

Abstract
Stress granules (SGs) are non-membrane bound organelles that form in response to multiple different stress stimuli, including exposure to sodium arsenite. SGs are postulated to support cells during periods of stress and provide a protective effect, allowing survival. Gle1 is a highly conserved, essential modulator of RNA-dependent DEAD-box proteins that exists as at least two distinct isoforms in human cells. Gle1A is required for proper SG formation, whereas Gle1B functions in mRNA export at the nuclear pore complex. Since Gle1A is required for SG function, we hypothesized that SG-dependent survival responses would also be Gle1-dependent. We describe here an experimental system for quantifying and testing the SG-associated survival response to sodium arsenite stress in HeLa cells. Gle1A was required for the sodium arsenite survival response, and overexpression of Gle1A supported the survival response. Overexpression of the SG-component G3BP also enabled the response. Next, we analyzed whether cells undergoing multiple rounds of stress yield a subpopulation with a higher propensity for SG formation and an increased resistance to undergoing apoptosis. After ten doses of sodium arsenite treatment, cells became resistant to sodium arsenite and to diclofenac sodium (another SG-inducing drug). The sodium arsenite-resistant cells exhibited changes in SG biology and had an increased survival response that was conferred in a paracrine manner. Changes in secreted factors occurred including a significantly lower level of MCP-1, a known regulator of stress granules and stress-induced apoptosis. This study supports models wherein SGs play a role in cell evasion of apoptosis and further reveal Gle1A and SG functions as targets for clinical approaches directed at chemoresistant/refractory cells.
Keywords: Stress Granules, Gle1, MCP-1, cancer, survival, evasion of apoptosis
Introduction
Stress granules (SGs) are non-membrane bound cytosolic structures that form transiently in response to a variety of extracellular challenges, including oxidative, osmotic, ultraviolet light, heat, and chemotoxic stress (Fournier et al., 2010; Mahboubi and Stochaj, 2017; Szaflarski et al., 2016). During such cellular stresses, SGs are important contributors to the reprogramming of gene expression through the sequestering of mRNA-protein complexes (mRNPs) and associated preinitation complexes when translation is repressed and polysomes disassemble. Initial SG assembly occurs through a bi-phasic process, wherein translation-stalled mRNPs oligomerize into a dense and stable core surrounded by a phase-separated shell of proteins and mRNPs in flux with the cytosol (Amandine et al., 2015; Jain et al., 2016; Wheeler et al., 2016). These initial SGs then fuse into larger mature assemblies through a microtubule-dependent process, incorporating multiple cores around a combined shell (Fujimura et al., 2009; Kolobova et al., 2009; Wheeler et al., 2016). In addition to translation factors, mRNA, and RNA-binding proteins, numerous signaling factors and scaffolding proteins localize to SGs. In this way, SGs are thought to serve as powerful, highly dynamic, signaling hubs that mediate rapid and temporal changes for redirecting cellular function and promoting cell survival (Kedersha et al., 2013; Mahboubi and Stochaj, 2017). Following the release of stress, SGs disassemble as translation is resumed, presumably triggered by the recruitment of SG components back to polysomes (Panas et al., 2016).
The dynamics of SG assembly and disassembly require RNA-dependent ATPase proteins of the DEAD-box family (DDXs) (Hilliker, 2012; Jain et al., 2016). DDX proteins perform critical roles throughout the gene expression pathway by unwinding RNA duplexes and/or remodeling the complement of RNA-binding proteins bound to mRNPs (Jarmoskaite and Russell, 2014; Singh et al., 2015). Several reports show that DDX3 and DDX6 localize directly to SGs and play critical roles in translational repression (Wilczynska et al., 2005; Shih et al., 2012), and ten DDXs are in the SG interactome (Jain et al., 2016). Moreover, the highly conserved and essential regulator of DEAD-box proteins, Gle1, is required for SG dynamics (Aditi et al., 2015). While both of the two major human Gle1 isoforms are recruited to SGs, only the predominantly cytoplasmic Gle1A isoform modulates DDX3, alters translation activity and drives proper SG assembly and disassembly. Furthermore, overexpression of Gle1A increases the size of SGs assembled in response to heat shock (Aditi et al., 2015). In contrast, the Gle1B isoform plays a critical role at the nuclear pore complex, in coordination with inositol hexakisphosphate (IP6), by regulating DDX19B functions during mRNA export (Alcázar-Román et al., 2006; Folkmann et al., 2013; Aditi et al., 2015; Adams et al., 2017). Overall, the function of DDX proteins and the regulator Gle1 are critical for SG biology.
Rigorous control of SG dynamics is critical for a cell to survive stress and disease. However, in some disease states, the cellular stress response can be hijacked such that SGs amass aggregation-prone proteins or bring together signaling molecules that further the disease state. Indeed, recent proteomic studies find that SG composition is variable and both stress-dependent and disease-specific (Aulas et al., 2017; Markmiller et al., 2018). Misregulation of SGs and the cellular stress response pathways may lead to inappropriate mRNA metabolism and induce untimely cell death and/or result in propagation of disease (Li et al., 2013). For example, SG dysfunction is implicated in diseases such as ALS (Aditi et al., 2015), Fragile X syndrome (Kim et al., 2006) and cancer (Panas et al., 2016; Anderson et al., 2015). SG perturbations are also connected with lingering mRNP aggregations that contribute to premature motor neuron death (Li et al., 2013).
The connections between SG function and cancer cell survival are especially intriguing. During cancer treatments, chemotherapeutics are extensively used to disrupt mitosis and induce apoptosis of rapidly dividing cancer cells. However, these effects on cellular metabolism also induce a chemotoxic stress response that frequently triggers SG assembly (Fournier et al., 2010; Gareau et al., 2011; Kaehler et al., 2014; Adjibade et al., 2015; Szaflarski et al., 2016). In numerous tumor types, increased presence of SGs is associated with poor patient outcomes (Somasekharan et al., 2015; Grabocka and Bar-Sagi, 2016; Gupta et al., 2017). Several studies suggest that SG formation allows cells to evade apoptosis by sequestration of signaling molecules that are potent inducers of apoptosis, such as tumor necrosis factor (TNF) receptor-associated factor 2 (TRAF2) (Kim et al., 2005) and ribosomal protein RACK1 (Arimoto et al., 2008). Alternatively, SGs can recruit factors such as the tyrosine kinase Syk, which was shown to increase autophagy and thus enhance the survival of cancer cells in the presence of metabolic and therapeutic stress (Krisenko et al., 2015). Lastly, it has been suggested that SG assembly enhances cancer cell survival by activating the antioxidant activity of ubiquitin-specific protease 10 (USP10), which results in reduced ROS-dependent apoptosis (Takahashi et al., 2013).
Several other observations suggest that SG assembly factors themselves might promote increased tumor survival. High levels of the SG nucleator G3BP2 are associated with poor clinical outcomes of breast cancer patients, whereas G3BP2 depletion from breast cancer cell lines reduces the tumor initiating population and expression of oncogenes SART3, Oct4 and Nanog (Gupta et al., 2017). Further, G3BP1 downregulation in sarcoma xenografts correlates with blocked SG formation and reduced tumour invasion and metastases, whereas higher G3BP1 expression is strongly linked with poorer overall survival in Ewing sarcoma patients (Somasekharan et al., 2015). Numerous medulloblastoma-linked mutations are found in the sequence encoding the helicase domain of DDX3 (Epling et al., 2015; Pugh et al., 2012), a Gle1A-regulated DEAD-box ATPase required for proper translation initiation and SG assembly (Aditi et al., 2015; Bolger and Wente, 2011). Disruption of DDX3 catalytic activity results in hyper-assembly of SGs and translation impairment, contributing to the overall survival advantage to tumor cells (Valentin-Vega et al., 2016).
Further insights into potential controls on tumor cell growth through SGs might also come from Gle1’s dependence on proper inositol phosphate signaling (York et al., 1999; Alcázar-Román et al., 2010). In addition to Gle1 requiring IP6 for regulation of DEAD-box proteins during mRNA export (Alcázar-Román et al., 2006; Weirich et al., 2006; Montpetit et al., 2011; Adams et al., 2017) and translation (Bolger et al., 2008; Alcázar-Román et al., 2010), inositol phosphates (IPs) are also implicated in the regulation of a multitude of biological processes, including metabolism regulation, transcriptional control and environmental stress response (Lee et al., 2012; Hatch al., 2017; Michell, 2008). Indeed, both prokaryotic and eukaryotic organisms utilize inositol derivatives to provide highly effective cytoprotection under multiple types of cellular stress (Perera et al., 2004; Yancey et al., 2005; Michell, 2008). Importantly, IPs are also critical in Ca2+ signaling and can be exploited in cancer cells to alter the apoptosis response and contribute to an overall survival advantage of these cells (Ando et al., 2018). For example, oncogenes such as K-Ras and Akt function in coordination with IP3 to reduce Ca2+ release and consequently induce apoptotic resistance (Marchi et al., 2008; Szado et al., 2008). Additionally, tumor suppressors, such as PTEN, also utilize IP3-receptor signaling to sensitize mitochondria for Ca2+ release and to induce apoptosis (Szado et al., 2008). Disruption of the IP-regulation pathway is proposed as a novel therapeutic strategy against cancer (Ando et al., 2018), and such altering of the IP pathway might impact Gle1, DDX, and SG functions.
With SG misregulation and disease pathogenesis linked, it is important to define the precise mechanisms underlying SG function. More so, it is not fully understood how SGs contribute to increased chemotherapeutic resistance and cancer cell survival through apoptotic evasion or whether SG assembly is necessary and sufficient for tumor cell survival. Given the role for Gle1A in SG dynamics and regulating DDX3, we sought to determine whether chemotoxic SG assembly and tumor cell survival are Gle1-dependent processes. Here, we directly demonstrate that SG formation confers a survival advantage to cells undergoing chemotoxic stress, and that Gle1A is necessary for this survival response.
