

Abstract
Pre-clinical studies have shown that anesthesia might accelerate the clinical progression of Alzheimer’s disease (AD) and can have an impact on tau pathology, a hallmark of AD. Although benzodiazepines have been suggested to increase the risk of incident dementia, their impact on tau pathology in vivo is unknown. We thus examined the impact of midazolam, a benzodiazepine that is often administered perioperatively as an anxiolytic on tau hyperphosphorylation in non-transgenic and in hTau mice, the latter a model of AD-like tau pathology.
The acute administration of midazolam in C57BL/6 mice was associated with downregulation of Protein Phosphatase-1 (PP1) and a significant and persistent increase in brain tau phosphorylation. In hTau mice, tau hyperphosphorylation was also observed; however, midazolam was neither associated with pro-aggregant changes nor spatial reference memory impairment.
In C57BL/6 mice, chronic midazolam administration immediately increased hippocampal tau phosphorylation, and this effect was more pronounced in older mice. Interestingly, in young C57BL/6 mice, chronic midazolam administration induced hippocampal tau hyperphosphorylation, which persisted for 1 week. In hTau mice, chronic midazolam administration increased hippocampal tau phosphorylation and, although this was not associated with pro-aggregant changes, this correlated with a decreased capacity of tau to bind to preassembled microtubules.
These findings suggest that midazolam can induce significant tau hyperphosphorylation in vivo, which persists well beyond recovery from its sedative effects. Moreover, it can disrupt one of tau’s critical functions. Hence, future studies should focus on the impact of more prolonged or repeated benzodiazepine exposure on tau pathology and cognitive decline.
Keywords: benzodiazepines, tau pathology, protein phosphatase-1, hTau mice, Barnes maze, spatial memory
1. Introduction
The two major neuropathological hallmarks of AD are extracellular senile plaques comprised of aggregates of β-amyloid protein and intraneuronal neurofibrillary tangles composed of aggregated, hyperphosphorylated tau protein. Tau is microtubule-associated protein, abundantly present in neuronal axons (Binder, et al., 1985), whose main function is to bind to microtubules (MTs) and promote microtubule assembly and stabilization (Drechsel, et al., 1992,Drubin, et al., 1988,Drubin, et al., 1985,Weingarten, et al., 1975). In AD and other tauopathies, tau can become aberrantly hyperphosphorylated, leading to its detachment from microtubules, subsequent aggregation, and ultimately the progression of neurofibrillary tangle pathology (Brion, et al., 1985,Grundke-Iqbal, et al., 1986a,Grundke-Iqbal, et al., 1986b,Kosik, et al., 1986,Wood, et al., 1986).
Previous studies have demonstrated that anesthetics can profoundly increase tau phosphorylation by inducing hypothermia-mediated inhibition of protein phosphatase 2A (PP2A) (Planel, et al., 2007). However, more recent studies have shown that certain inhalational anesthetics and intravenous sedative agents, including sevoflurane (Le Freche, et al., 2012), dexmedetomidine (Whittington, et al., 2015), and propofol (Whittington, et al., 2011), can directly increase tau phosphorylation even in the absence of hypothermia.
Benzodiazepines are commonly used anesthetic adjuncts that are administered clinically in order to produce sedative-hypnotic and anxiolytic effects. However, the clinical use of benzodiazepines has been associated with cognitive side effects (Acil, et al., 2004,Buffett-Jerrott and Stewart, 2002,Ghoneim, et al., 1984a,Ghoneim, et al., 1984b,Santana Santos, et al., 2005) and, more recently, an increased risk of incident dementia in the elderly (Billioti de Gage, et al., 2012,Billioti de Gage, et al., 2014). Despite this, there is a dearth of information related to the direct impact of benzodiazepines on tau hyperphosphorylation in vivo. Given that benzodiazepines mediate their effects through the allosteric potentiation of GABAergic neurotransmission and that other sedatives that activate this receptor subtype have been shown to increase tau phosphorylation (Le Freche, et al., 2012,Whittington, et al., 2011), we hypothesized that this class of sedative agents induces tau phosphorylation, accelerates tau aggregation, and disrupts the capacity of tau to bind to microtubules. Midazolam is a water-soluble benzodiazepine that is often administered perioperatively as an anxiolytic as well as on a prolonged basis for long-term sedation (Rhoney and Murry, 2003) in patients requiring mechanical ventilatory support. To test the aforementioned benzodiazepine-tau pathology hypothesis, we examined the impact of midazolam on tau phosphorylation, aggregation, and function in both non-transgenic and transgenic mice.
We observed that the acute administration of midazolam in non-transgenic mice, under normothermic conditions resulted in PP1 downregulation and produced tau hyperphosphorylation in the brain that persisted up to 24h in the cortex of non-transgenic mice. In hTau transgenic mice solely expressing non-mutant human tau, midazolam induced tau hyperphosphorylation in a slightly different pattern than that observed in non-transgenic mice; however, this hyperphosphorylation was neither associated with a change in soluble tau levels in the cortex nor a downstream change in spatial reference memory. Chronic administration of midazolam induced tau hyperphosphorylation in the brain of non-transgenic mice, and this effect increased as a function of age. Moreover, this midazolam-induced tau hyperphosphorylation was observed to persist for at least 1 week. Although chronic midazolam did not have pro-aggregant effects on tau in hTau mice, the capacity of tau to bind to microtubules was diminished.
2. Materials and Methods
2.1. Anesthetics and chemical reagents
Midazolam hydrochloride (Lipomed Inc., Cambridge, MA) was purchased from Lipomed Inc., (Cambridge, MA). All chemicals used in the preparation of mouse brain protein samples were purchased from Sigma-Aldrich (St. Louis, MO) with the exception of the protease inhibitors (Cocktail set III, Calbiochem, EMD Biosciences Inc., La Jolla, CA) and the bicinchoninate (BCA) protein assay reagents (Thermo Fisher Scientific Inc., Waltham, MA).
2.1. Antibodies
Tau phosphorylation levels were determined using antibodies directed at tau phosphorylated at the following epitopes: AT8 (pSer202/pThr205), CP13 (pSer202), PHF-1 (pSer396/pSer404), pSer262, AT180 (pThr231), AT270 (pThr181), 12E8 (pSer262/pSer356) and pSer199. Total tau levels were measured with the monoclonal antibodies Tau46, TG5, or Tau A0024. These particular tau antibodies were selected, as the phosphoepitopes they recognize have been associated with pre-tangle formation (CP13), paired helical filament and NFT formation (AT8, PHF-1) (Augustinack, et al., 2002,Goedert, 1993,Goedert, et al., 1994) as well as the microtubule binding domain (pSer262, 12E8) (Buee, et al., 2000). Hence, they were indeed suitable to provide a more comprehensive examination of the effects of midazolam on tau phosphorylation.
Furthermore, in order to dissect the mechanism underlying the tau phosphorylation changes following midazolam administration, the expression and activation of several major tau kinases were examined using the following antibodies purchased from Cell Signaling Technology, Inc. (Danvers, MA): CaMKII, , phospho-CaMKII (Thr286), GSK-3β, phospho-GSK-3β (Ser9), SAPK/JNK, phospho-SAPK-JNK (Thr183/Tyr185), p44/42 MAPK (ERK 1/2), phospho-p44/42 MAPK (Thr202/Tyr204), CDK5 and P35/P25. The changes in the expression of the tau phosphatases were determined using antibodies directed at the catalytic subunit of protein phosphatase 2A (PP2A-C, Sigma-Aldrich, St. Louis, Mo) as well as the demethylated PP2A-C subunit (Cell Signaling), protein phosphatase 1 (PP1), protein phosphatase 2B (PP2B), and protein phosphatase 5 (PP5). Each immunoblot was normalized for gel loading with β-actin or β-tubulin depending on the molecular weight of the protein of interest. A detailed summary of all of the antibodies used in these studies is shown in Table 1.
Table 1.
