

Abstract
The primary route for development of azole resistance in the fungal pathogen Candida glabrata is acquisition of a point mutation in the PDR1 gene. This locus encodes a transcription factor that upon mutation drives high level expression of a range of genes including the ATP-binding cassette transporter-encoding gene CDR1. Pdr1 activity is also elevated in cells that lack the mitochondrial genome (ρ0 cells), with associated high expression of CDR1 driving azole resistance. To gain insight into the mechanisms controlling activity of Pdr1, we expressed a tandem affinity purification (TAP)-tagged form of Pdr1 in both wild-type (ρ+) and ρ0 cells. Purified proteins were analyzed by multidimensional protein identification technology (MudPIT) mass spectrometry identifying a protein called Bre5 as a factor that co-purified with TAP-Pdr1. In Saccharomyces cerevisiae, Bre5 is part of a deubiquitinase complex formed by association with the ubiquitin-specific protease Ubp3. Genetic analyses in C. glabrata revealed that loss of BRE5, but not UBP3, led to an increase in expression of PDR1 and CDR1 at the transcriptional level. These studies support the view that Bre5 acts as a negative regulator of Pdr1 transcriptional activity and behaves as a C. glabrata-specific modulator of azole resistance.
Graphical Abstract
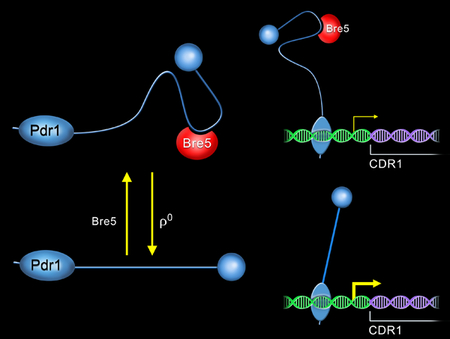
Bre5 is a known deubiquitinase subunit in Saccharomyces cerevisiae. We demonstrate that Bre5 is a negative regulator of Pdr1-dependent transcription in Candida glabrata, although this function is not conserved in S. cerevisiae. Induction of Pdr1 transcriptional activation is associated with decreased association of Bre5 with Pdr1 and we suggest that this represents a ubiquitin independent role for this protein.
Introduction
Loss of efficacy of antimicrobial agents is a pressing problem for modern medicine. Especially critical classes of chemotherapeutics are those effective against pathogenic fungi. There are only four antifungal drug classes currently available for use in the clinic consisting of the polyenes, pyrimidine analogues, azoles and echinocandins (reviewed in (Sanglard & Odds, 2002, Mazu et al., 2016)). Azole drugs represent the most commonly used antifungal compound and act by inhibiting biosynthesis of the fungal-specific sterol ergosterol (recently discussed in (Perlin et al., 2017)). Azole drugs also are administered orally, greatly facilitating their use compared to other antifungal agents.
While azole drugs have been very effective in their use as antifungals, this same success has also led to the development of azole tolerant organisms. An especially problematic fungus is the yeast Candida glabrata. This organism, which once constituted a small proportion of cases of candidiasis, now has risen to >20% of cases (see (Wiederhold, 2017) for a recent review). Unlike the primary fungal pathogen Candida albicans, C. glabrata is not well-controlled by azole drugs and resistant organisms are often found. Infections associated with azole resistant organisms have a poor prognosis and their frequency has led to echinocandins supplanting azole drugs as the primary treatment for C. glabrata-associated candidiasis (Bassetti et al., 2016). The basic mechanisms of resistance to azole and echinocandin drugs are very different but in troubling recent findings, echinocandin resistant C. glabrata isolates were found to frequently acquire azole resistance (reviewed in (Healey et al., 2016)). This is a true multidrug resistant pathogenic yeast for which the two most important classes of antifungal drugs are no longer effective in treatment. Most echinocandin resistance alleles involve mutations in the β-glucan synthase enzyme encoded by FKS1 and FKS2 and represent a change in the direct drug target protein (Perlin, 2007). Azole resistance most typically involves the mutational activation of a transcription factor called Pdr1 (Vermitsky & Edlind, 2004, Tsai et al., 2006) rather than mutation in the drug target gene called ERG11 (see for example (Garnaud et al., 2015)).
Pdr1 is a positively acting transcription factor that controls the expression of a variety of different target genes (reviewed in (Shahi & Moye-Rowley, 2009, Whaley & Rogers, 2016)). The major contributor to Pdr1-regulated azole resistance is the ATP-binding cassette (ABC) transporter-encoding gene called CDR1 (Sanglard et al., 1999). Azole resistant alleles of PDR1 exhibit high level overproduction of CDR1 mRNA along with many other Pdr1 transcriptional targets (Vermitsky et al., 2006, Tsai et al., 2010, Caudle et al., 2011). Exposure of wild-type C. glabrata cells to fluconazole induces CDR1 transcription in a Pdr1-dependent manner, indicating that Pdr1 provides inducible transcription upon drug challenge (Vermitsky et al., 2006). Experiments on the Saccharomyces cerevisiae homologues ScPdr1 and ScPdr3 provide evidence that the azole drugs can bind directly to this protein and trigger inducible transcription (Thakur et al., 2008). Both C. glabrata and S. cerevisiae strains that lack their mitochondrial genome (ρ0 cells) exhibit constitutive activation of both Pdr1 (Pdr3 in S. cerevisiae) function and CDR1 (PDR5 in S. cerevisiae) transcription (Hallstrom & Moye-Rowley, 2000, Bouchara et al., 2000, Sanglard et al., 2001).
These data suggest that C. glabrata maintains Pdr1 in a relatively low state of activity under basal conditions. Pdr1 activity can be stimulated acutely by addition of azole and other drugs or chronically by substitution mutations or loss of the mitochondrial genome. Essentially nothing is known of how Pdr1 activity is restrained in the absence of these stimulatory changes, although our recent work has identified the central domain of this transcription as essential for negative regulation (Khakhina et al., 2018). To determine if other proteins are associated with and potentially regulate Pdr1 activity, we used a chimeric tandem affinity purification (TAP)-tagged form of Pdr1 that we have previously characterized (Paul et al., 2014). This TAP-Pdr1 responds normally to loss of the mitochondrial genome and was purified from both ρ+ and ρ0 cells. Purified protein fractions were analyzed by multidimensional protein identification technology (MudPIT) that led to the identification of a Pdr1-associated protein called Bre5. Bre5 has been extensively studied in Saccharomyces cerevisiae as a component of a deubiquitinase enzyme complex with the ubiquitin-specific protease Ubp3 (Cohen et al., 2003). Our data indicate that Bre5 acts to negatively regulate Pdr1 function in a Ubp3 independent manner, consistent with a ubiquitin independent mode of action for Bre5. We find no role for the S. cerevisiae Bre5 in control of drug resistance in that organism, arguing that the Pdr1 modulatory role of Bre5 in C. glabrata represents a unique role for Bre5 in this pathogenic yeast.
Results
Identification of proteins associated with Pdr1
We have previously characterized a tandem affinity purification (TAP)-tagged form of Pdr1 that retains normal regulation and function (Paul et al., 2014). To identify proteins that might act as regulators of Pdr1 activity, we used two different strains expressing TAP-Pdr1 to generate protein extracts from which we could recover co-purifying factors. Whole cell protein extracts were prepared from either a ρ+ or ρ0 strain expressing TAP-Pdr1. Transcriptional activity of Pdr1 target genes was found to be strongly elevated in ρ0 cells compared to ρ+ (Paul et al., 2011). We purified TAP-Pdr1 from both these genetic backgrounds using standard IgG and calmodulin affinity chromatography as described (Puig et al., 2001). Aliquots of purified protein were then analyzed using multidimensional protein identification technology (MudPIT) (MacCoss et al., 2002). While several different proteins were found specifically to co-purify with TAP-Pdr1, we will only consider two in this manuscript: Bre5 and Ubp3.
