positions overlapping domain match states whose binding frequencies resulted in a precision of at least 0.5 in cross-validation testing (i.e. confident interactions, see ‘Materials and Methods’ section). Shown on the y-axis (top) is the fold change between the observed (K) and expected (E[K]) numbers of InteracDome-inferred putative binding sites (any type, DNA, RNA, peptide, ion, metabolite or small molecule) and other sites of interest (common naturally varying, Mendelian disease mutated, cancer somatically mutated). We compute the significance of this overlap (y-axis, bottom) using the Poisson binomial distribution. (A) Putative ligand-binding sites exhibit a significant lack of overlap with commonly varying sites across human proteins. (B) Conversely, putative ligand-binding sites overlap significantly with sites harboring Mendelian disease mutations. (C) Protein sites harboring missense cancer somatic mutations also overlap significantly with putative ligand-binding sites, suggesting that these sites are preferentially altered in human cancers.