Results
Sodium arsenite chemotoxicity induces a survival response and SG formation in HeLa cells
To investigate the mechanisms underlying connections between SG formation and increased tumor cell fitness under chemotoxic conditions, we established a defined cell culture system that recapitulates a chemotoxic stress-induced survival response. To do this, we tested a panel of known SG-inducing chemotoxic agents across a range of doses for their effect on the viability of HeLa cervical cancer cells (data not shown). Based on this analysis and prior studies (Takahashi et al., 2013; Chang and Huang, 2014), we focused on use of sodium arsenite (NaAsO2). HeLa cells were treated for 72 hours with a range of soluble sodium arsenite doses from 6 nM to 25 mM. Cell viability was assessed using the luminescence-based Cell Titer Glo® assay to measure cellular ATP levels. Between 0.001 μM and 10 μM sodium arsenite, a canonical inverse sigmoidal dose-response curve was observed, indicating loss of cell viability with increasing concentrations of sodium arsenite (Fig. 1A). However, higher doses of sodium arsenite produced a biphasic effect on cell viability, with increasing survival from 100 μM to 3 mM followed by decreasing viability from 4 mM to 25 mM sodium arsenite. The viability at the concentration range from 100 μM to 25 mM was termed the survival response, peaking at 3 mM with around 50% of cells surviving at this dose. Overall, treatment with sodium arsenite resulted in highly reproducible dose-response curves wherein very high doses induced a distinct survival response. This phenomenon was also detected when cell metabolic activity was measured via the colorimetric cell metabolism MTT assay (data not shown). A similar biphasic effect of sodium arsenite treatment on cell viability was found with DLD-1 colorectal adenocarcinoma cells, although the proportion of viable DLD-1 cells during the survival response at high dosage was limited to approximately 10% (data not shown).
Figure 1. High doses of sodium arsenite induce SG formation and a survival response in HeLa cells.
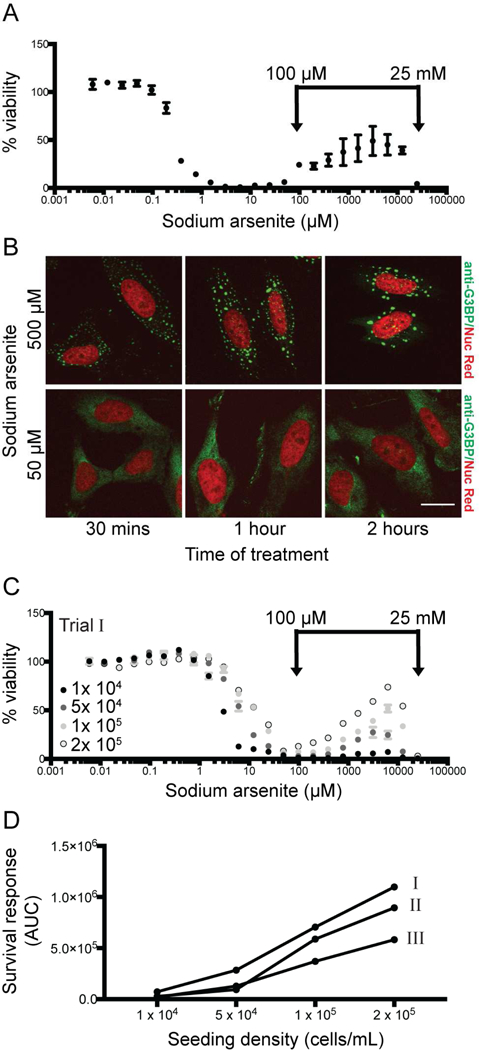
(A) HeLa cells (seeded at 1×105 cells/mL) were treated with sodium arsenite at concentrations ranging from 6 nM to 25 mM for 72 hours and viability was measured. A representative viability curve is shown, where each data point represents the average of 2 technical replicates normalized to duplicate untreated wells. Error bars denote range. Survival response is defined as the biphasic effect on cell viability induced at sodium arsenite concentrations > 100 μM. (B) SG formation was assessed by anti-G3BP indirect immunofluorescence, with Nuc Red® stain marking nuclear material. Scale bar represents 15 μm. (C) Viability and survival response were assessed at different seeding densities (1×104, 5×104, 1×105 or 2×105 cells/mL) Representative viability curves are shown. (D) The impact of seeding density on survival response (C) was quantified as area under the curve (AUC) from 100 μM-25 mM sodium arsenite, and shown for three independent trials. Data shown in (C) is indicated in (D) as “Trial I”.
To determine if the survival response observed in sodium arsenite-treated HeLa cells was coincident with SG assembly, we performed indirect immunofluorescence microscopy for localization of the SG nucleator protein G3BP. Large anti-G3BP-positive cytoplasmic foci, indicative of mature SGs, were present in HeLa cells treated with 500 μM sodium arsenite. In contrast, lower sodium arsenite concentrations only stimulated the formation of numerous small cytoplasmic foci typical of immature SGs (Fig. 1B).
We also tested the effect on the survival response when the number of cells in the starting culture changed (seeding density; cells/mL) (Fig. 1C). Across a twenty fold range of seeding densities, the respective survival responses were then quantified as the area under the curve (AUC) for each dose response curve from 100 μM to 25 mM. The survival response (AUC) versus seeding density was analyzed for three independent experiments (Fig. 1D). Increasing seeding density correlated with an apparent increase in the survival response of the given cell population (Fig. 1C, D). This data suggested that cell density is critically important in the survival response, possibly mediated through cell-cell contacts or paracrine signaling between neighboring cells. Taken together, this analysis established sodium arsenite-induced HeLa cell viability as a reproducible model of SG-associated chemotoxic survival and AUC as a robust and quantifiable measure of the survival response.
Gle1 is required for sodium arsenite-mediated survival response and SG dynamics
In our prior studies, we discovered a role for Gle1A in regulating the heat-shock induced SG response of HeLa cells (Aditi et al., 2015). Thus, we speculated that Gle1A is also required for the SG-associated survival response found under high sodium arsenite dose conditions. Using our cell culture model, the effect of Gle1 depletion was directly assayed. HeLa cells were subjected to either non-targeting (CTRL) or GLE1 siRNA treatment for 72 hours prior to sodium arsenite exposure. Given the essential roles for Gle1 in mRNA export and translation (Folkmann et al., 2013), reduced cell viability following GLE1 siRNA treatment was anticipated and, indeed, an overall 40–50% loss of viability was observed in GLE1 siRNA treated samples. Each experimental condition was normalized to duplicate untreated wells, allowing comparison of the survival response as a proportion of the overall cell viability in each condition. Comparison of the resulting dose response curves revealed that the sodium arsenite stress-induced survival response was abrogated in Gle1-depleted cells (Fig. 2A). This effect was quantified by AUC analysis of the normalized viability curves for four independent experiments. We observed a statistically significant reduction in survival response following Gle1 depletion (Fig. 2B).
Figure 2. Gle1 is required for survival response.
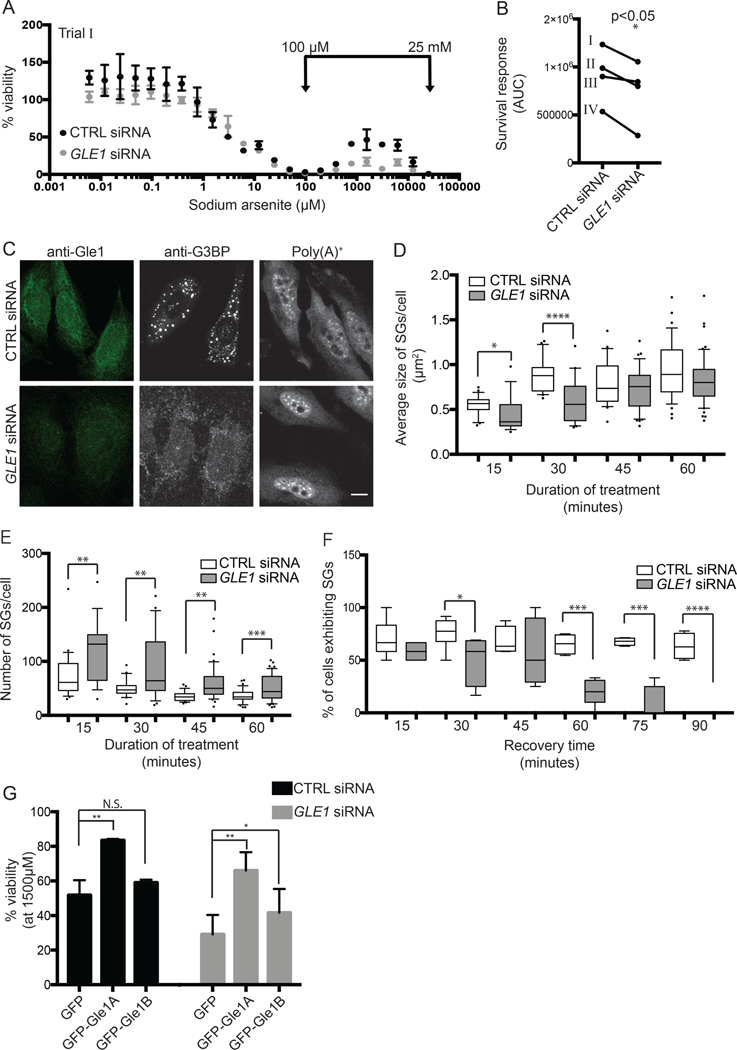
(A) Dose response curves of sodium arsenite on cell viability were assessed for nontargeting (CTRL) or GLE1 siRNA-treated HeLa cells exposed to 6 nM to 25 mM arsenite for 72 hours. Representative viability curves are shown, where each data point represents the average of 2 technical replicates normalized to duplicate untreated wells. Error bars denote range. (B) The sodium arsenite survival response was quantified as described, and plotted for four independent trials. Data shown in (A) is indicated in (B) as “Trial I”. (C) SG formation and mRNA export were evaluated in CTRL or GLE1 siRNA-treated HeLa cells exposed to 500 μM sodium arsenite for 1 hour. SGs were detected by anti-G3BP indirect immunofluorescence, and poly(A)+ localization was determined by in situ hybridization to Cy3-oligo dT. Scale bar represents 10 μm. (D, E) Size and number of SGs were determined for CTRL and GLE1 siRNA-treated HeLa cells after 500 μM sodium arsenite treatment 15 to 60 minutes. G3BP immunofluorescence was quantified using ImageJ 3D objects counter, where surface areas of 3D objects was reported. Data was collected at each time point for three independent experiments, and plotted onto a box and whiskers graph where whiskers denote 10th-90th percentile of data. (F) The percentage of cells exhibiting SGs was monitored over recovery time following 500 μM sodium arsenite treatment for 1 hour. SG-positive cells were quantified at each time point for three independent experiments, and plotted onto a box and whiskers graph. (G) Survival response to 1500 μM sodium arsenite for 72 hours was assessed in CTRL or GLE1 siRNA-treated HeLa cells exogenously expressing GFP, GFP-Gle1A or GFP-Gle1B. *p<0.05; **p<0.01; ***p<0.001; statistical significance was determined by Student’s paired t-test.