Bre5 and Ubp3 have been extensively studied in Saccharomyces cerevisiae as essential components of a protein complex that controls the ubiquitination status of client proteins. Ubp3 is a member of a family of ubiquitin-specific proteases that cleave the ubiquitin from client proteins (reviewed in (Wilkinson, 1997, Amerik & Hochstrasser, 2004)) while Bre5 appears to serve as an adaptor (Li et al., 2005) protein for Ubp3 delivery to these clients. In S. cerevisiae, loss of Bre5 appears to phenocopy ubp3 null mutants in almost every case, consistent with the notion that these factors work as deubiquitinase complex (Auty et al., 2004, Bilsland et al., 2007, Kvint et al., 2008, Kraft et al., 2008, Kang et al., 2016, Nostramo et al., 2016). However, there is no available evidence that either ScBre5 or ScUbp3 influence the function of ScPdr1 or ScPdr3 in particular or pleiotropic drug resistance in general in S. cerevisiae. Since our biochemical purification had recovered both Bre5 and Ubp3 as co-purifying factors with Pdr1, we carried out genetic analyses of the genes encoding these proteins to examine the impact on Pdr1 activity.
Bre5 but not Ubp3 influences function of Pdr1 in C. glabrata
We constructed isogenic disruptions of BRE5 and UBP3 in an otherwise wild-type C. glabrata strain. Representative isolates were grown to mid-log phase and then 1000 cells of each strain were spotted along a plate containing a gradient of fluconazole (FLC) with a maximum concentration of 45 mg/ml. We also compared the behavior of S. cerevisiae strains lacking BRE5 and UBP3 to determine if similar effects were caused by loss of these genes in this well-characterized yeast using the same spot test assay (although the maximum FLC concentration was 20 μg/ml). These plates were incubated at 30°C and then photographed (Figure 1A).
Figure 1. Phenotypic analysis of BRE5 and UBP3 in C. glabrata and S. cerevisiae.

A. Isogenic C. glabrata (left side) or S. cerevisiae (right side) strains containing the indicated alleles of BRE5 and UBP3 were grown to mid-log phase and then 1000 cells/spot placed on rich medium containing gradients of fluconazole (FLC). The maximum concentration of FLC is noted at the top of each panel with increasing drug concentration indicated by the bar of increasing width. B. The same strains from above were streaked on rich medium (YPD) or this same medium containing 250 μg/ml brefeldin A (BFA). C. Complementation of drug resistant phenotype of a bre5Δ null. A low-copy-number plasmid (pBM16 (Zordan et al., 2013)) was used as an empty vector (EV) or a full-length BRE5-containing plasmid (pBM16-BRE5) and transformed into a bre5Δ strain. Transformants were grown to mid-log phase and spotted onto YPD medium containing 50 μg/ml nourseothricin (selectable marker in pBM16) and a gradient of fluconazole as indicated. Plates were incubated at 30° C and then photographed.
Loss of BRE5 but not UBP3 led to an increase in FLC resistance in C. glabrata. Loss of either BRE5 or UBP3 decreased FLC resistance in S. cerevisiae. We also tested a previously described phenotype, sensitivity to the Golgi anterograde transport inhibitor brefeldin A (Muren et al., 2001), for the effect of this drug on BRE5 and UBP3 mutants in C. glabrata and S. cerevisiae. BRE5 was originally identified in S. cerevisiae on the basis of null alleles exhibiting strong sensitivity to Brefeldin A (Muren et al., 2001). Isogenic wild-type, bre5Δ and ubp3Δ strains of C. glabrata and S. cerevisiae were streaked on rich medium lacking or containing Brefeldin A at the indicated concentration (Figure 1B). Only loss of BRE5 caused brefeldin A hypersensitivity in C. glabrata while lack of either BRE5 or UBP3 prevented growth at this brefeldin A concentration in S. cerevisiae as reported previously (Ossareh-Nazari et al., 2010). We also confirmed that loss of BRE5 is the event triggering the observed increase in fluconazole resistance as reintroduction of a low-copy-number plasmid containing a wild-type copy of BRE5 eliminated the increased azole resistance seen in the bre5Δ strain (Figure 1C). We interpret these data as supportive of a unique role for Bre5 in FLC resistance in C. glabrata while this protein is required for the common phenotype of brefeldin A tolerance in both yeasts.
To ensure that this change in FLC tolerance occurred via a pathway requiring Pdr1 and Cdr1, we constructed double mutant strains using either pdr1Δ or cdr1Δ strains and then varied the gene dose of BRE5. These double mutant strains were compared to either single mutant as well as the isogenic wild-type using the gradient plate assay as above (Figure 2).
Figure 2. Epistatic relationship of BRE5 with PDR1 and CDR1.

An isogenic set of wild-type or bre5Δ strains were further modified where noted by disruption of the PDR1 or CDR1 genes. Representative transformants were grown to mid-log phase and compared for fluconazole (FLC) resistance by gradient plates as described above. Note the large differences in the gradients used for testing cells that contain Pdr1 and Cdr1 versus those that lack either of these drug resistance loci. The bottom panel shows the relationship between Bre5, Pdr1 and Cdr1. The T-bar line indicates inhibition of Pdr1 by Bre5 while the arrow indicates the stimulation of CDR1 gene expression by Pdr1.
Loss of either Pdr1 or Cdr1 caused a dramatic increase in FLC sensitivity as reported by several labs (Sanglard et al., 1999, Vermitsky et al., 2006, Tsai et al., 2010, Caudle et al., 2011). The increase in FLC resistance that was seen in a bre5Δ strain was prevented by removal of either Pdr1 or Cdr1 from the genetic background. This result is most consistent with Bre5 acting to increase FLC resistance in a Pdr1/Cdr1-dependent manner. Taken together, these phenotypic data suggest that Bre5 acts as a negative regulator of drug resistance, possibly at the level of control of Pdr1 activity.
Bre5 interacts with Pdr1 in C. glabrata
To confirm that Bre5 interacts with Pdr1 as suggested by its co-purification with TAP-Pdr1, we constructed a strain containing an epitope-tagged allele of BRE5 by appending 13 Myc tags to the C-terminus of Bre5. This allele of BRE5 exhibited wild-type FLC resistance (data not shown), consistent with the tagged form of Bre5 retaining normal activity. Isogenic strains expressing either the tagged or native forms of Bre5 were grown to mid-log phase and whole cell protein lysates prepared. Aliquots of each lysate were retained as input controls with the remaining fraction subjected to immunoprecipitation using our previously described anti-Pdr1 antibody (Paul et al., 2014). After immunoprecipitation, input controls and immunopurified fractions were resolved on SDS-PAGE and subjected to western blotting with both anti-Pdr1 antisera as well as anti-Myc antibodies (Figure 3).
Figure 3. Bre5 associates with Pdr1 in vivo.

Isogenic strains containing a wild-type (Bre5) or C-terminally Myc-tagged form of Bre5 (Bre5-Myc) were grown to mid-log phase and non-denaturing total protein extracts prepared. Aliquots of these total protein extracts were reserved and run as controls (Input) while the remainder was subjected to immunoprecipitation using anti-Pdr1 antiserum. An equal fraction of each immunoprecipitate was electrophoresed through SDS-PAGE in parallel, along with input controls, and the gels were blotted with either anti-Pdr1 antiserum (Left panel) or an anti-Myc antibody (Right panel). Prior to western blot analysis, the transferred proteins were visualized by staining the membranes with Ponceau S (Top panels). Molecular mass standards are indicated at the edge of both western blots.
The Bre5-Myc protein was recovered from anti-Pdr1 immunoprecipitates prepared from the strain containing the BRE5-13X Myc allele but no signal was seen from wild-type cells. This co-immunoprecipitation of Pdr1 and Bre5 supported our finding of the association of TAP-Pdr1 and Bre5 by mass spectrometry discussed above.
Bre5 is a negative regulator of Pdr1 transactivation
The above data are consistent with the hypothesis that the presence of Bre5, but not Ubp3, is required to lower the ability of Pdr1 to activate gene expression. To directly examine this proposal, we measured expression of several Pdr1-regulated genes in response to loss of BRE5. Total RNA was prepared from isogenic wild-type and bre5Δ strains and then analyzed for mRNA from various Pdr1 target genes using reverse transcription-quantitative PCR (RT-qPCR) analyses as done before (Paul et al., 2011, Khakhina et al., 2018).