List of antibodies used
| Antibody | Phospho-Epitope | Type | Source | Manufacturer | Location | Catalog # | WB Dilution |
|---|---|---|---|---|---|---|---|
| Tau | |||||||
| Tau46 | Total Tau | Monoclonal | Mouse | Cell Signaling | Danvers, MA | 4019 | 1:2500 |
| Anti-Human Tau A0024 | Total Tau 243-441 | Polyclonal | Rabbit | Dako Cytomation | Carpinteria, CA | A002401-2 | 1:10000 |
| TG5 | Total Tau 220-240 | Monoclonal | Mouse | Peter Davies | Bronx, NY | N/A | 1:1000 |
| AT8 | pSer202/pThr205 | Monoclonal | Mouse | Thermo Scientific | Waltham, MA | MN1020 | 1:10000 |
| CP13 | pSer202 | Monoclonal | Mouse | Peter Davies | Bronx, NY | N/A | 1:1000 |
| PHF-1 | pSer396/pSer404 | Monoclonal | Mouse | Peter Davies | Bronx, NY | N/A | 1:1000 |
| AT180 | pThr231 | Monoclonal | Mouse | Thermo Scientific | Waltham, MA | MN1040 | 1:1000 |
| PS422 | pSer422 | Polyclonal | Rabbit | Life Technologies | Carlsbad, CA | 44-764G | 1:1000 |
| PS262 | pSer262 | Polyclonal | Rabbit | Life Technologies | Carlsbad, CA | 44-750G | 1:1000 |
| PS199 | pSer199 | Polyclonal | Rabbit | Life Technologies | Carlsbad, CA | 44-734G | 1:1000 |
| AT270 | pThr181 | Monoclonal | Mouse | Thermo Scientific | Waltham, MA | MN1050 | 1:1000 |
| 12 E8 | pSer262/pSer356 | Monoclonal | Mouse | Pharmaceuticals | Francisco, CA | N/A | 1:1000 |
| Kinases | |||||||
| GSK-3β | Monoclonal | Rabbit | Cell Signaling | Danvers, MA | 9315 | 1:1000 | |
| Phospho-GSK-3 β (Ser9) | Ser9 | Polyclonal | Rabbit | Cell Signaling | Danvers, MA | 9336 | 1:1000 |
| SAPK/JNK | Monoclonal | Rabbit | Cell Signaling | Danvers, MA | 9258 | 1:1000 | |
| Phospho-SAPK/JNK | Thr183/Tyr185 | Monoclonal | Rabbit | Cell Signaling | Danvers, MA | 4671 | 1:1000 |
| p44/42 MAPK | Monoclonal | Mouse | Cell Signaling | Danvers, MA | 9107 | 1:2000 | |
| Phospho-p44/42 MAPK | Thr202/Tyr204 | Monoclonal | Mouse | Cell Signaling | Danvers, MA | 9106 | 1:2000 |
| CaMKII | Polyclonal | Rabbit | Cell Signaling | Danvers, MA | 3362 | 1:1000 | |
| Phospho-CaMKII | Thr286 | Polyclonal | Rabbit | Cell Signaling | Danvers, MA | 3361 | 1:1000 |
| p35/25 | Monoclonal | Rabbit | Cell Signaling | Danvers, MA | 2680 | 1:1000 | |
| Phosphatases | |||||||
| PP2A-C (catalytic subunit) | Monoclonal | Mouse | Sigma-Aldrich | St. Louis, MO | A5316 | 1:10000 | |
| demethylated PP2A-C | Monoclonal | Mouse | Cell Signaling | Danvers, MA | 4466 | 1:1000 | |
| PP1 (E-9) | Monoclonal | Mouse | Santa Cruz | Dallas, TX | sc-7482 | 1:1000 | |
| Pan-Calcineurin A (PP2B) | Polyclonal | Rabbit | Cell Signaling | Danvers, MA | 2614 | 1:1000 | |
| PP5 | Polyclonal | Rabbit | Cell Signaling | Danvers, MA | 2289 | 1:1000 | |
| Gel loading Controls | |||||||
| β-actin | Monoclonal | Mouse | Sigma-Aldrich | St.Louis, MO | A5316 | 1:10000 | |
| β3-tubulin | Monoclonal | Mouse | Cell Signaling | Danvers, MA | 4466 | 1:1000 | |
2.3. Mice treatment midazolam administration protocols for tau phosphorylation studies
The Columbia University Animal Care and Use Committee approved the experimental protocol, and appropriate measures were taken to minimize pain and discomfort as per National Institutes of Health (NIH) guidelines. Nontransgenic 8 to 10-week-old C57BL/6 male mice were purchased from a commercial vendor (Taconic, Germantown, NY). Three-month-old male hTau mice were purchased from the Jackson Laboratory (Bar Harbor, ME) and were aged accordingly in the Columbia University animal care facility. hTau mice, a model of AD-like tauopathy, express non-mutant human tau on a murine tau knock-out background, and develop age-related increases in tau hyperphosphorylation and aggregation (Andorfer, et al., 2003). All mice were housed at 22°C in a temperature-controlled room and kept on a 12h alternating light/dark cycle. Food and water were made available ad libitum, and all mice underwent an acclimatization period of at least 24h prior to being used in the experiments.
In experiments examining whether midazolam had a dose response-related effect on tau phosphorylation, C57BL/6 mice were treated with either midazolam 10 mg/kg, 25 mg/kg, or an equivalent volume of 0.9% saline (control; 100 μL per 10 g of body weight) via intraperitoneal (i.p.) injection. These doses of midazolam were based on a previous study demonstrating antinociception without any reported adverse effects (Chiba, et al., 2009). In studies examining remote tau phosphorylation following acute midazolam administration, the mice were treated with either midazolam 25 mg/kg or 0.9% saline (100 μL per 10g body weight) i.p. and sacrificed 30 min, 6h, or 24h following treatment.
In the chronic midazolam administration studies, midazolam or 0.9% saline were administered subcutaneously using an Alzet 2001D osmotic pump (DURECT, Cupertino, CA), which was implanted in the interscapular region under 1-2% isoflurane anesthesia (~ 5-10 min total anesthesia exposure) in 30% O2. The pumps delivered the midazolam solution or saline at a rate of 8 μL/h. The mice received preemptive analgesia with carprofen 5 mg/kg s.c. and, at the end of surgery, bupivacaine hydrochloride (2 mg/kg max) was applied to the surgical site by local subcutaneous infiltration. Midazolam was subcutaneously administered at a rate of 3 or 5 mg/kg/h.
Upon induction of sedation, normothermia was maintained in the midazolam-treated mice by transferring the mice to a water-jacketed incubator unit (Thermocare, Incline Village, NV), which was set to target temperature of ~37°C throughout the study. The control mice were returned to their home cage at room temperature after receiving their treatment injections. Rectal temperature was monitored using an electronic thermometer (Thermalert TH-5, Physitemp, Clifton, NJ) to confirm normothermia.
2.4. Preparation of mouse brain protein samples
All mice were killed by cervical dislocation followed by immediate decapitation at 30 min, 6h, 24h, or 1 week following midazolam or saline treatment. The brains were immediately harvested, and both hippocampi and the cerebral cortex were dissected in ice-cold Tris-EDTA buffer (100 mM Tris HCl, 1 mM EDTA, pH 7.4). These brain tissues were then immediately flash frozen in liquid nitrogen and stored at −80°C until they were used in the analyses. Tissue homogenates were prepared as previously described (Planel, et al., 2008). Briefly, hippocampal or cortical tissues were homogenized in 5× vol/w RIPA buffer containing 50 mM Tris-HCl (pH 7.4), 1 mM EDTA, 150 mM NaCl, 0.1% SDS, 0.5% sodium deoxycholate, 1% Nonidet P-40, phosphatase inhibitors (Cocktail 1 and 2, 1:100 dilution), and protease inhibitors (Cocktail set III, 1:200 dilution). All homogenates were incubated on ice for 30 min, sonicated for 30s in pulse mode, and then centrifuged at 11,000 × g for 10 min at 4°C. Total protein content was determined in the supernatant using a bicinchoninic acid protein assay (Smith, et al., 1985).
2.5. SDS-PAGE and western blot analysis
The protein expressions of phosphorylated tau and total tau were determined using SDS-polyacrylamide gel electrophoresis (SDS-PAGE) coupled with Western blot analysis, as we have previously described (Whittington, et al., 2011). For monoclonal tau antibodies, we used a goat anti-mouse light chain specific secondary antibody to avoid nonspecific signals (Petry, et al., 2014). Immunoreactive band signal intensity was visualized by enhanced chemiluminescence (ECL Plus, GE Healthcare Life Sciences, Piscataway, NJ) using an ImageQuant LAS 4000 imaging system (GE Healthcare Life Sciences, Piscataway, NJ). ImageQuant® TL 8.1 software (GE Healthcare Life Sciences, Piscataway, NJ) was used to perform densitometric quantification of the immunoreactive bands.
2.7. Tau solubility analyses
Assessment of tau solubility was performed using a modification of the protocol of Greenberg and Davis (Greenberg and Davies, 1990) as we have previously described (Planel, et al., 2009,Whittington, et al., 2015). Briefly, the cortices were homogenized with a mechanical homogenizer in 5:1 volume/weight RIPA buffer containing 50 mM Tris-HCl, pH 7.4; 1% Nonidet P-40; 0.25% Na-deoxycholate; 150 mM NaCl; 1 mM EDTA; 1 mM PMSF; 1 mM Na3VO4; 1 mM NaF; 1:100 dilution of a protease inhibitor cocktail (P8340; Sigma-Aldrich, St. Louis, MO, USA). The samples were next centrifuged at 20,000 × g for 20 min at 4°C. One aliquot of supernatant was used to analyze the total tau fraction. A second aliquot of supernatant was used to obtain the heat-stable, soluble, aggregate free fraction. This was achieved by boiling the aliquot for 5 min, followed by subsequent centrifugation at 20,000 × g for 20 min at 4°C to remove the protein aggregates. The remaining supernatants were adjusted to the same protein content with RIPA buffer to a final concentration of 1% sarkosyl (N-lauroyl sarcosinate). The supernatants were subsequently incubated for 30 min at room temperature with constant shaking, followed by centrifugation at 100,000 × g for 1 h at 20°C. The pellets containing the sarkosyl-insoluble aggregated tau were re-suspended in an appropriate volume of Laemmli buffer and analyzed using western blotting. Aliquots of the supernatant were analyzed by western blotting of the sarkosyl soluble tau fraction.