To analyze the impact of Gle1 depletion on sodium arsenite-induced SGs, we performed anti-G3BP indirect immunofluorescence microscopy. We previously reported that the heat shock-induced SG dynamics in HeLa cells are Gle1-dependent (Aditi et al., 2015), with perturbations of both SG assembly and disassembly. Here, in sodium arsenite treated cells, Gle1 depletion also reduced the average size of sodium arsenite-induced SGs (Fig. 2C & D) and increased the absolute number of SGs (Fig. 2E), indicative of incomplete SG assembly. Furthermore, SG disassembly following removal of sodium arsenite was completely resolved after 90 minutes of recovery in Gle1-depleted cells, whereas negligible SG disassembly was observed in CTRL-treated HeLa cells (Fig. 2F).
Overexpression of Gle1A or G3BP increase the survival response to chemotoxic stress
We next coupled our established GLE1 knockdown:addback approach (Aditi et al., 2016, 2015; Folkmann et al., 2013) with sodium arsenite treatment to determine which isoforms of Gle1 are capable of supporting the survival response. From assessments across the range of sodium arsenite doses (Fig. A), quantitation of viability was compared at 1500 μM in the high-dose survival response range (Fig. 2G). Exogenous expression of either GFP-Gle1A or GFP-Gle1B in GLE1 siRNA treated cells rescued the survival response during sodium arsenite stress, although Gle1A was more robust (Fig. 2G). However, only exogenous GFP-Gle1A overexpression increased the sodium arsenite stress-induced survival response in CTRL siRNA-treated cells. GFP-Gle1B overexpression had no effect on the sodium arsenite stress-induced survival response compared to GFP alone (Fig. 2G). These data were consistent with our prior demonstration that Gle1A specifically mediates SG dynamics and supported our hypothesis that SG assembly is necessary for chemotoxic survival.
Our knockdown:addback analysis suggested that Gle1A mediates the SG response to drive cell survival following sodium arsenite chemotoxicity. However, Gle1 depletion targets both Gle1A and Gle1B isoforms and thereby also inhibits mRNA export (Gle1B is specifically required for mRNA export (Folkmann et al., 2013); Fig. 2C). Therefore, as a control, we tested whether the loss of the survival response upon Gle1 depletion was a function of impaired mRNA export. The mRNA export factor Nxf1 (TAP) functions as an mRNA export receptor, binding to mature mRNA and transporting it across the central channel of the nuclear pore complex for subsequent translation in the cytoplasm (Grüter et al., 1998). In NXF1 siRNA treated cells, Nxf1 depletion was validated by anti-Nxf1 indirect immunofluorescence microscopy and in situ poly(A)+ hybridization (Fig. 3A). Decreased anti-Nxf1 signal and accumulation of nuclear poly(A)+ RNA in NXF1 siRNA-treated cells confirmed a block to mRNA export. The NXF1 siRNA treatment also reduced overall cell viability by approximately 60–70%; however, it did not significantly alter the sodium arsenite dose response curve for cell viability or the chemotoxic survival response at high sodium arsenite doses (Fig. 3B & C). Therefore, we concluded that Gle1A enhances the sodium arsenite stress-induced survival response through its role in SG dynamics.
Figure 3. Sodium arsenite survival response is mediated by stimulation of SG assembly, not mRNA export inhibition.
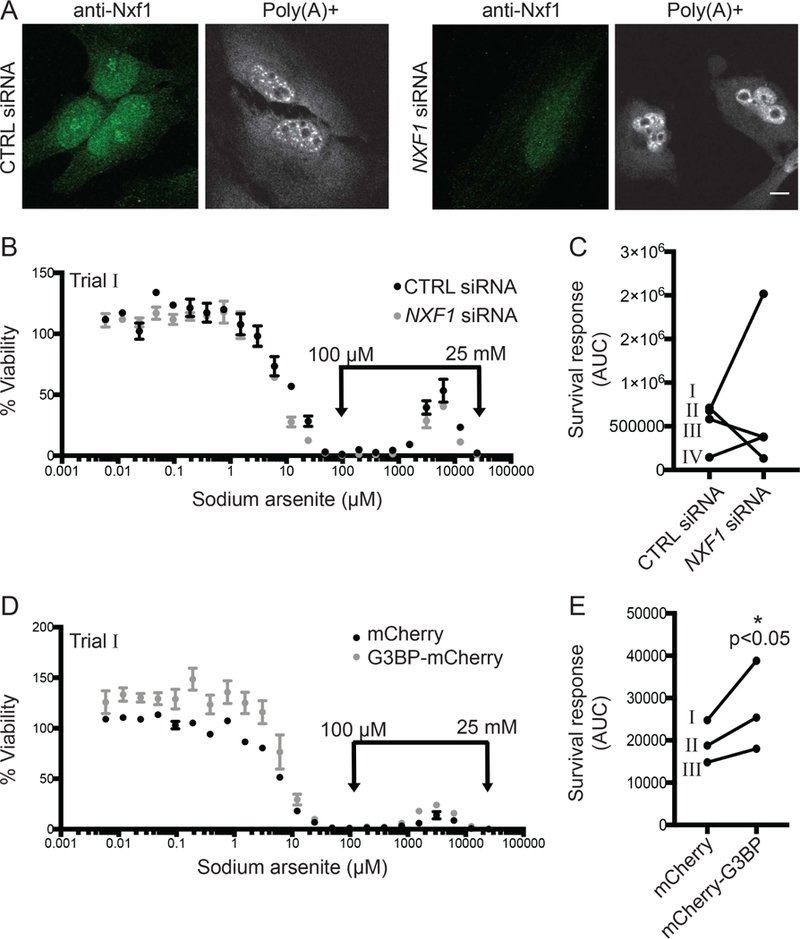
(A) Nxf1 localization and mRNA export were evaluated in control (CTRL) or NXF1 siRNA-treated HeLa cells by anti-Nxf1 indirect immunofluorescence and in situ hybridization to Cy3-oligo d(T), respectively. Scale bar represents 10 μm. (B, C, D, E) Dose response curves of sodium arsenite on cell viability and the sodium arsenite survival response were assessed as described for (B, C) CTRL or NXF1 siRNA-treated HeLa cells, or (D, E) HeLa cells exogenously expressing mCherry or mCherry-G3BP. Data shown in (B) and (D) are indicated in (C) and (D) as “Trial I”. *p<0.05; statistical significance was determined by Student’s paired t-test.
Overexpression of the SG nucleator G3BP induces the formation of SGs in the absence of stress conditions (Matsuki et al., 2013; Tourrière et al., 2003). We independently tested the influence of SG formation on our chemotoxic survival response model by overexpressing G3BP, an SG nucleator. G3BP overexpression, increased viability across the sodium arsenite dose response curve and reproducibly enhanced the survival response (Fig. 3D & E). We attempted to determine the impact of G3BP depletion by siRNA on the sodium arsenite stress-induced survival response; however, G3BP depletion resulted in greater than 90% reduced cell viability. The impact of G3BP overexpression on sodium arsenite stress-induced survival indicated that SG assembly is a driver of the cell’s capacity to survive chemotoxic stress.
Repeated sodium arsenite stress results in a population of cells with decreased sensitivity to sodium arsenite and altered SG dynamics
Next we established a model for a resistant sub-population of cells that exhibited a higher propensity of SG formation and a stronger survival response during sodium arsenite treatment. HeLa cells were exposed to either 500 μM sodium arsenite or vehicle for 10 rounds of treatment in order to generate a resistant population of cells (Fig. 4A). The resulting sodium arsenite-resistant cell population exhibited a distinct change in morphology (Fig. 4B), altered viability in different seeding densities (despite a comparable growth rate) (Fig. 4C; Fig. B), increased survival response to sodium arsenite-stress (Fig. 4D), and an increased IC50 to sodium arsenite treatment (Fig. 4E). When these sodium arsenite-resistant cells were allowed to recover over 10 further passages, the resistance phenotype was lost (data not shown). Anti-G3BP staining revealed that the SGs in the resistant cells were also altered. Resistant cells subjected to high doses to sodium arsenite (>500 μM) exhibited a significant decrease in the number of SGs per cell and an increase in SG size (Fig. 4F & G). Together, these metrics revealed an overall increase in the SG response for resistant cells. These results suggested that HeLa cells increase their propensity for SG formation following multiple exposures to sodium arsenite.