We analyzed the response of three different ABC transporter-encoding genes (CDR1, CDR2 and SNQ2) as well as the expression of the PDR1 gene itself to the loss of Bre5 (Figure 4A). CDR1 and CDR2 transcription was elevated by nearly 3- or 2-fold, respectively while levels of PDR1 mRNA increased by a more modest 40%. Another ABC transporter-encoding gene (SNQ2) did not significantly respond to loss of Bre5. These data support the view that some but not all Pdr1 target genes are transcriptionally repressed by the presence of Bre5.
Figure 4. Loss of Bre5 stimulates Pdr1-regulated gene expression.

A. Total RNA was isogenic wild-type and bre5Δ cells grown to mid-log phase. Levels of mRNA for genes of interest was determined using gene-specific primers and reverse transcription-quantitative PCR (RT-qPCR) analyses. All mRNA levels were normalized to expression of TEF1 mRNA which served as an unaffected control. Values listed inside each bar are the averages of at least 3 independent determinations of mRNA and represent a ratio of transcription in bre5Δ cells/transcription in the wild-type strain. B. Isogenic strains corresponding to the wild-type, bre5Δ and ubp3Δ were grown to early log phase and then either treated with fluconazole (+FLC) or allowed to continue to grow for 90 minutes with no treatment (-FLC). Total RNA was prepared after this time and analyzed for expression of either PDR1 or CDR1 by RT-qPCR. Levels of these transcripts were normalized to TEF1 as above. Values presented are a ratio of expression in presence of fluconazole relative to expression in the absence of drug.
We also compared the effect of loss of Bre5 and Ubp3 on the ability of Pdr1-regulated genes to respond to fluconazole challenge. PDR1 is transcriptionally induced by the presence of fluconazole and subsequently upregulates its target genes (Vermitsky et al., 2006, Thakur et al., 2008). Isogenic wild-type, bre5Δ or ubp3Δ cells were grown to mid-log phase, left untreated or exposed to fluconazole and total RNA prepared. Ratios of mRNA produced in the presence or the absence of fluconazole were then calculated after normalization to an internal control.
In the wild-type and ubp3Δ cells, PDR1 mRNA was 400% higher in the presence of fluconazole than in its absence (Figure 4B). However, bre5Δ cells only exhibited 200% increase upon fluconazole exposure. This same trend was seen for the Pdr1 target gene, the ABC transporter-encoding CDR1 locus. CDR1 transcription was 10- to 12-fold higher in the presence of fluconazole when assayed in wild-type and ubp3Δ cells but only 3-fold elevated when cells lacked Bre5. We suggest this reduced induction seen in bre5Δ cells was due to the increased basal expression found in cells lacking this negative regulator of Pdr1. Note in Figure 4A, PDR1 mRNA levels are nearly 50% higher upon loss of Bre5. To determine the basis of the increased Pdr1 activation of gene expression in bre5Δ cells, we examined levels of Pdr1 protein using the anti-Pdr1 antiserum as above.
Bre5 represses Pdr1 protein expression
The PDR1 gene has a bifunctional role in C. glabrata drug resistance as this locus both encodes the key regulator of this phenotype and is itself a Pdr1 target gene, owing to its positive autoregulatory circuit (Vermitsky et al., 2006, Tsai et al., 2006, Paul et al., 2011, Khakhina et al., 2018). To assess if the presence of Bre5 modified expression of the Pdr1 protein, we grew isogenic wild-type, bre5Δ, and ubp3Δ cells to mid-log phase and prepared total protein extracts from these strains. Steady-state levels of Pdr1 were then compared by western blotting using the anti-Pdr1 antiserum.
Loss of Bre5, but not Ubp3, led to a roughly 2-fold increase in expression of Pdr1 (Figure 5A). This is consistent with the presence of Bre5 acting to reduce Pdr1 transactivation by reducing activity of the positive autoregulation of PDR1 gene expression.
Figure 5. Pdr1 polypeptide responds to Bre5 and drug induction.

A. Isogenic PDR1 and pdr1Δ cells containing wild-type or null versions of BRE5 and UBP3 as indicated were grown to mid-log phase in rich medium. Total protein extracts were prepared and equal amounts of protein separated on SDS-PAGE. Levels of proteins were analyzed by western blot using either α-Pdr1 antiserum or α-tubulin (α-Tub1) antibody. The band labeled NS is a non-specific protein that cross reacts with the α-Pdr1 antiserum. The position of Pdr1 is indicated by the arrow at the right-hand side of the figure. Molecular mass standards are labeled on the right-hand side. Quantitation of the western blot is shown on the right-hand side. B. Isogenic wild-type and bre5Δ cells were grown to early log phase and then induced with the addition of fluconazole (FLC) as described above. After 90 minutes of growth in the presence (+) or absence (−) of fluconazole, total protein extracts were prepared and analyzed for levels of Pdr1 protein by western blot as above.
Previous work demonstrated that the presence of fluconazole leads to an induction in expression of genes under control of Pdr1 in C. glabrata (Vermitsky et al., 2006, Tsai et al., 2006, Culakova et al., 2015). To determine the influence of fluconazole on expression of the Pdr1 protein, isogenic wild-type and bre5Δ cells were grown to mid-log phase and then either left untreated or exposed to fluconazole. Levels of Pdr1 protein were determined by western blot analysis as above.
Exposure to fluconazole induced the expression of Pdr1 by approximately 2-fold (Figure 5B). As expected, levels of Pdr1 were elevated in bre5Δ cells while induction via fluconazole challenge was still detected. Bre5 is required to maintain the typically low level of Pdr1 in the absence of induction but its presence is not necessary for drug induction.
Synthesis of Pdr1 protein is induced in the absence of Bre5
The data above indicate that the steady-state level of Pdr1 protein is elevated upon removal of the BRE5 gene. To examine the step at which this elevation occurs, we carried out a pulse chase experiment using 35S-methionine. Isogenic wild-type and bre5Δ strains were grown to early log phase and then labeled with the addition of 35S-methionine for 10 minutes. After this step, a large excess of unlabeled methionine was added and time points taken. Pdr1 was recovered from each time point by immunoprecipitation and then analyzed by SDS-PAGE (Figure 6).
Figure 6. Synthesis of Pdr1 increases in the absence of Bre5.

A. The indicated strains were grown to early log phase and then labeled with the addition of 35S-methionine for 12 minutes. After this labeling period, a large excess of unlabeled methionine was added and time points removed immediately (0 time) or at 35 and 70 minutes. All time points were processed simultaneously to prepared a cell-free protein extract. Anti-Pdr1 immunoprecipitations were then carried out on each time point.
The synthesis rate of Pdr1 can be estimated from the 0 time point, taken immediately after onset of the chase. The level of incorporated 35S-methionine in Pdr1 is roughly 2-fold greater in bre5Δ cells than in the wild-type strain. We found no significant different in the turnover rate of Pdr1 in these two isogenic strains. These data further strengthen support for the view that loss of Bre5 induces synthesis of Pdr1.
Bre5:Pdr1 association responds to mitochondrial DNA status
These data suggest that Bre5 repression of Pdr1 activity is an important feature of control of the transcriptional output of this factor. Since our original identification of Bre5 association with Pdr1 utilized comparison of the proteins that co-purified with TAP-Pdr1 in ρ+ and ρ0 cells, we wondered if Bre5 binding to Pdr1 responded to loss of the mitochondrial genome. To assess this question, we generated ρ+ and ρ0 strains that contained the BRE5-myc allele. Cells were grown to mid-log phase and protein lysates prepared. Pdr1 was recovered using the anti-Pdr1 antiserum and these immunoprecipitates were resolved on SDS-PAGE and analyzed by western blotting. (Figure 7).
Figure 7. Mitochondrial status regulates Pdr1:Bre5 association.