2.8. Barnes maze testing
Barnes maze testing was used to assess short-term spatial reference memory following acute midazolam administration as we have previously described (Whittington, et al., 2015). Briefly, the Barnes maze apparatus (Stoelting, Inc., Wood Dale, IL) consisted of a circular metal platform that was 91 cm in diameter, with 20 holes around the perimeter of the platform. Each hole was 5 cm in diameter, and the platform was elevated 90 cm from the ground. All of the holes, with the exception of one, were blocked with a piece of metal as a means to prevent the mouse from entering and escaping the platform. The unblocked hole, referred to as the target hole, had a chamber through which the mouse was able to escape from the platform. Spatial cues were placed around the maze and incandescent bright light (580 lux) as well as white noise (85dB), generated by the Anymaze ™ software (Stoelting, Inc., Wood Dale, IL), were used as reinforcers. The movements of the mice were continuously tracked and recorded using the Anymaze ™ video tracking software for subsequent “offline” analysis.
The Barnes maze testing consisted of four phases. During an initial adaptation phase (Day 0), the mice were acclimated to the testing platform, target area, and the escape box. This was followed by the spatial acquisition phase (Days 1-4), where the mice were trained (3 trials a day for 4 days) to locate the target escape hole within 180 sec. At the end of the 4th day of the spatial acquisition phase, the hTau mice that reached asymptotic performance were randomized to receive either midazolam 25 mg/kg or 0.9% saline i.p.
On the 5th day, 18-20 hours after receiving the midazolam or saline injection, the mice underwent Probe Test I, which assessed short-term memory as previously described (Whittington, et al., 2015). During Probe Test 1, the mice were placed in the center of the Barnes maze, however the target escape hole was now blocked. Probe Test I lasted 90 sec, and the latency to reach the target hole, total and primary errors (number of errors committed before the first attempted entry into the target hole), total and primary (distance travelled before reaching the target hole for the first time), distance travelled as well as the mean speed (m/s) were measured. Following the completion of Probe Test I, the mice were sacrificed via cervical dislocation, and the hippocampal tissues were subsequently analyzed for total tau and phosphorylated tau levels.
2.9. Microtubule binding assay
We performed a microtubule (MT) binding assay to measure the binding of tau to preassembled, taxol-stabilized microtubules, to determine whether midazolam disrupted this critical function of tau. The assay was performed as we have described previously (Planel, et al., 2008) based on a modification of a method originally described by Maas et al. (Maas, et al., 2000). Briefly, brain hemispheres were homogenized in 5× weight/volume of BRB80 (Brinkley Reassembly Buffer; 80 mM PIPES/KOH pH 6.8, 1 mM EGTA, 1mM MgCl2) with protease and phosphatase inhibitors. The homogenates were then incubated on ice for 15 min and centrifuged at 20,000 × g for 20 min at 4°C. The resulting supernatants were then centrifuged again at 100,000 × g for 1 h at 4°C. These high-speed supernatant fractions were then adjusted to 1mM GTP, 10 μM taxol, and incubated with taxol-stabilized microtubules (30 μM, Cytoskeleton, Inc., Denver, CO) in a final volume of 50 μl for 10 min at 37°C. The mixtures were then centrifuged through 100 μl of 30% (w/v) sucrose cushions in BRB80 containing 1mM GTP and 10 μM taxol, at 100,000 × g for 30 min at room temperature. The supernatant, which contained the MT free fraction, was collected and diluted with a modified O+ buffer (O’Farrell, 1975) (62.5 mM Tris-HCl, pH 6.8; 10% glycerol; 5% 2-mercaptoethanol; 2.3% SDS; 1 mM EGTA; 1 mM EDTA; 1mM PMSF; 1 mM Na3VO4; 1 mM NaF; 10 l/ml Protease Inhibitor Cocktail P8340, Sigma-Aldrich). The pellet, which contained the MT bound fraction, was then resuspended in O+ buffer. The two fractions were then analyzed by Western blot using Total-tau and μ-tubulin antibodies for quantification.
3.0. Statistical analysis
Group comparisons of immunoblot relative band intensities were performed using either a one- or two-way analysis of variance (ANOVA) with Tukey’s post hoc test applied when appropriate or by an unpaired t-test. Statistical calculations were performed using Prism® 5 software (GraphPad Software, Inc., San Diego, CA), and all biochemical data are reported as mean ± SD with a value of P < 0.05 considered statistically significant.
Analysis of the Barnes Maze acquisition testing variables was performed by means of a two-way repeated measures ANOVA with Newman-Keuls Multiple Comparison post hoc test used when appropriate. An unpaired t-test was used to compare group analysis of probe test I variables. Prism® 5 software was also used to complete all of the Barnes Maze statistical analyses, with data reported as mean ± SEM and again P < 0.05 deemed statistically significant.
3. Results
3.1. Acute Midazolam administration increases tau phosphorylation under normothermic conditions in the mouse hippocampus
Using 8 to 10-week-old C57BL/6 male mice, we initially examined the impact of two intraperitoneally administered sedative doses of midazolam, 10 mg/kg and 25 mg/kg, versus saline (control) on hippocampal tau phosphorylation under normothermic conditions. These doses were selected as they both produced mild gait impairment and decreased locomotion but did not affect the loss of righting reflex; moreover, these motor effects resolved within 1h of its administration. Thirty min following the administration of midazolam 10 mg/kg and 25 mg/kg (n = 5 for each dose) significant increases (expressed as % of control) were observed at the AT8 (pSer202/pThr205; 247 ± 70 and 225 ± 89% of Ctl, respectively) and CP13 (pSer202; 215 ± 65 and 223 ± 81% of Ctl, respectively) phosphoepitopes (Fig. 1.1A,B), when compared to the saline-treated mice (Ctl, n = 4). Both doses of midazolam increased tau phosphorylation to a similar degree; however, neither midazolam dose had an effect on phosphorylated tau (p-tau) levels at PHF-1 (pSer396/pSer404) or total tau levels (Fig. 1.1C,D). Rectal temperatures at the end of the experiment, were similar in all of the study groups: Ctl 37.4 ± 0.3, Midazolam 10 mg/kg 37.3 ± 0.3, and Midazolam 25 mg/kg 37.4 ± 0.2°C.
Figure 1. Acute of administration of midazolam increases hippocampal tau phosphorylation in C57BL/6 mice.
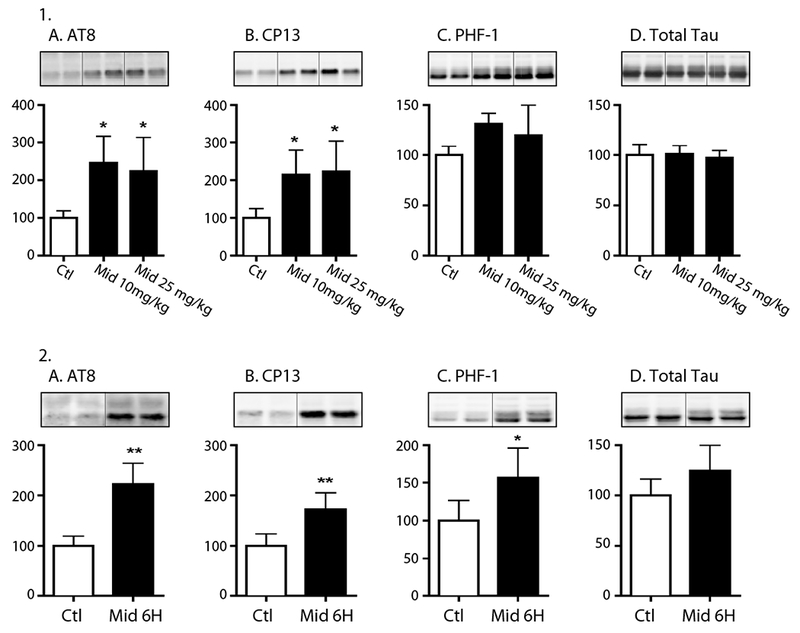
1. Hippocampal tau phosphorylation (% Ctl) at the AT8 (pSer202/pThr205), CP13 (pSer202), and PHF-1 (pSer396/Ser404) phosphoepitopes (1A-C) in 8-10-week-old C57BL/6 treated with midazolam (Mid) 10 mg/kg (n = 5) or 25 mg/kg (n = 5) i.p. Control (Ctl) mice (n = 4) were treated with 0.9% saline (100 μL per 10 g of body weight). All mice were sacrificed 30 min after treatment. No statistically significant difference in rectal temperatures (°C) was observed at the time of tissue harvest: Ctl 37.4 ± 0.3°C, Mid 10 mg/kg 37.3 ± 0.3°C, and Mid 25 mg/kg 37.4 ± 0.2°C. 2. Hippocampal tau phosphorylation (% Ctl) at the AT8, CP13, and PHF-1 phosphoepitopes (2A-C) in 8-10-week-old C57BL/6 mice treated with midazolam 25 mg/kg (MID 6h, n = 5) or 0.9% saline (Ctl, n = 4) i.p. and sacrificed 6h later. Normothermia was maintained throughout the study and rectal temperatures were similar at the time of hippocampal tissue harvest: Ctl 36.4 ± 0.3°C and MID 6h 36.9 ± 0.2 °C. Phosphorylated tau was normalized to total tau (1D and 2D) after controlling for gel loading with β-actin. Relative immunoreactive band intensities are expressed as a percent of Ctl and are displayed for each phosphoepitope and total tau. For each condition, 2 representative immunoblot bands are displayed. Data are expressed as mean ± SD and were analyzed using ANOVA with Tukey’s post-hoc test. * and ** denote P < 0.05 and P < 0.01 vs. Ctl, respectively.