Figure 4. Sodium arsenite-resistant cells exhibit altered cell morphology and SG biology.
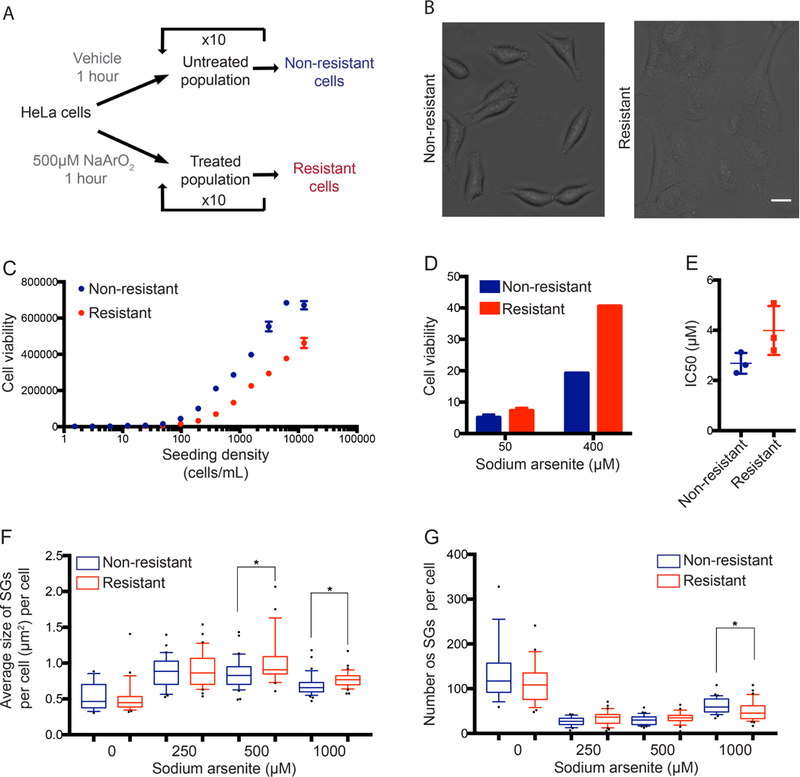
(A) Schematic depicting the process taken to generate arsenite-resistant HeLa cells. (B) Following 10 rounds of sodium arsenite treatment, cell morphology was evaluated by bright field microscopy. Scale bar represents 20 μM. (C) Cell viability was measured for non-resistant and arsenite-resistant HeLa cells seeded at increasing cell densities. (D, E) Dose response curves of sodium arsenite on cell viability were generated as described for non-resistant and arsenite-resistant HeLa cells. Survival responses and IC50 values were calculated from 3 independent experiments. (F, G) Non-resistant and arsenite-resistant HeLa cells were subjected to 0, 250, 500 or 1000 μM sodium arsenite treatment for 60 minutes and SG phenotypes were analyzed. Size (F) and number of SGs (G) were calculated as previously described. Data was collected for three independent experiments and plotted onto a box and whiskers. *p<0.05 using Student’s unpaired t-test.
To confirm that the enhanced SG assembly observed in the resistant cell model was reflective of a general increase in SG responsiveness, we tested whether the sodium arsenite resistant cell population also exhibited resistance to other chemotoxic agents. Measurement of the IC50s for non-resistant and resistant cells was conducted with other known SG-inducing agents (Fig. 5). The sodium arsenite resistant cells displayed decreased sensitivity to diclofenac sodium, increased sensitivity to paclitaxel (PCX) and no change in sensitivity to bortezomib (BTZ) or emetine (Fig. 5). Thus, increasing the propensity to form SGs in response to one chemotoxic agent did not universally increase resistance to all other SG-inducing agents.
Figure 5. Sodium arsenite resistance confers differential sensitivity to SG-inducing chemotoxic agents.
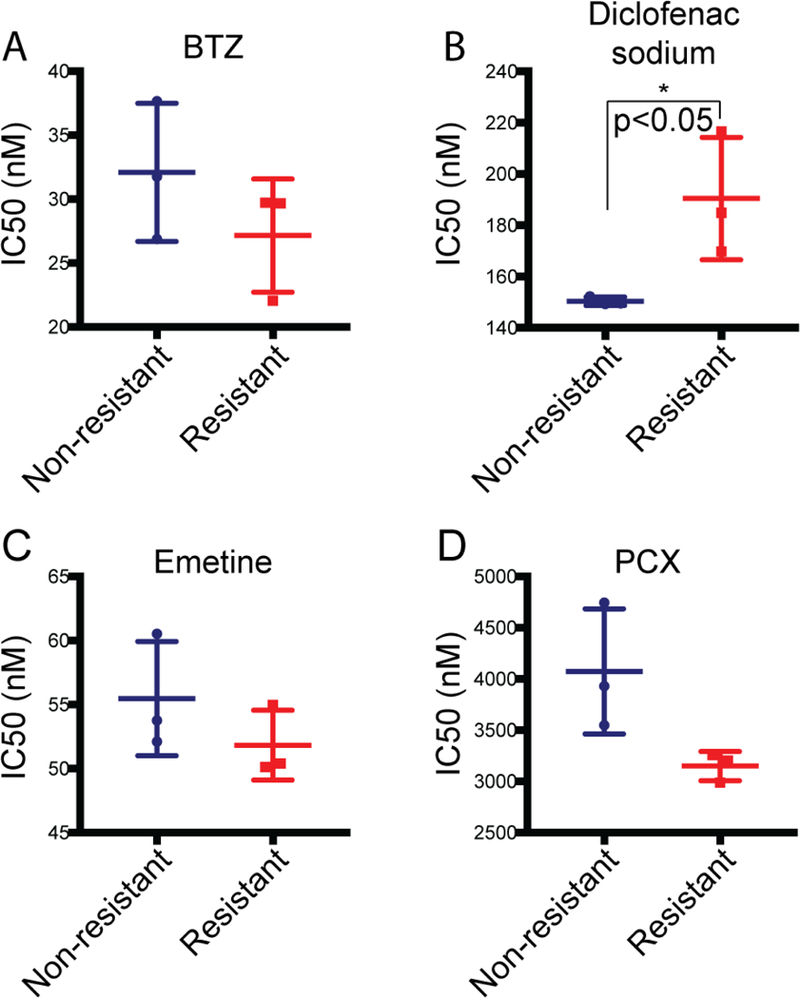
IC50s of non-resistant and arsenite resistant cell viability were determined after exposure to the indicated chemotoxic agents for 72 hours. Cells were treated with (A) 0.2 pM to 1 μM bortezomib (BTZ); (B) 9.5 nM to 40 mM diclofenac sodium; (C) 1 pM to 5 mM emetine; or (D) 2 pM to 8 mM paclitaxel (PCX). IC50s were calculated from 3 independent experiments. *p<0.05 using Student’s unpaired t-test.
Sodium arsenite-resistant cells secrete altered levels of MCP-1 and Serpin E1
Studies by others link secreted factors to pro-tumorigenic behavior, and there is an emerging body of evidence demonstrating that autocrine and paracrine signaling can promote SG formation and increase tumor cell survival (Fonseca et al., 2016; Quail and Joyce, 2013; Tan et al., 2018). The prostaglandin 15-d-PGJ2 increases SGs in mutant KRAS tumour cells, conferring protection (Grabocka and Bar-Sagi, 2016). Based on this, and our observation of a seeding density effect (Fig. 1C & D), we speculated that the sodium arsenite-induced survival response and the sodium arsenite-resistant cell line were dependent on secreted factor(s). To test this, conditioned media (CM) was collected from cultures of the non-resistant (NR) and the sodium arsenite-treated resistant (R) HeLa cell populations and analyzed for their capacities to induce a change in respective survival responses. We found that when non-resistant cells were incubated with CM from resistant cells, the non-resistant cells exhibited an increased survival response at high doses of sodium arsenite (Fig. 6A & B). Further, sodium arsenite resistant cells incubated with CM from non-resistant cells also retained a high survival response (Fig. 6A & B). This indicated that sodium arsenite-resistant cells alter the pool of secreted factor(s).
Figure 6. Arsenite-resistance is mediated by MCP-1 and Serpin E1.