Isogenic wild-type (BRE5) or a derivative carrying the BRE5-13X Myc fusion gene (Bre5-Myc) were generated that contained (ρ+) or lacked (ρ0) the mitochondrial genome. A ρ+ pdr1Δ strain (pdr1Δ) was also processed as a control for the location of the immunoprecipitated Pdr1. All strains were grown to mid-log phase in rich medium and whole cell protein extracts prepared. Aliquots (5%) of the whole cell extract were retained as input controls (Input for BRE5 and BRE5-Myc strains, In for the pdr1Δ strain). The remaining whole cell protein extracts were subjected to immunoprecipitation using the anti-Pdr1 antiserum. After washing, immunoprecipitates were electrophoresed along with the input controls through 8% SDS-PAGE and analyzed by western blotting using anti-Pdr1 (α-Pdr1) and anti-Myc (α-Myc) antisera. Locations of the immunoprecipitated proteins are indicated by the arrows at the left.
The amount of co-precipitated Bre5-Myc was found to be higher in samples of immunoprecipitated Pdr1 from ρ+ cells compared to ρ0 cells (compare lanes 17 and 18). Note that the expression level of Pdr1 is much higher in ρ0 cells (lane 8) than in ρ+ (lanes 7), yet less Bre5-Myc co-precipitates with the increased Pdr1. The steady-state level of Bre5-Myc was not affected by mitochondrial status (compare lanes 13 and 14). This decreased Pdr1:Bre5 association seen in ρ0 cells is consistent with Bre5 acting to negatively regulate Pdr1-dependent transcription.
Bre5 associates with Pdr1 target promoters
Since we could detect binding of Bre5 to Pdr1, we wanted to determine if the association of these proteins would be located on target promoters. We carried out chromatin immunoprecipitation (ChIP) using the Bre5-Myc fusion protein to evaluate which promoters were bound by this protein and if binding changed in response to the presence of Pdr1 or in strains with different mitochondrial statuses. We introduced the BRE5-13X Myc fusion gene into three different strain backgrounds: pdr1Δ ρ+, PDR1 ρ0 and PDR1 ρ+. Strains were grown to mid-log phase, and then processed for chromatin immunoprecipitation using the anti-Myc antibody as we have described before (Shahi et al., 2010). Immunoprecipitated DNA was recovered and analyzed by quantitative PCR using primers that detect the promoters of PDR1, CDR1, ERG11 or ACT1 (Figure 8)
Figure 8. Promoter association of Bre5 responds to mitochondrial genome status.

The strains indicated at the right all contain an integrated copy of the BRE5-13X Myc allele. These strains were grown to mid-log phase and then processed for chromatin immunoprecipitation using an anti-Myc antibody. Immunoprecipitated promoter fragments were analyzed by quantitative PCR using primer sets specific for the promoters of the genes listed at the bottom. Levels of immunoprecipitated DNA are plotted on the ordinate. Neither ACT1 or ERG11 (encodes the lanosterol α−14 demethylase target of azole drugs) is responsive to Pdr1 under these conditions.
Both PDR1 and CDR1 exhibited a similar pattern of Bre5-Myc association in the three different strains. Promoter-associated Bre5-Myc levels were highest in ρ+ cells on PDR1 and CDR1. Loss of the mitochondrial genome produced levels of Bre5-Myc association that were indistinguishable from those seen in the absence of Pdr1. Bre5-Myc promoter binding did not change in response to these different genetic backgrounds when association with either the ERG11 or ACT1 promoters were examined. The data indicate that Bre5 association is inversely correlated with the degree of expression of Pdr1 target genes. These data support the argument that increased association of Bre5 with Pdr1 is linked with decreased transcriptional activation mediated by this regulatory protein.
Discussion
Azole resistant isolates of C. glabrata are found with increasing frequency in clinics around the world (for examples see (Glockner & Cornely, 2015, Ostrosky-Zeichner et al., 2017)). Utilization of molecular techniques has led to the characterization of the majority of these isolates as containing single amino acid substitution mutations in their PDR1 locus (recently reviewed in (Whaley & Rogers, 2016)). Our working model is that these substitution mutations interfere with negative regulatory mechanisms that otherwise restrain Pdr1 in a low activity state. Direct support for this model has been difficult to obtain although earlier experiments in S. cerevisiae led to the discovery of an Hsp70 protein as a negative regulator of ScPdr1 homologue ScPdr3 (Shahi et al., 2007). In C. glabrata, mutational alterations in PDR1 (Ferrari et al., 2009), direct binding of azoles and other drugs (Thakur et al., 2008) or loss of the mitochondrial genome (Sanglard et al., 2001) all lead to an increase in function of Pdr1. Together, these data are consistent with multiple means to convert Pdr1 from a low activity state to one of significantly higher transactivation capability.
Here we identify Bre5, a negative regulator of Pdr1 that appears to be unique to C. glabrata. The lack of a role for Bre5 and/or Ubp3 in control of azole resistance in S. cerevisiae also illustrates the importance of investigating C. glabrata Pdr1 regulation directly in this pathogen. While there are clearly important similarities between these two related yeasts, each has its own circuitry providing regulation of Pdr1-controlled drug resistance. As we found for Bre5, others demonstrated that histone deacetylation catalyzed by the Hst1 deacetylase impacts azole and stress resistance in C. glabrata (Orta-Zavalza et al., 2013) but does not detectably influence these phenotypes in S. cerevisiae. The different lifestyles of these yeasts are likely at the root of why these very similar proteins exhibit such dramatically different physiological connections.
During our purification of Pdr1, we also recovered Ubp3 as a protein that co-purified with Pdr1 and Bre5. Ubp3 is well characterized as a deubiquitinating enzyme in S. cerevisiae and requires Bre5 for its normal activity (Cohen et al., 2003). Extensive work in S. cerevisiae has provided many examples of phenotypes that require both Bre5 and Ubp3 including trafficking through the secretory pathway (Cohen et al., 2003), DNA repair and resistance to drugs like brefeldin A (Dodgson et al., 2016) and rapamycin (Kraft et al., 2008). Roles in transcriptional regulation have also been found for the Ubp3:Bre5 complex as this deubiquitinase is required for control of ubiquitination of TFIID (Auty et al., 2004) and RNA polymerase II (Kvint et al., 2008). These data suggested the possibility that Pdr1 activity might be regulated by the action of the Ubp3:Bre5 deubiquitinase complex modulating ubiquitin modification of this transcription factor.
Our data do not support a role for ubiquitin modification of Pdr1 in the Bre5-dependent regulation of the function of this transcription factor. Unlike the cases described above in S. cerevisiae, loss of BRE5 or UBP3 did not result in the same phenotype for fluconazole resistance in C. glabrata. We also attempted to directly visualize levels of ubiquitin-modified Pdr1 using anti-ubiquitin antibodies but were unable to detect significant levels of this post-translational modification (data not shown). We recognize that, while the lack of a fluconazole resistance phenotype or detectable ubiquitin-modified Pdr1 are consistent with no role for ubiquitin in regulation of this transcription factor, we cannot entirely exclude this possibility. The ubiquitin modification may be labile and lost during isolation. Other ubiquitin-specific proteases may replace Ubp3 in control of Pdr1.
We hypothesize that negative regulation of Pdr1 by Bre5 represents a Ubp3 independent role for this protein. Experiments in Schizosaccharomyces pombe have revealed the presence of a Bre5 homologue designated Nxt3 that is also believed to interact with a Ubp3 homologue in this organism (Wang et al., 2012). Similar to the situation in C. glabrata, null mutants of nxt3 and ubp3 in S. pombe have different resistance phenotypes. The identification of Nxt3 in S. pombe was driven by the association of this protein with stress granules in this fission yeast (Wang et al., 2012). Roles for Bre5 in processes as varied as protein trafficking in the secretory pathway (Cohen et al., 2003), mitophagy (Muller et al., 2015), pseudohyphal growth (Kang et al., 2016), DNA damage (Bilsland et al., 2007) and ribosome function (Kraft et al., 2008) have been described in S. cerevisiae. Clearly, Bre5 and its homologues have a broad range of client proteins and impinge on many different cellular phenotypes. Its role in control of Pdr1 function represents a new and unique action for this protein.