We next examined whether tau phosphorylation persists in the mouse hippocampus following its acute administration. Given that we recently reported that acute administration of dexmedetomidine, a sedative-analgesic agent, produces tau hyperphosphorylation that lasts up to 6h (Whittington, et al., 2015), we first examined hippocampal tau phosphorylation 6h after the administration of midazolam 25 mg/kg. In these studies, the C57BL/6 mice received either midazolam 25 mg/kg (n = 5) or saline (control, n = 4) i.p. and were sacrificed 6h following treatment. Six hours following midazolam 25 mg/kg, tau hippocampal hyperphosphorylation was still present at the AT8, and CP13 phosphoepitopes (Fig. 1.2A,B). Interestingly, the PHF-1 epitope was also increased (Fig. 1.2C) while it was not at 30min (Fig. 1.1C), indicating a different kinetic phosphorylation profile than that observed with the other epitopes. There were no observed changes in total tau levels at 6h (Fig. 1.2D).
Overall our results indicate that a single injection of midazolam can lead to rapid (30 min) and prolonged (6 h) tau hyperphosphorylation in the hippocampus.
3.2. Midazolam induces cortical tau phosphorylation for up to 24 hours after its acute administration
As robust tau phosphorylation in the hippocampus was still evident 6h following midazolam administration, which was long after its sedative effects had dissipated, we next examined tau phosphorylation in the cortex of C57BL/6 mice that received midazolam 25 mg/kg i.p. and were sacrificed 30 min or 24h later (n = 6 for each treatment group). For these studies, tau phosphorylation was measured using a wider panel of phosphoepitope antibodies to more fully assess the degree of phosphorylation across several domains of tau, including the microtubule binding domain (i.e., pSer262 with 12E8).
At 30 min, increases in tau phosphorylation were again observed at AT8 and CP13 (Fig. 2A,B), with no change of phosphorylation at PHF-1 or total tau, confirming our results in the hippocampus (Fig. 1.1C). In addition, significant increases in tau phosphorylation were observed at 30 min at AT180 (pThr231; Fig. 2E) and 24h at pSer199 (Fig. 2G). No change in tau phosphorylation was observed at the 12E8 (pSer262/pSer356) or AT270 (pThr181) phosphoepitopes at either time point.
Figure 2. Cortical tau hyperphosphorylation following acute midazolam administration persists up to 24h in C57BL/6 mice.
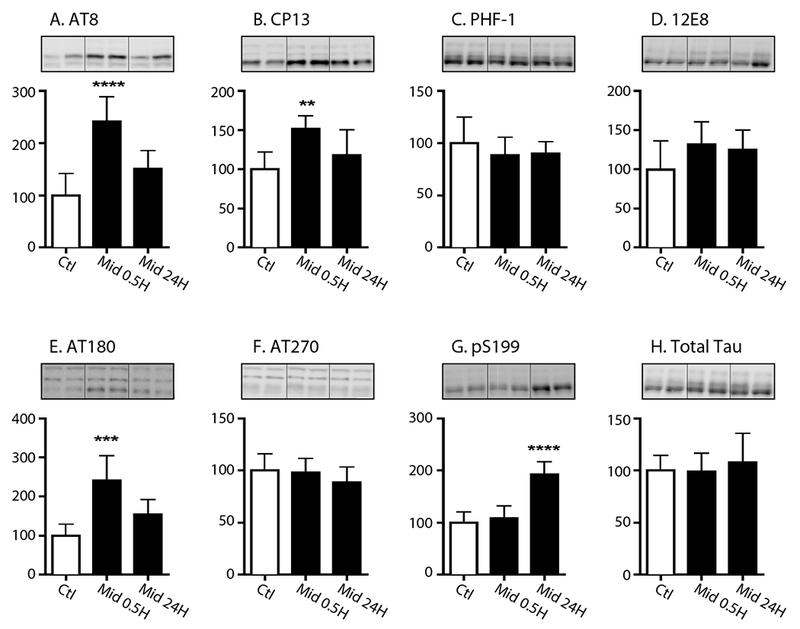
Cortical tau phosphorylation (% Ctl) at the AT8 (A), CP13 (B), PHF-1 (C), 12E8 (D), AT180 (E), AT270 (F) and pS199 (G) phosphoepitopes in 8-10-week old C57BL/6 mice treated with midazolam (Mid) 25 mg/kg i.p and sacrificed 30 min (0.5h, n = 6) or 24h (n = 6) later. Control mice (n = 6) were treated with 0.9% saline i.p. and sacrificed 30 min later. Normothermia was maintained throughout and rectal temperatures at the end of the study were 37.4 ± 0.2°C (Ctl), 37.0 ± 0.3°C (Mid 0.5h), and 37.0 ± 0.4°C (Mid 24h). Phosphorylated tau levels were normalized to total tau (H) after controlling for gel loading with β-actin. Data are expressed as mean ± SD and **,***, **** denote P < 0.01, P <0.001, and P < 0.0001 vs. Ctl., respectively using ANOVA with Tukey’s post-hoc test. Two representative immunoblot bands are displayed for each condition.
Overall, these data indicate that the acute administration of midazolam, under normothermic conditions, quickly increases tau phosphorylation in the hippocampus and cortex following its administration in non-transgenic mice. However, the tau hyperphosphorylation response observed following acute midazolam administration is complex, with some epitopes responding immediately, such as AT8 and CP13 (30min), while other epitopes have a more delayed onset, such as PHF-1 (6h) and pS199 (24h).
3.3. Mechanisms of tau hyperphosphorylation after midazolam administration
The tau phosphorylation state is dependent on a balance between the activity of several major tau protein kinases such as glycogen synthase kinase-3β (GSK-3β), stress-activated protein kinase (SAPK)/Jun-amino terminal kinase (JNK), extracellular signal-regulated kinase (ERK or MAPK1), calmodulin-dependent kinase II (CaMKII), cyclin dependent kinase 5 (CDK5) and its neuron-specific activator P35. Tau phosphorylation also depends on the activity of several protein phosphatases, including protein phosphatase 1 (PP1), protein phosphatase 2B (PP2B), and protein phosphatase 2A (PP2A), the latter being the main tau phosphatase in the brain (Ferrer, et al., 2002,Ferrer, et al., 2001,Ferrer, et al., 2005,Gong, et al., 2000,Planel, et al., 2007,Planel, et al., 2002,Tian and Wang, 2002). Therefore, we subsequently dissected the underlying tau phosphorylation mechanism by specifically determining the activation pattern of GSK-3β, ERK 1/2, SAPK/JNK, CAMKII, CDK5 as well as examining the protein expression of PP2A-C (catalytic subunit) and demethylated-PP2A-C in the same cortical samples obtained from the C57BL/6 mice at 30 min and 24h following acute midazolam or saline treatment.
At 30 min, a significant increase in phospho-GSK-3β (Ser9) levels (Fig. 3C) and a significant decrease in phospho-ERK 1/2 (p-ERK 1/2, Fig. 3G) levels were observed, when normalized to their respective total kinase levels (3D, 3H), in the midazolam-treated mice. This was consistent with inactivation of these enzymes and, consequently, could not explain the tau hyperphosphorylation observed at this time point. Moreover, no significant kinase expression changes consistent with hyperphosphorylation at 30 min were observed in phospho-CaMKII and phospho-SAPK/JNK (Fig. 3A, 3E), when normalized to their respective total kinase level (Fig. 3B, 3F) as well as in levels of CDK5 (Fig. 3I) and its activator P35 (Fig. 3J). Furthermore, at 24h, no changes in cortical kinase activation were observed that could explain the persistence of phosphorylation at pSer199.
Figure 3. The effect of acute midazolam administration on tau kinases in the mouse cortex at 0.5h and 24h.