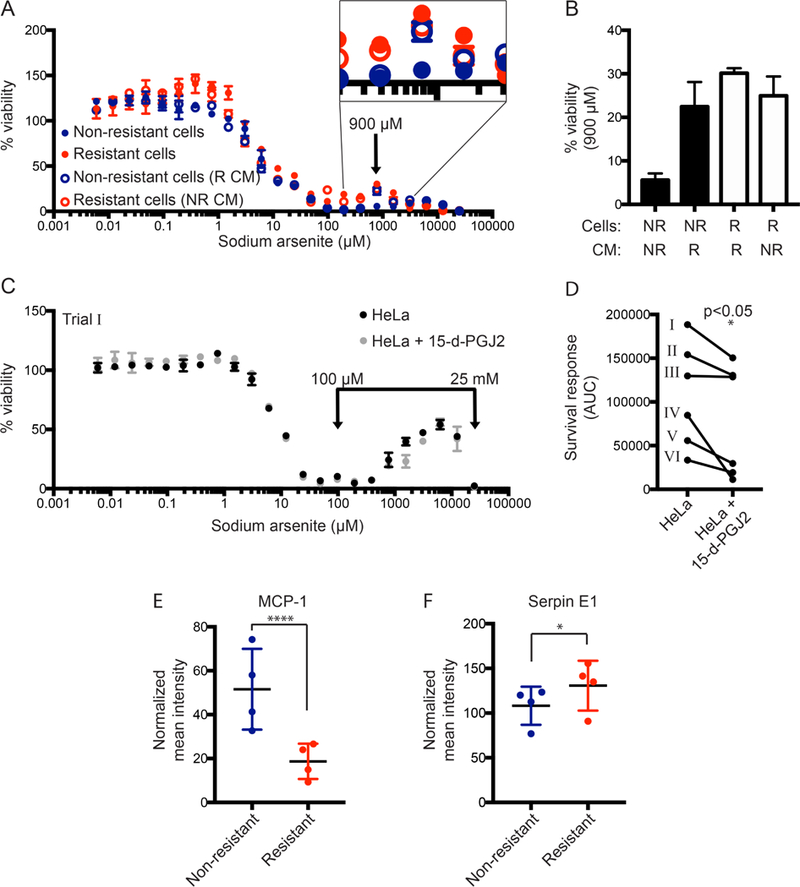
(A) The conditioned media of non-resistant and arsenite-resistant HeLa cells was swapped and dose responses of sodium arsenite on cell viability were measured and plotted as described. (B) Cell viability was compared for each experimental group at 900 μM sodium arsenite, denoted by an arrow on the viability curves in (A). (C) Dose responses of sodium arsenite on cell viability were determined and plotted as described for untreated HeLa cells or HeLa cells pre-incubated with 50 μM 15-d-PGJ2 for 1 hour. (D) The arsenite survival response was quantified as described, and plotted for six independent trials. Data from (C) is represented in (D) as “Trial I”. *p<0.05 using Student’s paired t-test. (E, F) Conditioned media samples from non-resistant and arsenite-resistant cells were probed for 105 human cytokines using a membrane-based sandwich immunoassay. Densitometry was performed for each cytokine and normalized to a loading control. Two-way ANOVA with multiple comparisons was performed to identify differentially secreted factors; *p=0.0255, ****p<0.0001.
Since the secreted prostaglandin 15-d-PGJ2 is known to mediate SG signaling (Grabocka and Bar-Sagi, 2016), we tested whether addition of this factor to naïve HeLa cells would increase their sodium arsenite stress-induced survival response. Following 1 hour pre-treatment at 50 μM 15-d-PGJ2, cells were subjected to sodium arsenite exposure as described. Surprisingly, the survival response was decreased compared with untreated cells (Fig. 6C & D). There were no differences in overall viability between cells treated with 15-d-PGJ2 and those treated with vehicle. Thus, this specific prostaglandin alone did not induce SG-formation and increase the survival response in all contexts.
Because the results from the conditioned media swap experiments (Fig. 6A and B) indicated that the sodium arsenite-induced survival response was being conferred by a secreted factor(s), we tested conditioned media from non-resistant and sodium arsenite-resistant cells for the presence of 104 human cytokines using a commercially available membrane-based antibody array. Four independent collections of CM from sodium arsenite-resistant and non-resistant cells were incubated with membranes spotted with capture antibodies for each cytokine. Signal was detected using streptavidin-HRP and chemiluminescent reagents. Densitometry of duplicate spots was normalized to an internal reference control. Mean signal from each cytokine is summarized in Fig. C. A two-way ANOVA statistical test of the resulting signal for each analyte revealed that MCP-1 levels were dramatically reduced in CM from sodium arsenite-resistant cells compared to non-resistant cells (Fig. 6E). Additionally, Serpin E1 concentration was increased in the CM from the sodium arsenite-resistant cells (Fig. 6F). Thus, we concluded that these factors are candidates for mediating the survival response to sodium arsenite.
Discussion
The major clinical obstacle in the treatment of cancer patients is the emergence of resistant or refractory disease (Zheng, 2017). Cancer cells can acquire resistance to therapeutic agents through a variety of mechanisms including mutations, compensatory pathways, or by hijacking stress response pathways (Anderson et al., 2015). There are several studies that implicate a role for SGs in increasing the fitness of tumor cells; however, the precise mechanisms remain unclear. For this study, we established experimental systems to test the role of SGs in acute chemotoxic cell survival and to analyze determinants of the survival response for a sodium arsenite-resistant cell culture model. By altering Gle1 levels and testing the Gle1A and Gle1B isoforms for functionality, we find that SG formation is essential for the sodium arsenite stress-induced survival response. Using this cell model, we further propose that the survival response of sodium arsenite resistant cells is conferred via a secreted factor.
Our work here supports models wherein SGs play a protective role during periods of stress that allow tumor cells to evade apoptosis. When HeLa cells properly form large SGs, they are more likely to survive treatment with sodium arsenite (Fig. 2A). We directly tested this premise through depletion of the key SG modulator, Gle1. Survival is significantly reduced in Gle1-depleted cells that are unable to form mature SGs, indicating that proper SG assembly is necessary for the survival response (Fig. 2A & B). Further, sodium arsenite stress-induced survival is dependent upon the presence of Gle1A and most enhanced by Gle1A overexpression (Fig. 2G) the isoform specifically required to drive SG maturation (Aditi et al., 2015). While a slight increase in sodium arsenite stress-induced survival is also seen with Gle1B expression, we attribute this effect to its rescue of DDX19B stimulation, which potentially plays roles in both mRNA export and translation (Fig. 2G). Additionally, overexpression of G3BP, which stimulates nucleation of new SGs (Tourrière et al., 2003; Matsuki et al., 2013), increases the survival response in our experimental system. Overall, these data provide direct evidence that SG assembly is sufficient to drive the survival response.
The effect of Gle1 depletion on the formation of sodium arsenite-induced SGs mirrors the phenotype reported for heat-shock induced SGs, where smaller and more numerous SGs are observed in the absence of Gle1 (Aditi et al., 2015). Interestingly, the effect on SG disassembly during sodium arsenite stress is distinct from what we previously reported for heat-shock induced SGs. Here, sodium arsenite-induced SGs disassemble more rapidly in Gle1-depleted cells, whereas in heat-shocked cells, the SGs are more stable without Gle1 (Aditi et al., 2015). The different phenotypes are likely due to the stress-specific mechanisms used to induce SG formation. A recent study of SG biology suggests that different forms of stress result in considerable differences in the structure and composition of SGs (Aulas et al., 2017) and in the mechanisms of SG clearance. For example, ZFAND1, which recruits the 26S proteasome to SGs, is specifically required in the clearance of sodium arsenite-induced SGs, but not for SGs induced by heat-shock, osmotic or oxidative stress (Turakhiya et al., 2018). The contrasting stabilities of Gle1-depleted sodium arsenite-induced versus heat shock-induced SGs provide a further example of stress-specific SG responses.
Our finding that the levels of secreted factors are altered in sodium arsenite-resistant cell conditioned media supports a hypothesis that paracrine signaling is responsible for SG-mediated resistance and survival. The transfer of conditioned media from sodium arsenite-resistant cells confers a survival response to the naive cells, demonstrating that cell non-autonomous signaling can induce a “resistance” phenotype in cells that were previously “non-resistant”. This echoes findings by others wherein a secreted factor or factors increase tumor cell survival by increasing SG formation (Anderson et al., 2015; Grabocka and Bar-Sagi, 2016). We also find that increased cell density during sodium arsenite treatment increases the survival response. This is consistent with the model that increased paracrine/non cell autonomous signaling contributes to a survival mechanism in the tumor microenvironment and leads to a higher proportion of cells evading apoptosis. Furthermore, following multiple rounds of sodium arsenite treatment, it appears that the sodium arsenite-resistant population of cells require a higher seeding density for equivalent growth than that for the population treated with vehicle, despite having comparable proliferation rates (Fig. 4C; Fig. B). Thus, the sodium arsenite-resistant cells might have an increased dependency on a secreted factor(s). This type of behavior might also be indicative of cancer cell quiescence, where cells are maintained in vivo at a reduced proliferative rate, and thus are resistant to anti-proliferating anti-cancer drugs (Chen et al., 2016).
Although the signaling lipid molecule (15-d-PGJ2) is implicated in enhanced SG formation and stress resistance (Grabocka and Bar-Sagi, 2016), it did not increase the survival response in our experimental system. Through analysis of the conditioned media from sodium arsenite-resistant cells, we instead pinpoint monocyte chemotactic protein-1 (MCP-1; CCL2) as a candidate negative regulator of chemotoxic survival. MCP-1 stimulates monocyte chemotactic protein-induced protein 1 (MCPIP1), which is a multifunctional nuclear factor responsible for apoptosis of cardiomyocytes (Zhou et al., 2006), angiogenesis (Yang et al., 2013) and inflammatory reactions (Jura et al., 2012). Importantly, MCPIP1 negatively regulates SG formation (Qi et al., 2011). In splenocytes from Mcpip1−/− mice, SGs form even in the absence of sodium arsenite stress and the cells display resistance to stress-induced apoptosis (Qi et al., 2011). Here, sodium arsenite-resistant cells that secrete reduced levels of MCP-1, the factor upstream of MCPIP1, assemble fewer and larger SGs and exhibit a marked increase in survival response (Fig. 6). Moreover, the reduced MCP-1 levels in resistant cell conditioned media convey a paracrine signal to naïve HeLa cells that increases their sodium arsenite stress-induced survival response, whereas resistant cells maintain chemoresistance through continued regulation of autocrine MCP-1 signaling (Fig. 6B). Therefore, we posit that MCP-1 and MCPIP1 act as negative regulators of the chemotoxic survival response, through inhibition of SG assembly (Fig. 7).
Figure 7. Model for the role of MCP-1 in differential response of non-resistant vs arsenite-resistant cell lines.