It is interesting to note that ScPdr3 has been found to be induced by specific defects in the folding of a subclass of mitochondrially-targeted proteins (Weidberg & Amon, 2018). These authors provide data that ρ0 cells also have issues in the import of these same mitochondrial proteins including the phosphatidylserine decarboxylase Psd1. We previously found that overproduction of a form of ScPsd1 corresponding to only the amino-terminal targeting information was sufficient to induce PDR3 expression and Pdr3 protein levels (Gulshan et al., 2008) and that overproduction of CgPsd1 in C. glabrata was able to induce CDR1 transcription in this pathogenic yeast (Paul et al., 2011). In both S. cerevisiae and C. glabrata, Psd1 control of its cognate transcription factor must only represent a part of the full spectrum of signals that are interpreted by these transcriptional regulatory proteins as either loss of the mitochondrial genome or gain-of-function mutations lead to a much greater effect on downstream gene expression and phenotypes in both yeasts.
The link between Bre5 and mitophagy in S. cerevisiae suggests that these signals might be conserved with C. glabrata but ScUbp3 is also associated with ScBre5-dependent effects on mitophagy (Muller et al., 2015). The lack of any demonstrable for the control of Pdr1 by Bre5 in C. glabrata is most consistent with the signaling pathways being different in these two yeasts. Further analyses of the connection between mitochondria and Pdr1 in C. glabrata are required to establish the components of this signaling pathway.
In mammalian cells, the Bre5 homologue G3BP1 has been observed to regulate transcription factors by directly binding target proteins. G3BP1 can bind to Smad factors and inhibit their activation in a manner that appears to involve a reduction of activating phosphorylation (Zhang et al., 2015). G3BP1 also binds to the p53 protein and is believed to control the subcellular distribution of this important tumor suppressor (Kim et al., 2007). The precise action of Bre5 in C. glabrata remains to be ascertained but this factor is clearly a negative modulator of Pdr1 activity. We believe that Bre5 is only one of the negative signals that restrain Pdr1 transcriptional activation. This belief is based on the observation that gain-of-function mutant forms of Pdr1 exhibit much higher expression level of target genes like CDR1 (Vermitsky & Edlind, 2004, Tsai et al., 2006, Vermitsky et al., 2006) than do single bre5Δ strains. We are currently evaluating the effect of these hyperactive alleles of PDR1 on the association with Bre5.
Experimental procedures
Strains, media, and spot test assay
The C. glabrata strains used for this study are listed in Table 1. Yeast cells were grown on rich YPD (2% yeast extract, 1% peptone, 2% glucose) medium or on synthetic complete medium lacking appropriate auxotrophic components (Sherman et al., 1979). The ρ0 strains used in this study were generated by growing the corresponding strain on YPD plates containing 25 μg/ml ethidium bromide for 48hrs at 300C. The ρ0 state was confirmed by inability to grow in a nonfermentable carbon source, such as YPGE (2% yeast extract, 1% peptone, 3% glycerol, 3% ethanol), as well as by a lack of mitochondrial nucleoids, as revealed by DAPI (4,6-diamidino- 2-phenylindole) staining. Yeast strains were transformed by the lithium acetate method (Ito et al., 1983). Drug resistance was measured either by spot test assay on gradient drug plates by growing mid-log-phase cells at 37°C or by streaking cells on plates with the indicated drug concentration.
Table 1.
List of strains used in this study.
| Name | Parent | Genotype | Reference |
|---|---|---|---|
| KKY2001 | CBS138 | his3Δ::FRT leu2Δ::FRT trp1Δ::FRT | (Schwarzmuller et al., 2014) |
| PSY95 | pdr1Δ::Nat KKY2001 | TAP-PDR1::LEU2 | (Paul et al., 2014) |
| SPG71 | PSY95 | ρ0 TAP-PDR1::LEU2 | (Paul et al., 2014) |
| bre5Δ | BY4742 | bre5Δ::kanMX4 | Open Biosystems |
| ubp3Δ | BY4742 | bre5Δ::kanMX4 | Open Biosystems |
| SPG86 | KKY2001 | bre5Δ::ScHIS3 | This study |
| SPG98 | KKY2001 | ubp3Δ::ScHIS3 | This study |
| SPG102 | pdr1Δ::natMX KKY2001 | bre5Δ::ScHIS3 pdr1Δ::natMX | This study |
| SPG104 | cdr1Δ::natMX KKY2001 | bre5Δ::ScHIS3 cdr1Δ::natMX | This study |
| SPG94 | KKY2001 | BRE5-13xMyc- HIS3MX6 | This study |
| SPG118 | SPG94 | ρ0 BRE5-13xMyc-HIS3MX6 | This study |
Plasmids
For this study, all plasmid cloning was done using a Gibson Assembly cloning kit (NEB #E5510S) unless otherwise specified. Primer design and reaction conditions were performed according to manufacturer’s recommendations. To generate the BRE5 disruption cassette, 550 bp upstream and downstream of the BRE5 gene were PCR amplified from the KKY2001 strain and cloned by Gibson assembly at either end of a ScHIS3 marker cassette PCR amplified from pRS313 and inserted at the pUC19 BamHI site. This plasmid was named pSP71. The BRE5 disruption cassette was released from the pUC19 vector backbone by EcoR1 and SalI digestion of pSP71 for transformation into C. glabrata. Similarly, the UBP3 knockout cassette was generated by PCR amplifying 550 bp upstream and downstream of the UBP3 gene from the KKY2001 strain and cloned on either side of a ScHIS3 marker cassette PCR amplified from pRS313 and inserted at the pUC19 BamHI site. This plasmid was named pSP70. The ubp3::ScHIS3 disruption cassette was released from the vector backbone by Sac1 and XbaI digestion of pSP70 for transformation into C. glabrata. To generate the 13xMyc tag at the carboxy-terminus terminus of BRE5, the fragment containing a 13xMyc-ScADH1 terminator-HIS3MX6 was released from the pFA6a-13Myc-His3MX6 (Longtine et al., 1998) plasmid by SmaI and EcoRV digestion. This fragment was flanked upstream by 550 bp long PCR amplified DNA corresponding to the region encoding the C-terminus of BRE5 but lacking the stop codon, and flanked downstream by 550bp corresponding to DNA downstream of the BRE5 gene. The above 3 fragments were cloned by Gibson assembly into a pUC19 vector that was linearized at its BamH1 site. The final plasmid was named pSP85. The BRE5-13xMyc cassette was released from pSP85 by KpnI and SalI digestion for transformation into C. glabrata.
TAP tag purification
Wild-type ρ+ and ρ0 C. glabrata cells carrying an integrated copy of TAP-PDR1 were grown to mid-log phase in YPD medium. Cells were collected by centrifugation and then resuspended in lysis buffer. Whole cell protein extracts were prepared by grinding these cell suspensions in a Bead Beater. Lysates were cleared by centrifugation and the supernatant loaded onto an IgG-sepharose column. Binding was allowed to occur for 2 hours at 4° C, followed by extensive washing with column buffer. Bound TAP-Pdr1 fusion proteins were eluted by cleavage with TEV protease overnight at 37° C. Eluates from this IgG column were then applied to calmodulin-sepharose column in the presence of 10 mM calcium chloride. Binding proceeded at 4° C for 2 hours, followed by extensive washing in the same column buffer. Bound TAP-Pdr1 proteins were eluted with the addition of EGTA to 10 mM. Eluates were collected and precipitated with acetone. After washing, these precipitates were used for mass spectrometric analyses.