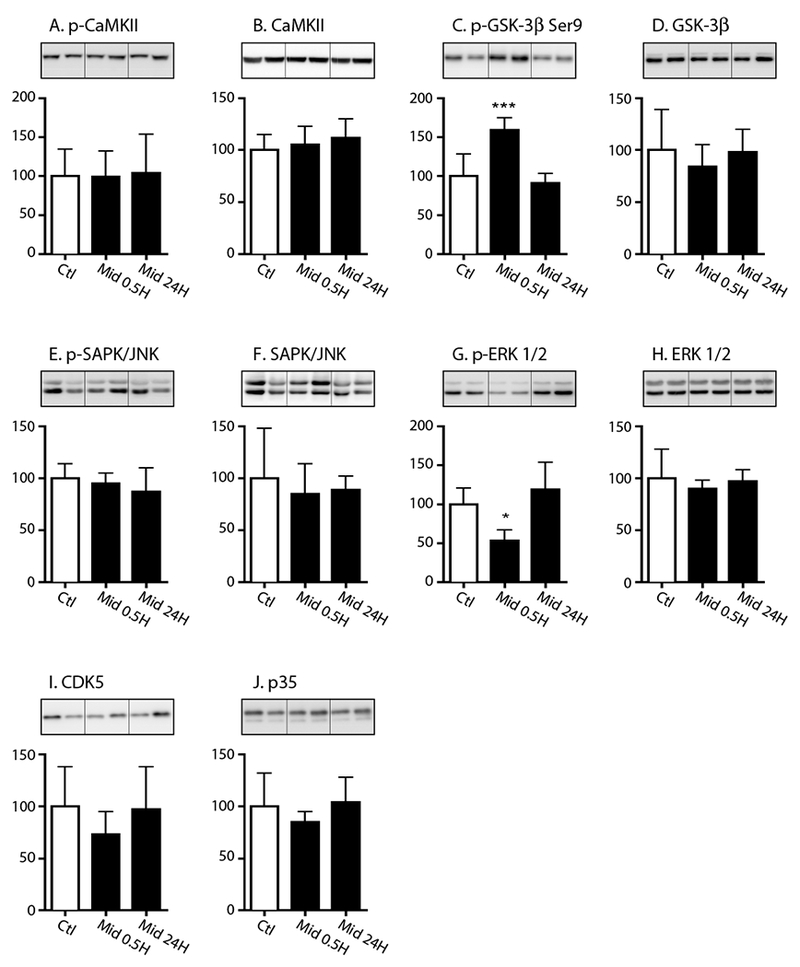
Cortical proteins from the in 8-10 week old C57BL/6 mice that received either saline (Ctl) or midazolam 25 mg/kg i.p. and were sacrificed 30 min (Mid 0.5) or 24h (Mid 24) were separated by SDS-PAGE and protein levels of kinases were determined using antibodies directed at the following proteins: (A) phospho-CaMKU, (B) total CaMKII, (C) phospho-GSK-3β Serine (Ser) 9, (D) total GSK-3p, (E) phospho-SAPK/JNK, (F) total SAPK/JNK, (G) phospho-ERK 1/2 (phospho-p44/42 MAPK), (H) total ERK 1/2 (p44/42 MAPK), (I) CDK5, and (J) P35 (activator of CDK5). Relative immunoreactive band intensities are expressed as a percent of saline control (Ctl) after appropriately normalizing for gel loading with β-actin or β-tubulin based on the molecular weight of the protein of interest. Phosphorylated proteins were also normalized to each respective non-phosphorylated total protein. For each condition, 2 representative immunoblot bands are displayed with Ctl (n = 6), 0.5h (n = 6), and 24h (n = 6). Data are expressed as mean ± SD. *, *** denote P < 0.05, P < 0.001, vs. Ctl, respectively; ANOVA with Tukey’s post hoc test.
Given the absence of kinase activation at these time points, levels of relevant tau protein phosphatases were also examined at 30 min and 24h in these same cortical tissues. At 30 min, midazolam induced a decrease in demethylated protein phosphatase 2A-C levels (Fig. 4A), which again was inconsistent with increased tau phosphorylation, as it reflects increased activity. Overall, there were no changes in the protein expression of the catalytic subunit of PP2A (Fig. 4B), protein phosphatase 2B (PP2B, Fig. 4D), and protein phosphatase 5 (PP5, Fig. 4E). Interestingly, a decrease in protein phosphatase 1 (PP1, Fig. 4C) levels was observed at 30 min, suggesting that perhaps this enzyme may play a role in midazolam-induced tau phosphorylation. At 24h, there was still an equivalent decrease of PP1; however, it did not reach statistical significance.
Figure 4. The effect of acute midazolam administration on phosphatase levels in the mouse cortex at 0.5h and 24h.
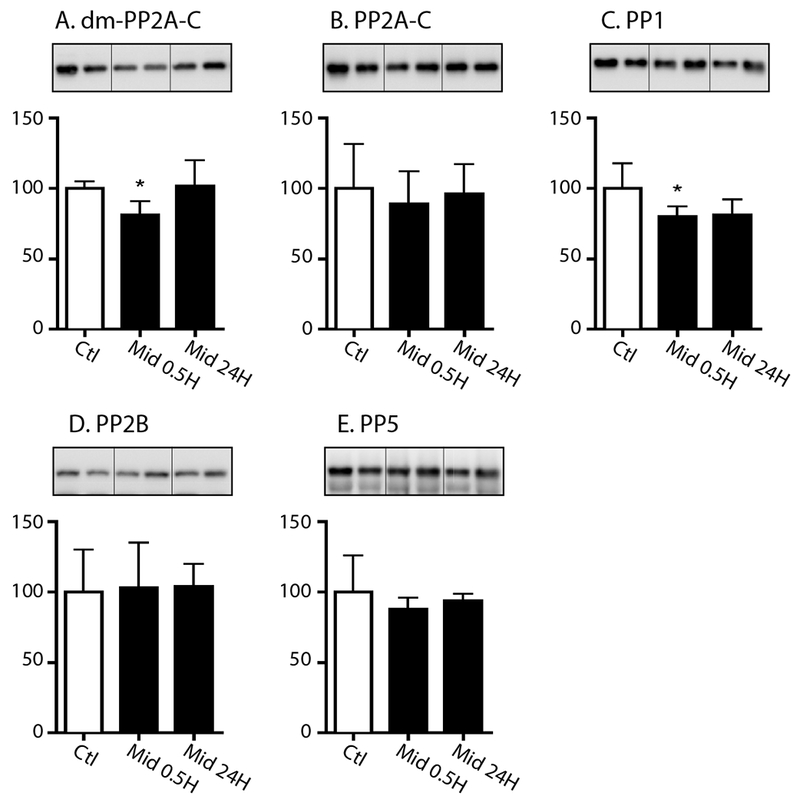
Cortical proteins from the in 8-10 week old C57BL/6 mice that received either saline (Ctl) or midazolam 25 mg/kg i.p. and were sacrificed 30 min (Mid 0.5) or 24h (Mid 24) were separated by SDS-PAGE and protein levels of several protein phosphatases were determined using antibodies directed at (A) the demethylated protein phosphatase catalytic subunit (dm-PP2A-C), (B) the PP2A catalytic subunit (PP2A-C), (C) protein phosphatase 1 (PP1), (D) protein phosphatase 2B (PP2B), and (E) protein phosphatase 5 (PP5). Relative immunoreactive band intensities are expressed as a percent of saline control (Ctl) after appropriately normalizing for gel loading with β-actin or β-tubulin. For each condition, 2 representative immunoblot bands are displayed with Ctl (n = 6), 0.5h (n = 6), and 24h (n = 6). Data are expressed as mean ± SD. * denotes P < 0.05 vs. Ctl using ANOVA with Tukey’s post hoc test.
Taken together, our data do not allow the identification of a definite mechanism. Nevertheless, they suggest that PP1 downregulation following midazolam administration might contribute to the observed hyperphosphorylation of tau.
3.4. Acute midazolam administration increases hippocampal tau phosphorylation levels 6h following its administration in hTau mice without altering cortical tau solubility
In order to determine whether the tau hyperphosphorylation response following midazolam administration is impacted by pre-existing tau pathology, the tau phosphorylation pattern was also examined in 3-month-old male hTau mice, a transgenic strain solely expressing non-mutant human tau that develops neurofibrillary pathology similar to that observed in AD (Andorfer, et al., 2003). These mice were studied at 3 months of age, as Andorfer et al. previously demonstrated that, by 3 months of age, accumulation of CP13 reactive tau occurs in the hippocampal cell bodies of this transgenic strain (Andorfer, et al., 2003).
The hTau mice received either midazolam 25 mg/kg (n = 6) or 0.9% saline (control, n = 6) i.p. and were sacrificed 6h following administration (Fig. 5). Midazolam produced a significant increase in hippocampal tau phosphorylation at 6h solely at CP13, 153 ± 20% of control (Fig. 5.1B); furthermore, unlike the C57BL/6 mice no persistence of tau hyperphosphorylation was observed at AT8 (Fig. 5.1A).
Figure 5. Hippocampal tau phosphorylation is present at the CP13 phosphoepitopes 6h following acute midazolam administration in hTau mice but is not associated with an increase in insoluble tau levels.