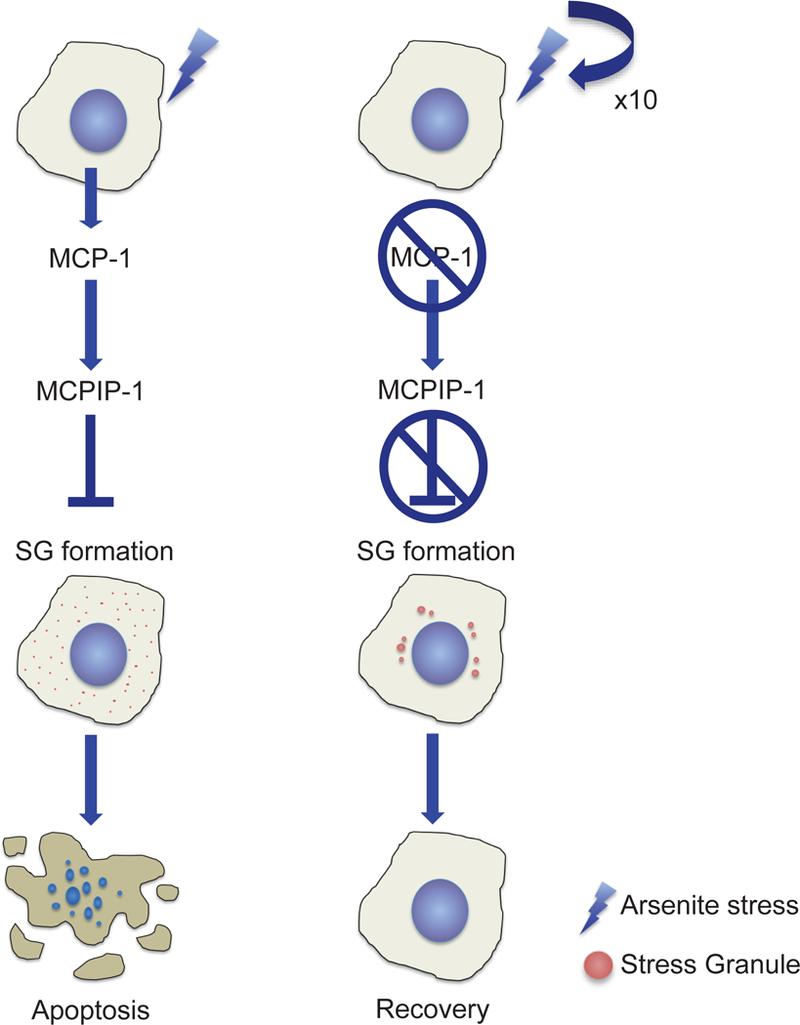
In non-resistant cells (left), MCP-1 is secreted, which in turn induces MCPIP-1; a known suppressor of SG formation. When cells cannot form SGs, apoptosis is more likely to be induced. However, in arsenite-resistant cells (right), MCP-1 secretion is suppressed. This prevents stimulation of MCPIP-1 and therefore relieves the blockage of SG formation. This leaves the cells with an increased propensity to form SGs and allows the cell to survive the stress and recover.
Our analysis also detects a modest increase in Serpin E1 levels in conditioned media from sodium arsenite resistant cells. (Fig. 6F). Serpin E1 is a component of SGs (Omer et al., 2018), and Serpin E1 mRNA-binding protein 1 (SERBP1) is recruited to SGs during sodium arsenite treatment (Lee et al., 2014). Additionally, elevated plasma levels of Serpin E1 and genetic polymorphisms are a diagnostic indicator of poor prognosis for certain cancers (Divella et al., 2015; McMahon and Kwaan, 2015). In sum, these data point toward a role for Serpin1 in promoting sodium arsenite-induced survival through enhanced SG assembly. Further studies will be required to clarify the exact contributions of MCP-1 and Serpin E1 secretion in SG formation and the regulation of chemotoxic survival.
Taken together, this study reveals that Gle1A-directed SG assembly offers a protective survival advantage to the cell and its neighbors during chemotoxic stress. If targeting the formation and function of SGs provides a clinical opportunity for increasing the susceptibility of cancer cells to undergo apoptosis, then our work suggests that Gle1A inhibitors are potential therapeutic candidates. As Gle1 function is impacted by IP6, altering the IP pathway might also impact SGs or shift the pools of Gle1A versus Gle1B that is available for function at SGs. In addition, modulating the levels of secreted factors like MCP-1 and Serpin E1 might impact in vivo chemoresistance. This work sets the stage for future investigations of Gle1A function, other SG assembly factors and components, secreted factors, and the IP pathway in combination with the effects of different chemotoxic stress inducers that will provide critical insights into the SG-associated survival response and the accompanying mechanisms of cell survival.
Materials and Methods
Cells culture and treatments
HeLa cells (ATCC) were cultured in DMEM (Thermo Fisher Scientific, Waltham, MA), supplemented with 10% Fetal Bovine Serum (FBS, Atlanta Biologicals, Flowery Branch, GA) at 37°C in 5% CO2. Sodium arsenite (NaAsO2, Sigma, St Louis, MO) was diluted in media (25 mM to 6 nM) before addition to cells. Cells were treated with dilutions of bortezomib (BTZ; 1 μM to 0.2 pM), diclofenac sodium (40 mM to 9.5 nM), emetine (5 mM to 1 pM) or paclitaxel (PCX; 8 mM to 2 pM). 15-d-PGJ2 (Sigma) was added to media at 50 μM (50 mM stock solution in dH2O).
Cell Viability assays
Cells were seeded in 96-well plates (Fisher Scientific, Pittsburgh, PA) and grown for 24 hr prior to drug incubation. Cells were incubated with either sodium arsenite, BTZ, PCX, emetine or diclofenac sodium at the indicated concentrations for 72 hr. Cell Titer-Glo reagent (Promega, Madison, WI) was added directly to wells according to manufacturer’s instructions, and luminescence was measured using a Synergy 2 plate reader (BioTek, Winooski, VT). Dose-response curves were normalized to untreated wells, and survival response was assessed by calculating the area under the curve (AUC) from 100 μM-25 mM sodium arsenite for each curve using Prism 7 (GraphPad Prism 7, La Jolla, CA). Alternatively, MTT assays were carried out after drug treatments. Briefly, following aspiration of media, MTT (12 mM in PBS) was added to wells and incubated at 37°C for 4 hr. The reaction was stopped with 0.04 N HCl then absorbance was assessed using a plate reader at wavelength 570 nm.
siRNAs and plasmids transfections
Transfections and our previously validated knockdown-add back approach were performed as previously described (Aditi et al., 2015; Folkmann et al., 2013) using AllStars Negative control siRNA (Qiagen, Valencia, CA), GLE1 siRNA (Qiagen), NXF1 siRNA (Dharmacon, Lafayette, CO) and G3BP Stealth RNAi siRNA (Thermo Fisher Scientific). Following manufacturer instructions, siRNAs were reverse transfected into cells using HiPerFect (Qiagen). Transfections of siRNA-resistant plasmids were performed using Fugene 6 (Promega) according to the manufacturer recommended protocol.
Immunofluorescence
Cells treated as indicated were processed for indirect immunofluorescence as previously described (Aditi et al., 2015). The following primary antibodies were used: anti-G3BP 1:300 (BD Transduction, San Jose, CA), anti-Gle1 1:300 (ASW48), and anti-NXF1 1:300 (Abcam, Cambridge, MA), incubated overnight at 4°C, and detected with Alexa Fluor-conjugated secondary antibodies (Thermo Fisher Scientific). Slides were mounted with ProLong Gold Antifade mountant (ThermoFisher Scientific), and cells were imaged using a 63X/1.4 NA oil-immersion objective on a Leica TCS SP5 confocal microscope (Leica Microsystems. Buffalo Grove, IL). For SG characterization, post-image processing to quantify the size of SGs per cell was performed for 10 fields of view per experimental condition using the plug-in 3D object counter for ImageJ (National Institutes of Health, Bethesda, MD) with the measurement parameters set to “surface” and minimum size filter set to 3. GraphPad Prism 7 was used to generate a box plot of data as median ± IQR, and to calculate the unpaired, two-tailed Students t test of statistical significance.
In situ hybridization
In situ hybridization of Cy3-conjugated oligo-dT was performed as previously described (Aditi et al., 2015). Cells were imaged using a 63X/1.4 NA oil-immersion objective on a Leica TCS SP5 confocal microscope (Leica Microsystems). After imaging, nuclear:cytoplasmic ratio was determined by mean intensity of the fluorescent signal in the respective compartments in ImageJ (National Institutes of Health, Bethesda, MD).
Generation of Conditioned Media
Conditioned Media (CM) was prepared as in Grabocka & Bar-Sagi (2016). Briefly, the indicated cell populations were seeded to 70% confluency, washed three times with serum-free DMEM, and cultured in serum-free media for 20 hr. CM was collected, centrifuged to remove cell debris and diluted 1:1 with fresh serum-free media prior to use.
Cytokine array
The Proteome profiler array (ARY022B; R&D systems, Minneapolis MN) was performed to evaluate the CM cytokine composition, according to manufacturer’s instructions. The mean intensity of each spot was determined using Quick Spots image analysis software (Western Vision Software, Salt Lake City, UT), and normalized to internal reference control spots. Data was analyzed with GraphPad Prism 7 using two-way ANOVA with multiple comparisons.
Supplementary Material
Acknowledgements
We thank members of the Wente laboratory for critical discussions regarding these projects and reading of the manuscript. This work was supported by a grant from the National Institutes of Health to S.R.W. (R37 GM051219).