Multidimensional Protein Identification Technology
The precipitated proteins were resolubilized in a solution of 8M urea 100 mM Tris pH 8.5, reduced with TCEP (tris-(2-carboxyethyl)-phosphine) (10 mM), and alkylated with iodoacetamide (20 mM). Samples were diluted back to 2M urea, trypsin added and allowed to digest overnight at 37° C. Resulting peptides were analyzed via MudPIT (Multidimensional Protein Identification Technology) essentially as described in (MacCoss et al., 2002, Martinez et al., 2012). Briefly, digested peptides were loaded onto a biphasic 100 micron ID pre-column consisting of 4 cm of reversed phase (RP) material followed by 4 cm of strong cation exchange (RP) material. Once loaded this column was placed in line with a 100 micron x 20 cm RP analytical column packed into a nanospray emitter tip directly coupled to a linear ion trap mass spectrometer (LTQ). A subset of peptides were eluted from the SCX material onto the RP analytical via a pulse of volatile salt (ammonium acetate), those peptides separated by an RP gradient, and then ionized via nanoelectrospray directly into the mass spectrometry where both the intact masses (MS) and peptide fragmentation spectra (MS/MS) of the peptides were collected.
Peptide MS/MS spectral data were searched against a C. glabrata protein database using Sequest (Yates et al., 1995) and the resulting identifications collated and filtered using Scaffold (http://www.proteomesoftware.com).
Real-time PCR
Three OD600 units of mid-log phase cells (0.5–0.8 OD600/ml) were harvested per sample. Total RNA was isolated according to the instructions in the RNeasy mini kit (Qiagen) including the on-column DNase digestion step. Synthesis of cDNA was performed using iScript cDNA Synthesis Kit (Bio-Rad) with 500 ng of RNA as a template. Quantitative real-time PCR (qRT-PCR) was accomplished using the iTaq Universal SYBR Green Supermix (Bio-Rad) in MyIQ 2 Two Color Real-Time PCR Detection System (Bio-Rad). Melting curve was analyzed with each run to evaluate nonspecific amplification. The threshold cycle (Ct) values were determined for each gene and the average Ct value for each sample was calculated from the triplicate. The average Ct value of the PGK1 gene was used for normalization of variable cDNA levels, and induction factors were determined for each gene and condition. The comparative 2–DDCt method was used to calculate the fold change of the gene of interest between samples (Livak & Schmittgen, 2001). All experiments were repeated in three technical and two biological replicates.
Western blot analysis
Cells were grown to mid-log phase (0.5–0.8 OD600/ml) in YPD. Five OD600 units of culture were harvested per sample. Protein extracts were prepared as previously described (Paul et al., 2011) with the following modifications: Protein pellets were resuspended in 50 μl/OD600 unit of urea sample buffer (8 M urea, 1% 2-mercaptoethanol, 40 mM Tris-HCl pH 8.0, 5% SDS, bromophenol blue). The resuspended proteins were boiled at 900C for 5 min and an aliquot was resolved on an 8% SDS-PAGE. The proteins were transferred to a nitrocellulose membrane, blocked for 1 hour with 5% nonfat dry milk, and then probed with the anti-Pdr1 antibody (Paul et al., 2014) diluted 1:2,000. All membranes were probed for α-tubulin as loading control with 12G10 anti-α-tubulin monoclonal antibody (Developmental Studies Hybridoma Bank at the University of Iowa) for 1 hr at room temperature. The membrane was probed with secondary Li-Cor antibodies IRD dye 680RD goat anti-rabbit (# 926–68071) and IRD dye 800CW goat anti-mouse (# 926–32210) diluted 1:15,000. Western blot signals were detected using a Li-Cor Odyssey infrared imaging system, application software version 3.0 and quantified using Image Studio Lite software (Li-Cor).
Immunoprecipitation-Western blot analysis
Ten OD600 units of cells grown to mid-log phase in YPD medium were harvested, washed in PBS, and resuspended in 500μl of RIPA lysis buffer (25 mM Tris-HCl pH 7.5, 150 mM NaCl, 1% sodium deoxycholate, 1% Nonidet, 0.1% SDS) complemented with 1 mM PMSF and 1x Complete protease inhibitor. Cells in RIPA buffer were lysed in the presence of glass beads by vortexing at maximum speed for 10 min, and the insoluble material was pelleted by centrifugation at top speed. After the polyclonal Pdr1 antibody was added to the supernatant in 1:150 dilution, the cell lysate was incubated overnight at 40C by rotation. Immunoprecipitation was performed using 50 μl of Protein A/G PLUS- Agarose Immunoprecipitation Reagent (Santa Cruz Biotechnology). Agarose beads were first equilibrated for 2 hrs at 40C with the RIPA buffer. After adding the cell lysate with Pdr1 antibody to the beads, the mixture was incubated for 3 h at 40C by rotation. Immunoprecipitates were collected by centrifugation and the pellet was washed four times with RIPA buffer. Pellets were incubated in 25 μl of urea sample buffer (8 M urea, 40 mM Tris pH 8.0, 5% SDS, 5% 2-mercapthoethanol, 10% glycerol, bromophenol blue) for 30 min at 370C and 12 μl of the eluate were loaded onto the 8% SDS-PAGE gel and separated. The immunoblotting procedure was performed as described above. Anti Pdr1 Ab was used at 1:1000 while monoclonal anti-myc Ab (9E10, Santa Cruz) was used at 1:5000 dilution.
Pulse chase analysis
Mid-log phase cells (0.5–0.8 OD600/ml) grown in complete synthetic media (including methionine) at 300C were harvested, washed in PBS, and resuspended in CSM minus methionine for 15 min at 300C with shaking. Expre35S35S Protein Labeling Mix (Perkin- Elmer #NEG072014MC) was added at 15 μCi/OD600 unit cells into the media and the suspension was incubated for 12 min at 300C with shaking. Termination of the labeling reaction was achieved by adding excess of cold methionine and cysteine (5 μg/ml each). Cells corresponding to the time point 0 was removed from the medium immediately after addition of cold methionine and cysteine and placed on ice in 50 μM sodium azide. Aliquots from 4 time points (0, 35 and 70 and 105 min) were collected. Immunoprecipitation was carried out as described above, with the following modifications: Cells were harvested, washed with 10 mM sodium azide and resuspended in 200 μl RIPA lysis buffer complemented with 1% SDS, 10 mM EDTA, 1 mM PMSF and 1x Complete protease inhibitor. Immunoprecipitates were resuspended in 25 μl of urea sample buffer and then loaded onto a 8% SDS-PAGE gel and separated. The gel was then incubated in fixing solution (40% methanol, 5% acetic acid) on a shaker for 20 min at room temperature, followed by incubation in 1 M sodium salicylate under the same conditions. The gel was dried for 30 min at 800C and the signal detected by a Typhoon phosphoimager. Protein densitometry was quantified by ImageJ 1.5. Half-lives of Pdr1 in wild-type and bre5Δ cells were calculated based on the equation t1/2 = ln(2)/k where k is the rate constant. Protein half-lives were calculated from two biologically independent experiments.
Chromatin Immunoprecipitation
50 ml cell cultures were grown to an A600 of 0.6 to 0.8 in YPD medium at 300C. 1.4 ml of 37% formaldehyde (Sigma) were added and incubated in a 300C shaker (150 rpm) for 15 min to fix the cells. 2.5 ml of 2.5 M glycine was added to stop fixing and incubated for another 5 min at 300C shaker (150rpm). Cells were washed in 25 ml PBS and the cells were resuspended in 500 μl ChIP lysis buffer (50 mM HEPES-KOH, 140 mM NaCl, 1 mM EDTA, 1% Triton-X, 0.1% sodium deoxycholate, supplemented with 1 mM PMSF and 1x Complete protease inhibitor. Cells were lysed in the presence of 0.5 mm glass beads (BioSpec Products Inc.) four times (5 min each) with 2-min pauses in between in a Vortex Genie 2 instrument at a speed setting of 10 in 1.5-ml microcentrifuge tubes. Shearing of chromatin was done in an E220 Focused ultrasonicator (Covaris) under the following conditions: peak incident power (W), 175; duty factor, 10%; cycles per burst, 200; treatment time, 420 s; temperature, 7°C; sample volume, 130 l. An AFA Fiber Pre-Slit Snap-Cap (6 by 15 mm) microTUBE (Covaris) was used for the shearing. For immunoprecipitation, Pdr1-specific polyclonal antibody (described above) was used at 1:150 while the monoclonal anti-myc Ab (9E10, Santa Cruz) was used at 1:250 dilution.