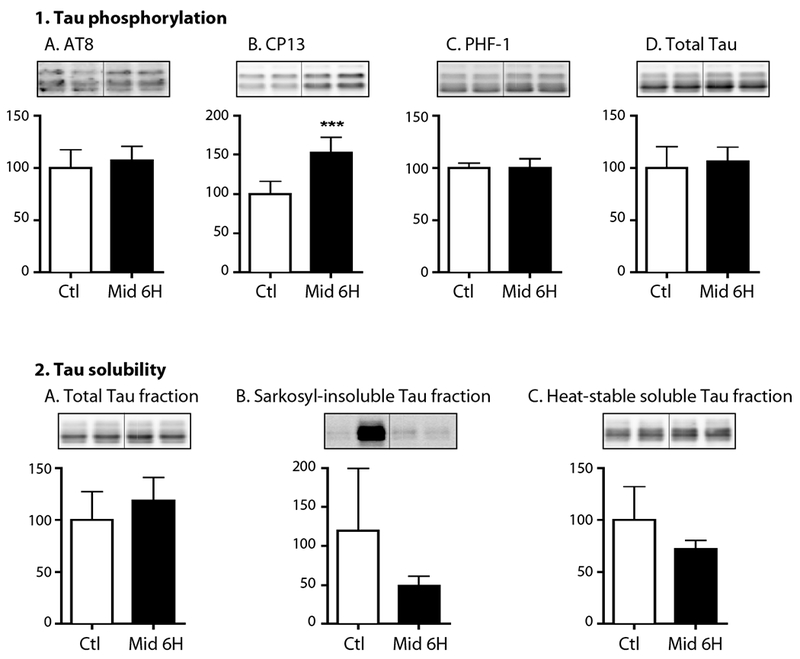
Hippocampal tau phosphorylation (% Ctl) at the AT8, CP13, and PHF-1 phosphoepitopes (1A-C) in 3-month-old hTau mice treated with midazolam 25 mg/kg (n = 6) or saline (Ctl, n = 6) i.p. and sacrificed 6h later. Normothermia was maintained throughout the study and rectal temperatures were similar at the time of hippocampal tissue harvest: Ctl 36.8 ± 0.9°C and Midazolam 36.8 ± 0.7 °C. Phosphorylated tau levels were normalized to total tau (1D), after controlling for gel loading with β-actin. Levels of tau in the total (2A), sarkosyl-insoluble (2B), sand heat stabilized fractions (2C) were measured using immunoblotting and total tau antibody. Relative immunoreactive band intensities are expressed as a percent of Ctl and are displayed for each phosphoepitope and total tau. For each condition, 2 representative immunoblot bands are displayed. Data are expressed as mean ± SD and *** P < 0.001 vs. Ctl using an unpaired t-test.
We also examined the impact of midazolam on cortical tau solubility 6h following its administration. Cortical sarkosyl-soluble tau levels were very variable, and no significant changes were detected at 6h (data not shown), with no change in unfractionated total tau (Fig. 5.2A) sarkosyl-insoluble (Fig. 5.2B), as well as the heat stabilized tau fraction (Fig. 5.2C). Rectal temperatures were similar in all groups at the end of this study: 36.8 ± 0.9 and 36.8 ± 0.7°C.
In summary, acute midazolam administration in hTau mice was associated with persistent phosphorylation at the CP13 phosphoepitope; however, this phosphorylation was not associated with any increase in cortical sarkosyl-insoluble tau levels. This suggests that despite the presence of persistent hyperphosphorylation at 6h in the hTau mice, midazolam was not associated with any tau-related pro-aggregant changes in this transgenic strain.
3.5. Acute midazolam administration does not impact short-term spatial reference memory in hTau mice
Barnes Maze testing was performed in hTau mice to assess short-term spatial reference memory following midazolam 25 mg/kg (n = 8) or 0.9% saline (10 uL/kg; n = 8) administration. Data from the acquisition phase (Days 1-4) demonstrate that both mice groups learned rapidly and reach asymptotic performance by Day 4 (Fig. 6). By Day 4, there were no significant between-group differences in total distance, total latency and errors (Fig. 6A-C). On Day 5, 18-20 hours following the administration midazolam or saline, a probe test (Fig. 6D-I) assessing spatial reference memory was performed and revealed that midazolam had no significant effect on primary and total distance (Fig. 6D,E), primary and total number of errors (Fig. 6G,H) as well as primary latency (Fig. 6I). The mean speed was similar in both groups demonstrating that midazolam produced no motor impairment at that time point (Fig. 6F).
Figure 6. Acute Administration of a midazolam dose known to induce tau hyperphosphorylation does not result in the impairment of spatial memory in hTau mice.
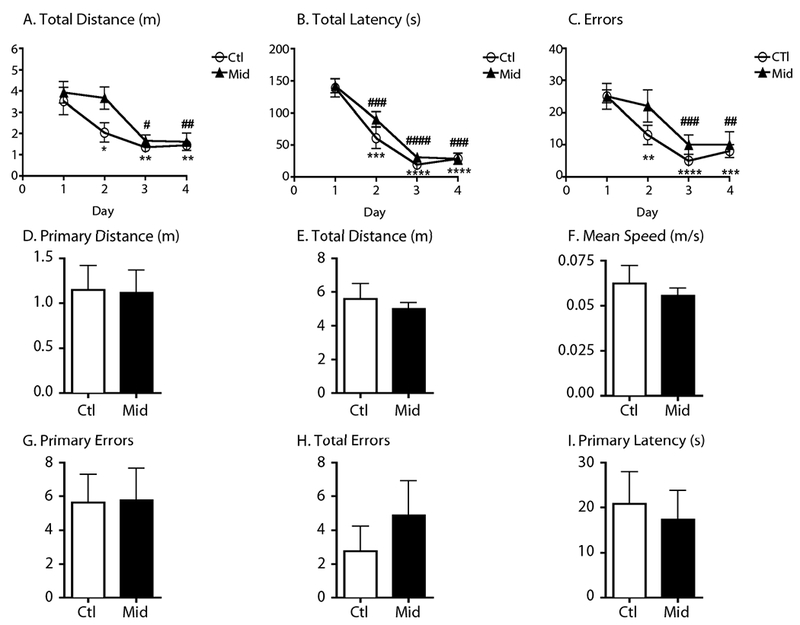
Acquisition phase testing data (mean ± SEM) in 6-month-old hTau mice demonstrate that asymptotic performance is readily achieved in the Barnes maze with 4 days of training (A-C), before treatment. By Day 4, total distance (m), total latency (s) and total errors in the saline and midazolam groups (n = 8 per group) were significantly decreased from their respective day 1 performance for each variable, denoting good learning performance. *, **, ***, and **** denote p < 0.05, p < 0.01, p < 0.001, and p < 0.0001 versus day 1 (saline), respectively and #, ##, ###, and #### denote p < 0.05, p < 0.01, p < 0.001, and p < 0.0001 versus day 1 (midazolam) using repeated-measures analysis of variance (ANOVA) with Newman-Keuls post hoc test. Probe test data (mean ± SEM) were obtained one day after the end of the acquisition training phase (Day 5). Primary and total distance (m), primary and total errors, primary latency (s) and mean speed (m/s) were obtained 18-20 hours following treatment with midazolam 25mg/kg or saline i.p. Midazolam-treated mice demonstrated no deterioration of spatial memory retention as measured by distance, errors, and latency. Moreover, there were no differences in terms of mean speed suggesting the absence of midazolam-induced motor impairment.
Thus, our results reveal that administration of midazolam, at a dose previously demonstrated to induce tau hyperphosphorylation, is not associated with short-term memory impairment in young hTau mice.
3.6. Chronic midazolam administration increases hippocampal tau phosphorylation in an age-dependent manner in non-transgenic mice
As midazolam is often administered as a prolonged infusion for long-term sedative purposes, we next examined the impact of chronic midazolam administration on hippocampal tau phosphorylation in 8-10-week-old as well as 11-month-old C57BL/6 mice. We initially measured hippocampal tau phosphorylation in 8-10-week-old C57BL/6 mice (n = 6 per group) following a 24h infusion of midazolam 3 mg/kg/h or 0.9% saline. These mice were sacrificed immediately upon termination of the infusion. Hippocampal tau phosphorylation solely increased at the CP13 phosphoepitope (150 ± 16% of Ctl; Fig. 7B), demonstrating that the CP13 phosphoepitope is again susceptible to midazolam-induced tau hyperphosphorylation. No significant increases in total tau as well as tau phosphorylation at AT8 and PHF-1, AT270, pSer262, AT180, pSer199 phosphoepitopes were observed (Fig. 7A, 7C-H). In the young mice, there was no significant difference in rectal temperatures between the Ctl and midazolam-treated groups: 36.5 ± 0.2 and 36.8 ± 0.8 °C, respectively. In contrast, when the 11-month-old C57BL/6 mice (n = 5) per group received similar midazolam or saline treatment, significant increases in hippocampal tau phosphorylation were observed in CP13, PHF-1, and AT180 in the midazolam group (Fig. 7J-L) while no significant changes were still not observed at AT270, pSer199, pSer262, or total tau (Fig. 7M-P). Again, in these aged mice, there was no significant difference in rectal temperatures between the Ctl and midazolam-treated groups: 36.9 ± 0.4 and 36.5 ± 0.04 °C, respectively.