Footnotes
Declarations of interest: none
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Adams RL, Mason AC, Glass L, Aditi, Wente SR, 2017. Nup42 and IP6 coordinate Gle1 stimulation of Dbp5/DDX19B for mRNA export in yeast and human cells.Traffic 18, 776–790. 10.1111/tra.12526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aditi, Folkmann AW, Wente SR, 2015. Cytoplasmic hGle1A regulates stress granules by modulation of translation. Mol. Biol. Cell 26, 1476–1490. 10.1091/mbc.E14-11-1523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aditi, Glass L, Dawson TR, Wente SR.,2016. An amyotrophic lateral sclerosis-linked mutation in GLE1 alters the cellular pool of human Gle1 functional isoforms. Adv. Biol. Regul. 62, 25–36. 10.1016/j.jbior.2015.11.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adjibade P, St Sauveur VGG, Quevillon Huberdeau M, Fournier MJJ, Savard A, Coudert L, Khandjian EW, Mazroui R, 2015. Sorafenib, a multikinase inhibitor, induces formation of stress granules in hepatocarcinoma cells. Oncotarget 6, 4392743943 10.18632/oncotarget.5980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alcázar-Román AR, Tran EJ, Guo S, Wente SR, 2006. Inositol hexakisphosphate and Gle1 activate the DEAD-box protein Dbp5 for nuclear mRNA export. Nat. Cell Biol. 8, 711–6. 10.1038/ncb1427 [DOI] [PubMed] [Google Scholar]
- Amandine M, Jamshid T, Jihun L, Maura C, Anderson PK, Hong Joo K, Tanja M, Taylor JP, 2015. Phase separation by low complexity domains promotes stress granule assembly and drives pathological fibrillization. Cell 163, 123–133. 10.1016/j.cell.2015.09.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson P, Kedersha N, Ivanov P, 2015. Stress granules, P-bodies and cancer. Biochim. Biophys. Acta. 1849, 861–870. 10.1016/j.bbagrm.2014.11.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ando H, Kawaai K, Bonneau B, Mikoshiba K, 2018. Remodeling of Ca 2+ signaling in cancer: Regulation of inositol 1,4,5-trisphosphate receptors through oncogenes and tumor suppressors. Adv. Biol. Regul. 68, 64–76. https://doi.org/10.1016Zj.jbior.2017.12.001 [DOI] [PubMed] [Google Scholar]
- Arimoto K, Fukuda H, Imajoh-Ohmi S, Saito H, Takekawa M, 2008. Formation of stress granules inhibits apoptosis by suppressing stress-responsive MAPK pathways. Nat. Cell Biol. 10, 1324–1332. 10.1038/ncb1791 [DOI] [PubMed] [Google Scholar]
- Aulas A, Fay MM, Lyons SM, Achorn CA, Kedersha N, Anderson P, Ivanov P, 2017Stress-specific differences in assembly and composition of stress granules and related foci. J. Cell Sci. 130, 927–937. 10.1242/jcs.199240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolger TA, Folkmann AW, Tran EJ, Wente SR, 2008. The mRNA Export Factor Gle1 and Inositol Hexakisphosphate Regulate Distinct Stages of Translation. Cell 134, 624–633. 10.1016/jxell.2008.06.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolger TA, Wente SR, 2011. Gle1 is a multifunctional DEAD-box protein regulator that modulates Ded1 in translation initiation. J. Biol. Chem. 286, 39750–9. 10.1074/jbc.M111.299321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang Y-WW, Huang Y-SS, 2014. Arsenite-activated JNK signaling enhances CPEB4-Vinexin interaction to facilitate stress granule assembly and cell survival. PloS One 9. 10.1371/journal.pone.0107961 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen W, Dong J, Haiech J, Kilhoffer M-CC, Zeniou M, 2016. Cancer stem cell quiescence and plasticity as major challenges in cancer therapy. Stem Cells Int, 1740936. 10.1155/2016/1740936 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Divella R, Daniele A, Abbate I, Savino E, Casamassima P, Sciortino G, Simone G, Gadaleta-Caldarola G, Fazio V, Gadaleta CD, Sabbá C, Mazzocca A, 2015. Circulating Levels of PAI-1 and SERPINE1 4G/4G Polymorphism are predictive of poor prognosis in HCC patients undergoing TACE. Transl. Oncol. 8, 273–278. https://doi.org/10.1016Zj.tranon.2015.05.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Epling LB, Grace CR, Lowe BR, Partridge JF, Enemark EJ, 2015. Cancer-associated mutants of RNA helicase DDX3X are defective in RNA-stimulated ATP hydrolysis. J. Mol. Biol. 427, 1779–1796. 10.1016/jjmb.2015.02.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Folkmann AW, Collier SE, Zhan X, Aditi, Ohi MD., Wente SR, 2013. Gle1 functions during mRNA export in an oligomeric complex that is altered in human disease. Cell 155, 582–593. 10.1016/j.cell.2013.09.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fonseca P, Vardaki I, Occhionero A, Panaretakis T, 2016. Metabolic and signaling functions of cancer cell-derived extracellular vesicles. Int. Rev. Cell Mol. Biol. 326, 175–199. 10.1016/bs.ircmb.2016.04.004 [DOI] [PubMed] [Google Scholar]
- Fournier M-J, Gareau C, Mazroui R, 2010. The chemotherapeutic agent bortezomib induces the formation of stress granules. Cancer Cell Int. 10, 12 10.1186/1475-2867-10-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujimura K, Katahira J, Kano F, Yoneda Y, Murata M, 2009. Microscopic dissection of the process of stress granule assembly. Biochim. Biophys. Acta. 1793, 1728–37. 10.1016/j.bbamcr.2009.08.010 [DOI] [PubMed] [Google Scholar]
- Gareau C, Fournier M-JJ, Filion C, Coudert L, Martel D, Labelle Y, Mazroui R, 2011. p21(WAF1/CIP1) upregulation through the stress granule-associated protein CUGBP1 confers resistance to bortezomib-mediated apoptosis. PloS One 6. 10.1371/journal.pone.0020254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grabocka E, Bar-Sagi D, 2016. Mutant KRAS enhances tumor cell fitness by upregulating 0stress granules. Cell 167, 1803 10.1016/j.cell.2016.11.035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grüter P, Tabernero C, von Kobbe C, Schmitt C, Saavedra C, Bachi A, Wilm M, Felber BK, Izaurralde E, 1998. TAP, the human homolog of Mex67p, mediates CTE-dependent RNA export from the nucleus. Mol. Cell 1, 649–659. 10.1016/S1097-2765(00)80065-9 [DOI] [PubMed] [Google Scholar]
- Gupta N, Badeaux M, Liu Y, Naxerova K, Sgroi D, Munn LL, Jain RK, Garkavtsev I, 2017. Stress granule-associated protein G3BP2 regulates breast tumor initiation. Proc. Natl. Acad. Sci. U. S. A. 114, 1033–1038. 10.1073/pnas.1525387114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatch AJ, Odom AR, York JD, 2017. Inositol phosphate multikinase dependent transcriptional control. Adv. Biol. Regul. 64, 9–19. https://doi.org/10.1016Zj.jbior.2017.03.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hilliker A, 2012. Analysis of RNA helicases in P-bodies and stress granules. Methods Enzym 511, 323–46. 10.1016/B978-0-12-396546-2.00015-2 [DOI] [PubMed] [Google Scholar]
- Jain S, Wheeler JR, Walters RW, Agrawal A, Barsic A, Parker R, 2016. ATPase-modulated stress granules contain a diverse proteome and substructure. Cell 164, 487–498. 10.1016/j.cell.2015.12.038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jarmoskaite I, Russell R, 2014. RNA helicase proteins as chaperones and remodelers. Annu. Rev. Biochem. 83, 697–725. 10.1146/annurev-biochem-060713-035546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jura J, Skalniak L, Koj A, 2012. Monocyte chemotactic protein-1-induced protein-1 (MCPIP1) is a novel multifunctional modulator of inflammatory reactions. Biochim. Biophys. Acta. 1823, 1905–1913. 10.1016/j.bbamcr.2012.06.029 [DOI] [PubMed] [Google Scholar]
- Kaehler C, Isensee J, Hucho T, Lehrach H, Krobitsch S, 2014. 5-Fluorouracil affects assembly of stress granules based on RNA incorporation. Nucleic Acids Res. 42, 6436–6447. 10.1093/nar/gku264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kedersha N, Ivanov P, Anderson P, 2013. Stress granules and cell signaling: more than just a passing phase? Trends Biochem. Sci. 38, 494–506. 10.1016/j.tibs.2013.07.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SH, Dong WK, Weiler IJ, Greenough WT, 2006. Fragile X mental retardation protein shifts between polyribosomes and stress granules after neuronal injury by arsenite stress or in vivo hippocampal electrode insertion. J. Neurosci. Off. J. Soc. Neurosci. 26, 2413–2418. 10.1523/JNEUROSCI.3680-05.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim WJ, Back SH, Kim V, Ryu I, Jang SK, 2005. Sequestration of TRAF2 into stress granules interrupts tumor necrosis factor signaling under stress conditions. Mol. Cell. Biol. 25, 2450–2462. 10.1128/MCB.25.6.2450-2462.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolobova E, Efimov A, Kaverina I, Rishi AK, Schrader JW, Ham AJ, Larocca MC, Goldenring JR, 2009. Microtubule-dependent association of AKAP350A and CCAR1 with RNA stress granules. Exp. Cell Res. 315, 542–55. 10.1016/j.yexcr.2008.11.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krisenko MO, Higgins RLL, Ghosh S, Zhou Q, Trybula JS, Wang W-HH, Geahlen RL, 2015. Syk is recruited to stress granules and promotes their clearance through autophagy. J. Biol. Chem. 290, 27803–27815. 10.1074/jbc.M115.642900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J-Y, Kim Y, Park J, Kim S, 2012. Inositol polyphosphate multikinase signaling in the regulation of metabolism. Ann. N. Y. Acad. Sci. 1271, 68–74. 10.1111/j.1749-6632.2012.06725.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee Y-JJ, Wei H-MM, Chen L-YY, Li C, 2014. Localization of SERBP1 in stress granules and nucleoli. FEBS J. 281, 352–364. 10.1111/febs.12606 [DOI] [PubMed] [Google Scholar]