Acknowledgements
This work was supported by National Institutes of Health GM49825. We thank Drs. Chris Macdonald, Bao Vu and Rob Piper for reagents and discussions.
References
- Amerik AY & Hochstrasser M, (2004) Mechanism and function of deubiquitinating enzymes. Biochim Biophys Acta 1695: 189–207. [DOI] [PubMed] [Google Scholar]
- Auty R, Steen H, Myers LC, Persinger J, Bartholomew B, Gygi SP & Buratowski S, (2004) Purification of active TFIID from Saccharomyces cerevisiae. Extensive promoter contacts and co-activator function. J Biol Chem 279: 49973–49981. [DOI] [PubMed] [Google Scholar]
- Bassetti M, Peghin M & Timsit JF, (2016) The current treatment landscape: candidiasis. J Antimicrob Chemother 71: ii13–ii22. [DOI] [PubMed] [Google Scholar]
- Bilsland E, Hult M, Bell SD, Sunnerhagen P & Downs JA, (2007) The Bre5/Ubp3 ubiquitin protease complex from budding yeast contributes to the cellular response to DNA damage. DNA Repair (Amst) 6: 1471–1484. [DOI] [PubMed] [Google Scholar]
- Bouchara JP, Zouhair R, Le Boudouil S, Renier G, Filmon R, Chabasse D, Hallet JN & Defontaine A, (2000) In-vivo selection of an azole-resistant petite mutant of Candida glabrata. J Med Microbiol 49: 977–984. [DOI] [PubMed] [Google Scholar]
- Caudle KE, Barker KS, Wiederhold NP, Xu L, Homayouni R & Rogers PD, (2011) Genomewide expression profile analysis of the Candida glabrata Pdr1 regulon. Eukaryot Cell 10: 373–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen M, Stutz F, Belgareh N, Haguenauer-Tsapis R & Dargemont C, (2003) Ubp3 requires a cofactor, Bre5, to specifically de-ubiquitinate the COPII protein, Sec23. Nat Cell Biol 5: 661–667. [DOI] [PubMed] [Google Scholar]
- Culakova H, Dzugasova V, Valencikova R, Gbelska Y & Subik J, (2015) Stress response and expression of fluconazole resistance associated genes in the pathogenic yeast Candida glabrata deleted in the CgPDR16 gene. Microbiol Res 174: 17–23. [DOI] [PubMed] [Google Scholar]
- Dodgson SE, Santaguida S, Kim S, Sheltzer J & Amon A, (2016) The pleiotropic deubiquitinase Ubp3 confers aneuploidy tolerance. Genes Dev 30: 2259–2271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrari S, Ischer F, Calabrese D, Posteraro B, Sanguinetti M, Fadda G, Rohde B, Bauser C, Bader O & Sanglard D, (2009) Gain of function mutations in CgPDR1 of Candida glabrata not only mediate antifungal resistance but also enhance virulence. PLoS Pathog 5: e1000268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garnaud C, Botterel F, Sertour N, Bougnoux ME, Dannaoui E, Larrat S, Hennequin C, Guinea J, Cornet M & Maubon D, (2015) Next-generation sequencing offers new insights into the resistance of Candida spp. to echinocandins and azoles. J Antimicrob Chemother 70: 2556–2565. [DOI] [PubMed] [Google Scholar]
- Glockner A & Cornely OA, (2015) Candida glabrata--unique features and challenges in the clinical management of invasive infections. Mycoses 58: 445–450. [DOI] [PubMed] [Google Scholar]
- Gulshan K, Schmidt J, Shahi P & Moye-Rowley WS, (2008) Evidence for the bifunctional nature of mitochondrial phosphatidylserine decarboxylase: role in Pdr3-dependent retrograde regulation of PDR5 expression. Mol Cell Biol 28: 5851–5864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hallstrom TC & Moye-Rowley WS, (2000) Multiple signals from dysfunctional mitochondria activate the pleiotropic drug resistance pathway in Saccharomyces cerevisae. J. Biol. Chem 275: 37347–37356. [DOI] [PubMed] [Google Scholar]
- Healey KR, Jimenez Ortigosa C, Shor E & Perlin DS, (2016) Genetic Drivers of Multidrug Resistance in Candida glabrata. Frontiers in microbiology 7: 1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito H, Fukuda Y, Murata K & Kimura A, (1983) Transformation of intact yeast cells treated with alkali cations. J. Bacteriol 153: 163–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang CM, Chang M, Park YS & Yun CW, (2016) Rck1 promotes pseudohyphal growth via the activation of Ubp3 phosphorylation in Saccharomyces cerevisiae. Biochem Biophys Res Commun 469: 333–339. [DOI] [PubMed] [Google Scholar]
- Khakhina S, Simonicova L & Moye-Rowley WS, (2018) Positive autoregulation and repression of transactivation are key regulatory features of the Candida glabrata Pdr1 transcription factor. Mol Microbiol 107: 747–764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim MM, Wiederschain D, Kennedy D, Hansen E & Yuan ZM, (2007) Modulation of p53 and MDM2 activity by novel interaction with Ras-GAP binding proteins (G3BP). Oncogene 26: 4209–4215. [DOI] [PubMed] [Google Scholar]
- Kraft C, Deplazes A, Sohrmann M & Peter M, (2008) Mature ribosomes are selectively degraded upon starvation by an autophagy pathway requiring the Ubp3p/Bre5p ubiquitin protease. Nat Cell Biol 10: 602–610. [DOI] [PubMed] [Google Scholar]
- Kvint K, Uhler JP, Taschner MJ, Sigurdsson S, Erdjument-Bromage H, Tempst P & Svejstrup JQ, (2008) Reversal of RNA polymerase II ubiquitylation by the ubiquitin protease Ubp3. Mol Cell 30: 498–506. [DOI] [PubMed] [Google Scholar]
- Li K, Zhao K, Ossareh-Nazari B, Da G, Dargemont C & Marmorstein R, (2005) Structural basis for interaction between the Ubp3 deubiquitinating enzyme and its Bre5 cofactor. J Biol Chem 280: 29176–29185. [DOI] [PubMed] [Google Scholar]
- Livak KJ & Schmittgen TD, (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 25: 402–408. [DOI] [PubMed] [Google Scholar]
- Longtine MS, McKenzie A, Demarini DJ, Shah NG, Wach A, Brachat A, Philippsen P & Pringle JR, (1998) Additional modules for versatile and economical PCR-based gene deletion and modification in Saccharomyces cerevisiae. Yeast 14: 953–961. [DOI] [PubMed] [Google Scholar]
- MacCoss MJ, McDonald WH, Saraf A, Sadygov R, Clark JM, Tasto JJ, Gould KL, Wolters D, Washburn M, Weiss A, Clark JI & Yates JR 3rd, (2002) Shotgun identification of protein modifications from protein complexes and lens tissue. Proc Natl Acad Sci U S A 99: 7900–7905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez MN, Emfinger CH, Overton M, Hill S, Ramaswamy TS, Cappel DA, Wu K, Fazio S, McDonald WH, Hachey DL, Tabb DL & Stafford JM, (2012) Obesity and altered glucose metabolism impact HDL composition in CETP transgenic mice: a role for ovarian hormones. J Lipid Res 53: 379–389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazu TK, Bricker BA, Flores-Rozas H & Ablordeppey SY, (2016) The Mechanistic Targets of Antifungal Agents: An Overview. Mini Rev Med Chem 16: 555–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller M, Kotter P, Behrendt C, Walter E, Scheckhuber CQ, Entian KD & Reichert AS, (2015) Synthetic quantitative array technology identifies the Ubp3-Bre5 deubiquitinase complex as a negative regulator of mitophagy. Cell reports 10: 1215–1225. [DOI] [PubMed] [Google Scholar]
- Muren E, Oyen M, Barmark G & Ronne H, (2001) Identification of yeast deletion strains that are hypersensitive to brefeldin A or monensin, two drugs that affect intracellular transport. Yeast 18: 163–172. [DOI] [PubMed] [Google Scholar]