Figure 7. Hippocampal tau phosphorylation following chronic midazolam administration increases as a function of age in C57BL/6 mice.
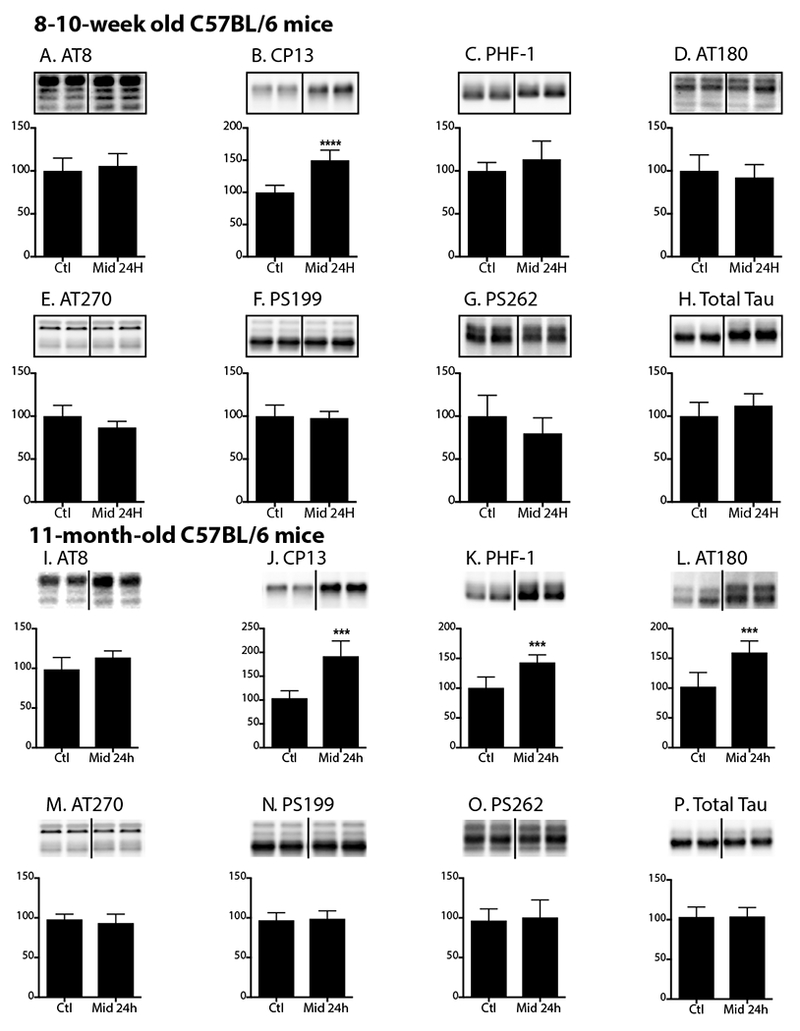
Hippocampal tau phosphorylation (% Ctl) at the AT8, CP13, PHF-1, AT180, AT270, pSer199, pSer262 phosphoepitopes in 8-10-week-old (A-G) or 11-month-old (I-O) C57BL/6 mice treated with a subcutaneous infusion of midazolam 3 mg/kg/h (n = 6 young; n = 5 old) or 0.9% saline (Ctl) (n = 6 young; n = 5 old) i.p. and sacrificed 24h after the start of the infusion. Normothermia was maintained throughout the administration of midazolam or saline, and rectal temperatures were similar at the time of hippocampal tissue harvest: young Ctl 36.5 ± 0.2°C and Midazolam 36.8 ± 0.8 °C and old Ctl 36.9 ± 0.4°C and Midazolam 36.5 ± 0.04 °C. Phosphorylated tau levels were normalized to each groups respective level of total tau (H,P), after controlling for gel loading with β-actin. Relative immunoreactive band intensities are expressed as a percent of Ctl and are displayed for each phosphoepitope and total tau. For each condition, 2 representative immunoblot bands are displayed. Data are expressed as mean ± SD and ***, **** denote P < 0.001 and P < 0.0001, respectively, vs. Ctl using an unpaired t-test.
Hence, these data suggest the susceptibility to tau phosphorylation following chronic midazolam administration increases as a function of age in this non-transgenic strain.
3.6. Chronic midazolam administration induces hippocampal tau hyperphosphorylation that persist for up to a 1 week in young non-transgenic mice
The persistence of hippocampal tau phosphorylation was also examined 1 week after the start of a 24h infusion of midazolam 3 mg/kg/h (n = 5) or 0.9% saline (n = 6) in 8-10-week-old C57BL/6 mice. Interestingly, 1 week following a chronic infusion of midazolam, increased hippocampal phosphorylated tau levels at AT8 and CP13 (152 ± 29 and 211 ± 69 % of Ctl, respectively; Fig. 8A,B) were observed, with no similar change detected in phosphorylation at PHF-1 or total tau levels (Fig. 8C,D) at this same time point.
Figure 8. Hippocampal tau phosphorylation following chronic midazolam administration persists as a function of age in young C57BL/6 mice.
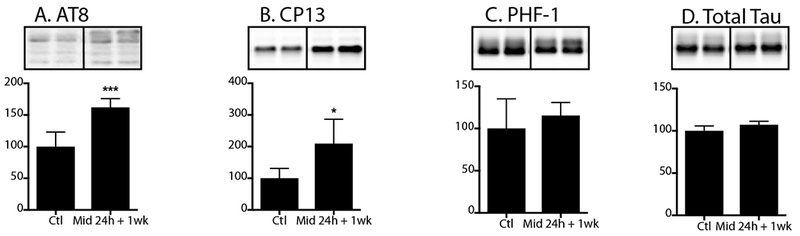
Hippocampal tau phosphorylation (% Ctl) at the AT8, CP13, PHF-1 phosphoepitopes in 8-10-week-old (A-C) C57BL/6 mice treated with a subcutaneous infusion of midazolam 3 mg/kg/h (n = 5) or 0.9% saline (Ctl; n = 6) and sacrificed 1 week after the start of the 24h infusion. Normothermia was maintained throughout the administration of midazolam or saline. Phosphorylated tau levels were normalized to total tau (D), after controlling for gel loading with β-actin. Relative immunoreactive band intensities are expressed as a percent of Ctl and are displayed for each phosphoepitope and total tau. For each condition, 2 representative immunoblot bands are displayed. Data are expressed as mean ± SD and *, *** denote P < 0.05 and P < 0.001, respectively, vs. Ctl using an unpaired t-test.
Therefore, these studies indicate that chronic midazolam administration, in young non-transgenic mice, results in persistent hippocampal tau phosphorylation for up to 1 week, which is a period long after the sedative effects of midazolam have dissipated.
3.7. The impact of chronic midazolam administration on tau phosphorylation, solubility, and microtubule binding
As we observed an increase in tau phosphorylation in the non-transgenic mice following chronic midazolam administration, the impact of this type of exposure on tau phosphorylation, solubility, and function were subsequently investigated in 7-month-old hTau mice.
Tau phosphorylation and solubility studies
7-month-old hTau mice received either midazolam 5 mg/kg/h or an equal volume of 0.9% saline (n = 6 per group) for 24h. Upon termination of the 24h infusion, hippocampal tau phosphorylation was increased at AT8 and CP13 phosphoepitopes in the midazolam-treated group with no change in phosphorylation at pSer262 or total tau levels (Fig 9.1A-D).
Figure 9. Chronic midazolam administration increases hippocampal tau phosphorylation in hTau mice without altering tau solubility.
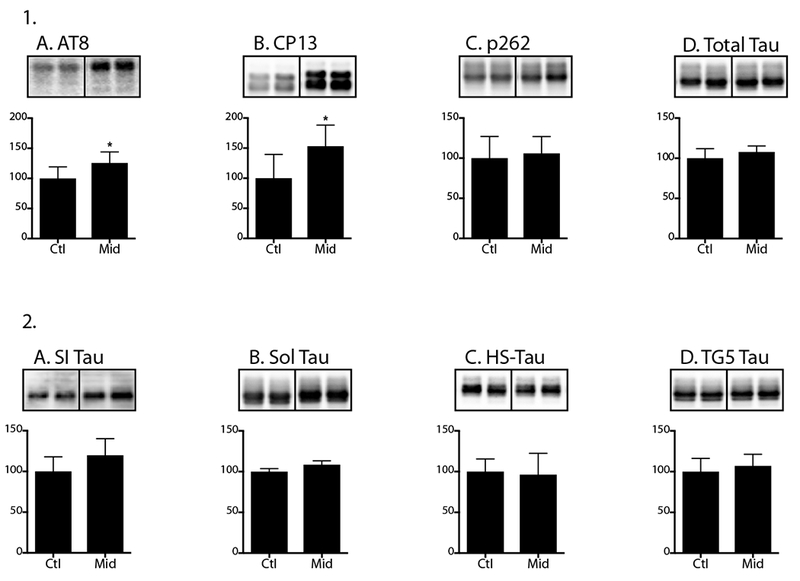
Hippocampal tau phosphorylation (% Ctl) at the AT8, CP13, and PHF-1 phosphoepitopes (1A-C) in 7-month-old hTau mice treated midazolam 5 mg/kg/h or 0.9% saline (n = 6 per group) and sacrificed 24h after the start of the chronic infusion. Normothermia was maintained throughout the study and rectal temperatures were similar at the time of hippocampal tissue harvest: Ctl 37.3 ± 1.0 °C and Midazolam 36.9 ± 0.3 °C. Phosphorylated tau levels were normalized to total tau (1D), after controlling for gel loading with β-actin. Levels of tau in the sarkosyl-insoluble (SI; 2A), sarkosyl-soluble (Sol; 2B), and heat-stabilized (HS; 2C) and total (TG5) fractions (2D) in cortical homogenates were measured using immunoblotting and total tau (TG5) antibody. Relative immunoreactive band intensities are expressed as a percent of Ctl and are displayed for each phosphoepitope and total tau. For each condition, 2 representative immunoblot bands are displayed. Data are expressed as mean ± SD and * denotes P < 0.05 vs. Ctl using an unpaired t-test.