- Li YR, King OD, Shorter J, Gitler AD, 2013. Stress granules as crucibles of ALS pathogenesis. J. Cell Biol. 201, 361–372. 10.1083/jcb.201302044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahboubi H, Stochaj U, 2017. Cytoplasmic stress granules: Dynamic modulators of cell signaling and disease. Biochim. Biophys. Acta. 1863, 884–895. https://doi.org/10.1016Zj.bbadis.2016.12.022 [DOI] [PubMed] [Google Scholar]
- Marchi S, Rimessi A, Giorgi C, Baldini C, Ferroni L, Rizzuto R, Pinton P, 2008. Akt kinase reducing endoplasmic reticulum Ca2+ release protects cells from Ca2+-dependent apoptotic stimuli. Biochem. Biophys. Res. Commun. 375, 501–505. 10.1016/j.bbrc.2008.07.153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markmiller S, Soltanieh S, Server KL, Mak R, Jin W, Fang MY, Luo E-CC, Krach F, Yang D, Sen A, Fulzele A, Wozniak JM, Gonzalez DJ, Kankel MW, Gao F-BB, Bennett EJ, Lecuyer E, Yeo GW, 2018. Context-dependent and disease-specific diversity in protein interactions within stress granules. Cell 172, 590–376537088. 10.1016/j.cell.2017.12.032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuki H, Takahashi M, Higuchi M, Makokha GN, Oie M, Fujii M, 2013. Both G3BP1 and G3BP2 contribute to stress granule formation. Genes Cells Devoted Mol. Cell. Mech. 18, 135–146. 10.1111/gtc.12023 [DOI] [PubMed] [Google Scholar]
- McMahon BJ, Kwaan HC, 2015. Components of the plasminogen-plasmin system as biologic markers for cancer. Adv. Exp. Med. Biol. 867, 145–156. 10.1007/978-94-017-7215-0_10 [DOI] [PubMed] [Google Scholar]
- Michell RH, 2008. Inositol derivatives: evolution and functions. Nat. Rev. Mol. Cell Biol. 9, 151–161. 10.1038/nrm2334 [DOI] [PubMed] [Google Scholar]
- Montpetit B, Thomsen ND, Helmke KJ, Seeliger MA, Berger JM, Weis K, 2011. A conserved mechanism of DEAD-box ATPase activation by nucleoporins and IP6 in mRNA export. Nature 472, 238–242. 10.1038/nature09862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Omer A, Patel D, Lian XJ, Sadek J, Di Marco S, Pause A, Gorospe M, Gallouzi IE, 2018. Stress granules counteract senescence by sequestration of PAI-1. EMBO Rep. 19 10.15252/embr.201744722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panas MD, Ivanov P, Anderson P, 2016. Mechanistic insights into mammalian stress granule dynamics. J. Cell Biol. 215, 313–323. 10.1083/jcb.201609081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perera NM, Michell RH, Dove SK, 2004. Hypo-osmotic stress activates Plc1p-dependent phosphatidylinositol 4,5-bisphosphate hydrolysis and inositol Hexakisphosphate accumulation in yeast. J. Biol. Chem. 279, 5216–5226. 10.1074/jbc.M305068200 [DOI] [PubMed] [Google Scholar]
- Pugh TJ, Weeraratne SD, Archer TC, Pomeranz Krummel DA, Auclair D, Bochicchio J, Carneiro MO, Carter SL, Cibulskis K, Erlich RL, Greulich H, Lawrence MS, Lennon NJ, McKenna A, Meldrim J, Ramos AH, Ross MG, RussC. Shefler, E., Sivachenko A, Sogoloff B, Stojanov P, Tamayo P, Mesirov JP, Amani V, Teider N., Sengupta, S. Francois, J.P., Northcott P.A., Taylor, Yu F, Crabtree GR, Kautzman AG, Gabriel SB, Getz G, Jager N, Jones DT, Lichter P, Pfister SM, Roberts TM, Meyerson M, Pomeroy SL, Cho Y-JJ, 2012. Medulloblastoma exome sequencing uncovers subtype-specific somatic mutations. Nature 488, 106–110. 10.1038/nature11329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi D, Huang S, Miao R, She Z-G, Quinn T, Chang Y, Liu J, Fan D, Chen EY, Fu M, 2011. MCP-induced protein 1 suppresses stress granule formation and determines apoptosis under stress. J. Biol. Chem. 10.1074/jbc.M111.276006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quail DF, Joyce JA, 2013. Microenvironmental regulation of tumor progression and metastasis. Nat. Med. 19, 1423–1437. 10.1038/nm.3394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shih JW, Wang WT, Tsai TY, Kuo CY, Li HK, Wu Lee YH, 2012. Critical roles of RNA helicase DDX3 and its interactions with eIF4E/PABP1 in stress granule assembly and stress response. Biochem. J. 441, 119–29. 10.1042/BJ20110739 [DOI] [PubMed] [Google Scholar]
- Singh G, Pratt G, Yeo GW, Moore MJ, 2015. The clothes make the mRNA: Past and present trends in mRNP fashion. Annu. Rev. Biochem. 84, 325–54. 10.1146/annurev-biochem-080111-092106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Somasekharan SP, El-Naggar A, Leprivier G, Cheng H, Hajee S, Grunewald TG, Zhang F, Ng T, Delattre O, Evdokimova V, Wang Y, Gleave M, Sorensen PH, 2015. YB-1 regulates stress granule formation and tumor progression by translationally activating G3BP1. J. Cell Biol. 208, 913–929. 10.1083/jcb.201411047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szado T, Vanderheyden V, Parys JB, Smedt HD, Rietdorf K, Kotelevets L, Chastre E, Khan F, Landegren U, Söderberg O, Bootman MD, Roderick HL, 2008. Phosphorylation of inositol 1,4,5-trisphosphate receptors by protein kinase B/Akt inhibits Ca2+ release and apoptosis. Proc. Natl. Acad. Sci. 105, 2427–2432. 10.1073/pnas.0711324105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szaflarski W, Fay MM, Kedersha N, Zabel M, Anderson P, Ivanov P, 2016. Vinca alkaloid drugs promote stress-induced translational repression and stress granule formation. Oncotarget 7, 30307–30322. 10.18632/oncotarget.8728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi M, Higuchi M, Matsuki H, Yoshita M, Ohsawa T, Oie M, Fujii M 2013. Stress granules inhibit apoptosis by reducing reactive oxygen species production. Mol. Cell. Biol. 33, 815–829. 10.1128/MCB.00763-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan C, Hu W, He Y, Zhang Y, Zhang G, Xu Y, Tang J, 2018. Cytokine-mediated therapeutic resistance in breast cancer. Cytokine 108, 151–159. https://doi.org/10.1016Zj.cyto.2018.03.020 [DOI] [PubMed] [Google Scholar]
- Tourrière H, Chebli K, Zekri L, Courselaud B, Blanchard JM, Bertrand E, Tazi J, 2003. The RasGAP-associated endoribonuclease G3BP assembles stress granules. J. Cell Biol. 160, 823–831. 10.1083/jcb.200212128 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Turakhiya A, Meyer SR, Marincola G, Cell B-S, 2018. ZFAND1 recruits p97 and the 26S proteasome to promote the clearance of arsenite-induced stress granules. Mol. Cell 70, 906–919. 10.1016/j.molcel.2018.04.021 [DOI] [PubMed] [Google Scholar]
- Valentin-Vega YA, Wang Y-DD, Parker M, Patmore DM, Kanagaraj A, Moore J, Rusch M, Finkelstein D, Ellison DW, Gilbertson RJ, Zhang J, Kim HJ, Taylor JP., 2016. Cancer-associated DDX3X mutations drive stress granule assembly and impair global translation. Sci. Rep. 6, 25996 10.1038/srep25996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weirich CS, Erzberger JP, Flick JS, Berger JM, Thorner J, Weis K, 2006. Activation of the DExD/H-box protein Dbp5 by the nuclear-pore protein Gle1 and its coactivator InsP6 is required for mRNA export. Nat. Cell Biol. 8, 668–676. 10.1038/ncb1424 [DOI] [PubMed] [Google Scholar]
- Wheeler JR, Matheny T, Jain S, Abrisch R, Parker R, 2016. Distinct stages in stress granule assembly and disassembly. eLife 5. 10.7554/eLife.18413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilczynska A, Aigueperse C, Kress M, Dautry F, Weil D, 2005. The translational regulator CPEB1 provides a link between dcp1 bodies and stress granules. J. Cell Sci. 118, 981–992. 10.1242/jcs.01692 [DOI] [PubMed] [Google Scholar]
- Yang Y-JJ, Wu L-SS, Shu B, Qian M-ZZ, 2013. MCPIP1 mediates MCP-1-induced vascular smooth muscle cell proliferation. Acta. Physiol. Sin. 65, 616–622. [PubMed] [Google Scholar]
- Yancey PH, 2005. Organic osmolytes as compatible, metabolic and counteracting cytoprotectants in high osmolarity and other stresses. J. Exp. Biol. 208, 2819–2830. 10.1242/jeb.01730 [DOI] [PubMed] [Google Scholar]
- York JD, Odom AR, Murphy R, Ives EB, Wente SR, 1999. A Phospholipase C-Dependent Inositol Polyphosphate Kinase Pathway Required for Efficient Messenger RNA Export. Science 285, 96–100. 10.1126/science.285.5424.96 [DOI] [PubMed] [Google Scholar]
- Zheng H-CC, 2017. The molecular mechanisms of chemoresistance in cancers. Oncotarget 8, 59950–59964. 10.18632/oncotarget.19048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou L, Azfer A, Niu J, Graham S, Choudhury M, Adamski FM, Younce C, Binkley PF, Kolattukudy PE, 2006. Monocyte chemoattractant protein-1 induces a novel transcription factor that causes cardiac myocyte apoptosis and ventricular dysfunction. Circ. Res. 98, 1177–85. 10.1161/01.RES.0000220106.64661.71 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.