- Nostramo R, Varia SN, Zhang B, Emerson MM & Herman PK, (2016) The Catalytic Activity of the Ubp3 Deubiquitinating Protease Is Required for Efficient Stress Granule Assembly in Saccharomyces cerevisiae. Mol Cell Biol 36: 173–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orta-Zavalza E, Guerrero-Serrano G, Gutierrez-Escobedo G, Canas-Villamar I, Juarez-Cepeda J, Castano I & De Las Penas A, (2013) Local silencing controls the oxidative stress response and the multidrug resistance in Candida glabrata. Mol Microbiol 88: 1135–1148. [DOI] [PubMed] [Google Scholar]
- Ossareh-Nazari B, Bonizec M, Cohen M, Dokudovskaya S, Delalande F, Schaeffer C, Van Dorsselaer A & Dargemont C, (2010) Cdc48 and Ufd3, new partners of the ubiquitin protease Ubp3, are required for ribophagy. EMBO Rep 11: 548–554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ostrosky-Zeichner L, Harrington R, Azie N, Yang H, Li N, Zhao J, Koo V & Wu EQ, (2017) A Risk Score for Fluconazole Failure among Patients with Candidemia. Antimicrob Agents Chemother 61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paul S, Bair TB & Moye-Rowley WS, (2014) Identification of Genomic Binding Sites for Candida glabrata Pdr1 Transcription Factor in Wild-Type and rho0 Cells. Antimicrob Agents Chemother 58: 6904–6912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paul S, Schmidt JA & Moye-Rowley WS, (2011) Regulation of the CgPdr1 transcription factor from the pathogen Candida glabrata. Eukaryot Cell 10: 187–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perlin DS, (2007) Resistance to echinocandin-class antifungal drugs. Drug Resist Updat 10: 121–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perlin DS, Rautemaa-Richardson R & Alastruey-Izquierdo A, (2017) The global problem of antifungal resistance: prevalence, mechanisms, and management. Lancet Infect Dis 17: e383–e392. [DOI] [PubMed] [Google Scholar]
- Puig O, Caspary F, Rigaut G, Rutz B, Bouveret E, Bragado-Nilsson E, Wilm M & Seraphin B, (2001) The tandem affinity purification (TAP) method: a general procedure of protein complex purification. Methods 24: 218–229. [DOI] [PubMed] [Google Scholar]
- Sanglard D, Ischer F & Bille J, (2001) Role of ATP-binding cassette transporter gene in high-frequency acquisition of resistance to azole antifungals in Candida glabrata. Antimicrob. Agents Chemother 45: 1174–1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanglard D, Ischer F, Calabrese D, Majcherczyk PA & Bille J, (1999) The ATP binding cassette transporter gene CgCDR1 from Candida glabrata is involved in the resistance of clinical isolates to azole antifungal agents. Antimicrob Agents Chemother 43: 2753–2765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanglard D & Odds FC, (2002) Resistance of Candida species to antifungal agents: molecular mechanisms and clinical consequences. Lancet Infect Dis 2: 73–85. [DOI] [PubMed] [Google Scholar]
- Schwarzmuller T, Ma B, Hiller E, Istel F, Tscherner M, Brunke S, Ames L, Firon A, Green B, Cabral V, Marcet-Houben M, Jacobsen ID, Quintin J, Seider K, Frohner I, Glaser W, Jungwirth H, Bachellier-Bassi S, Chauvel M, Zeidler U, Ferrandon D, Gabaldon T, Hube B, d’Enfert C, Rupp S, Cormack B, Haynes K & Kuchler K, (2014) Systematic phenotyping of a large-scale Candida glabrata deletion collection reveals novel antifungal tolerance genes. PLoS Pathog 10: e1004211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shahi P, Gulshan K & Moye-Rowley WS, (2007) Negative transcriptional regulation of multidrug resistance gene expression by an Hsp70 protein. J Biol Chem 282: 26822–26831. [DOI] [PubMed] [Google Scholar]
- Shahi P, Gulshan K, Naar AM & Moye-Rowley WS, (2010) Differential roles of transcriptional mediator subunits in regulation of multidrug resistance gene expression in Saccharomyces cerevisiae. Mol Biol Cell 21: 2469–2482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shahi P & Moye-Rowley WS, (2009) Coordinate control of lipid composition and drug transport activities is required for normal multidrug resistance in fungi. Biochim Biophys Acta 1794: 852–859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherman F, Fink G & Hicks J, (1979) Methods in Yeast Genetics. Cold Spring Harbor Laboratory: Cold Spring Harbor, New York. [Google Scholar]
- Thakur JK, Arthanari H, Yang F, Pan S-J, Fan X, Breger J, Frueh DP, Gulshan K, Li D, Mylonakis E, Struhl K, Moye-Rowley WS, Cormack BP, Wagner G & Naar AM, (2008) A nuclear receptor-like pathway regulating multidrug resistance in fungi. Nature 452: 604–609. [DOI] [PubMed] [Google Scholar]
- Tsai HF, Krol AA, Sarti KE & Bennett JE, (2006) Candida glabrata PDR1, a transcriptional regulator of a pleiotropic drug resistance network, mediates azole resistance in clinical isolates and petite mutants. Antimicrob Agents Chemother 50: 1384–1392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai HF, Sammons LR, Zhang X, Suffis SD, Su Q, Myers TG, Marr KA & Bennett JE, (2010) Microarray and molecular analyses of the azole resistance mechanism in Candida glabrata oropharyngeal isolates. Antimicrob Agents Chemother 54: 3308–3317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vermitsky JP, Earhart KD, Smith WL, Homayouni R, Edlind TD & Rogers PD, (2006) Pdr1 regulates multidrug resistance in Candida glabrata: gene disruption and genome-wide expression studies. Mol Microbiol 61: 704–722. [DOI] [PubMed] [Google Scholar]
- Vermitsky JP & Edlind TD, (2004) Azole resistance in Candida glabrata: coordinate upregulation of multidrug transporters and evidence for a Pdr1-like transcription factor. Antimicrob Agents Chemother 48: 3773–3781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang CY, Wen WL, Nilsson D, Sunnerhagen P, Chang TH & Wang SW, (2012) Analysis of stress granule assembly in Schizosaccharomyces pombe. RNA 18: 694–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weidberg H & Amon A, (2018) MitoCPR-A surveillance pathway that protects mitochondria in response to protein import stress. Science 360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whaley SG & Rogers PD, (2016) Azole Resistance in Candida glabrata. Current infectious disease reports 18: 41. [DOI] [PubMed] [Google Scholar]
- Wiederhold NP, (2017) Antifungal resistance: current trends and future strategies to combat. Infect Drug Resist 10: 249–259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkinson KD, (1997) Regulation of ubiquitin-dependent processes by deubiquitinating enzymes. FASEB J 11: 1245–1256. [DOI] [PubMed] [Google Scholar]
- Yates JR 3rd, Eng JK, McCormack AL & Schieltz D, (1995) Method to correlate tandem mass spectra of modified peptides to amino acid sequences in the protein database. Anal Chem 67: 1426–1436. [DOI] [PubMed] [Google Scholar]
- Zhang H, Ma Y, Zhang S, Liu H, He H, Li N, Gong Y, Zhao S, Jiang JD & Shao RG, (2015) Involvement of Ras GTPase-activating protein SH3 domain-binding protein 1 in the epithelial-to-mesenchymal transition-induced metastasis of breast cancer cells via the Smad signaling pathway. Oncotarget 6: 17039–17053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zordan RE, Ren Y, Pan SJ, Rotondo G, De Las Penas A, Iluore J & Cormack BP, (2013) Expression plasmids for use in Candida glabrata. G3 3: 1675–1686. [DOI] [PMC free article] [PubMed] [Google Scholar]