Using tissues from this same experiment, we then determined whether this midazolam-induced increase in tau phosphorylation was associated with any changes in tau solubility in cortical homogenates. Interestingly, there were no significant changes in the sarkosyl-insoluble tau, sarkosyl-soluble tau, heat-stabilized tau and total tau levels (Fig 9.2A-D) observed between the midazolam and saline-treated groups.
Thus, although this chronic infusion of midazolam produced gait impairment and significant hippocampal tau phosphorylation, these changes did not result in changes in tau aggregation.
Tau microtubule binding studies
We then examined whether chronic midazolam administration, affected the capacity of tau to bind to preassembled, taxol-stabilized microtubules (MTs) using tissues from the midazolam and saline-treated hTau mice. Compared to control mice, we observed a significant decrease in tau levels in the MT bound fraction, for equivalent amounts of μ-tubulin, in the midazolam-treated mice (Fig 10). Although a slight increase in tau levels in the MT free fraction was observed in the midazolam-treated mice versus control, this was not statistically significant.
Figure 10. Chronic midazolam administration decreases the capacity of tau to bind to taxol-stabilized preformed microtubules in hTau mice.
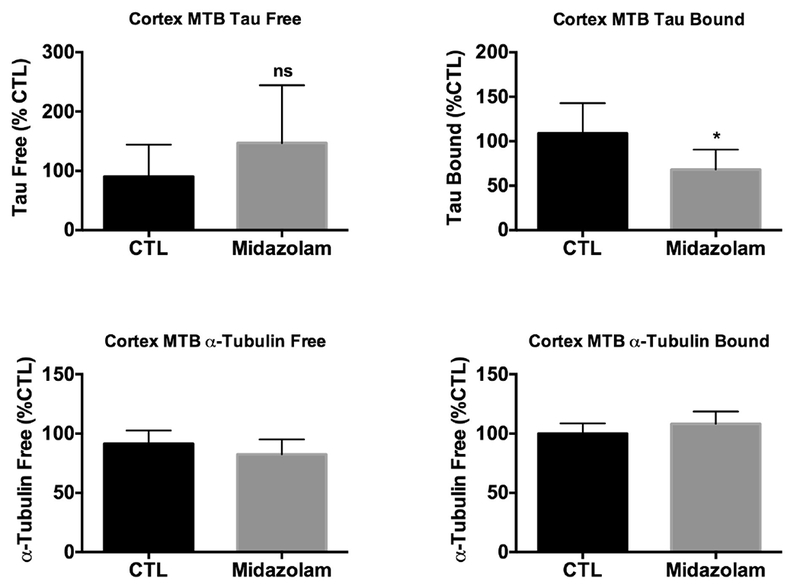
Neocortical brain proteins from 7-month-old hTau mice treated midazolam 5 mg/kg/h or 0.9% saline (n = 6 per group) for 24h were extracted and incubated with taxol-stabilized microtubules. Total tau and μ-tubulin levels from the microtubule free and bound fractions were evaluated by immunoblot analysis. Levels of total tau and μ-tubulin in microtubule free (A,C, respectively) and bound fractions (B,D, respectively) following midazolam treatment were compared to saline (Ctl) after controlling for gel loading with β-actin. Relative immunoreactive band intensities are expressed as a percent of Ctl and, for each condition, 2 representative immunoblot bands are displayed. Data are expressed as mean ± SD and * denotes P < 0.05 vs. Ctl using an unpaired t-test.
Nevertheless, these data indicate that the chronic administration of midazolam has the ability to impact the ability of tau to bind to preformed MTs in a transgenic mouse model expressing non-mutant human tau.
Discussion
Although it has been well established that anesthetics can induce profound tau hyperphosphorylation by producing anesthesia-induced hypothermia (Planel, et al., 2009,Planel, et al., 2008,Planel, et al., 2007), our current studies further support the notion that certain anesthetics can still increase tau phosphorylation in the absence of hypothermia. The present findings specifically demonstrate that midazolam, a commonly used benzodiazepine, directly increases tau phosphorylation in non-transgenic as well as a transgenic AD mouse model expressing non-mutant human tau. To our knowledge, it is the first study reporting tau hyperphosphorylation in vivo following acute and chronic administration of a benzodiazepine.
We initially observed that even a single dose of midazolam, in non-transgenic mice, was associated with increased cortical tau phosphorylation at AT8, CP13 and AT180 at 30 min, and pSer199 for up to 24h. Interestingly, compared to the 2h and 6h duration of tau phosphorylation that we previously reported following an acute dose of propofol (Whittington, et al., 2011) and dexmedetomidine (Whittington, et al., 2015), respectively, the duration of tau phosphorylation at pS199 following midazolam administration lasted up 24h. As those previous studies involving the acute administration of sedatives employed even more profound levels of sedation than that observed with midazolam, our present findings suggest that midazolam possesses a much greater capacity to directly increase the duration of tau phosphorylation than these two commonly used intravenous hypnotic agents, having an effect long after its sedative effect has disappeared.
As a benzodiazepine, midazolam produces many of its effects by allosterically binding to the benzodiazepine receptor on GABAA receptors, which in turn potentiates the action of GABA on this receptor subtype (Ehlert, et al., 1983,Sigel and Baur). Previous studies have demonstrated that activation of GABAA receptor using the benzodiazepine desalkylflurazepam (an active metabolite of flurazepam), increased AT8 tau phosphorylation in vitro, but had no effect on TG3 (pSer231) or PHF13 (pSer396) (Nykanen, et al., 2012). Interestingly, Nykanen et al. also observed persistent tau phosphorylation in vitro at AT8 after treatment following a washout period; in contrast, we solely observed persistent tau hyperphosphorylation at pS199 in vivo. Nevertheless, our findings are consistent with those of Nykanen et al., as the AT8 (pSer202/pThr205) and pSer199 phosphoepitopes are located in the same proline-rich region of tau (Augustinack, et al., 2002,Buee, et al., 2000), suggesting a propensity for benzodiazepines to induce persistent phosphorylation in this area.
Our studies examining tau kinases and phosphatases did not allow for the isolation of the mechanism underlying tau hyperphosphorylation but show that midazolam administration led to the downregulation of PP1. Interestingly, Shibasaki et al. have shown that in vivo administration of zolpidem, an imidazopyridine that binds and activates GABAA receptors at the same location than benzodiazepines, results in the downregulation of PP1 (Shibasaki, et al., 2013), suggesting that benzodiazepines might have the same effect and supporting our results. We did not find a downregulation of PP2A, but Nykanen et al. observed that GABAA receptor-mediated increases in tau phosphorylation in vitro were associated with decreased in PP2A binding to tau, and not a decrease in global PP2A level or activity per se (Nykanen, et al., 2012). Overall, and in light of the literature, our results suggest that tau hyperphosphorylation following midazolam treatment might be due in part to PP1 downregulation.
Importantly, acute midazolam-induced tau hyperphosphorylation in 3-month old hTau mice was not associated with increases in cortical insoluble tau levels or with impaired short-term spatial reference memory. This is in stark contrast to what we recently observed in hTau mice of the same age with dexmedetomidine, where increases in insoluble tau and impaired spatial memory were observed following its acute administration (Whittington, et al., 2015). differences in tau aggregation might be due to different epitopes being hyperphosphorylated. The example, acute dexmedetomidine administration immediately induced tau hyperphosphorylation at the PHF-1 site (Whittington, et al., 2015), while midazolam did not. In contrast to our results, midazolam administered before probe test impaired retrieval of spatial memory in rats tested with the Morris water maze (Timic, et al., 2013). However, the rats were tested 20 min after midazolam administration, while the probe test was performed 18-20 h after in our mice. However, other studies confirm our results since the administration of midazolam at different stages of learning did not affect acquisition, consolidation or recall of spatial memory (Valentim, et al., 2013a,Valentim, et al., 2013b). Overall, our results suggest that acute midazolam administration does not affect tau aggregation or spatial memory in our experimental design.
Given this persistence of tau phosphorylation following its acute administration, we also examined hippocampal tau phosphorylation in non-transgenic mice following a 24-hour midazolam infusion, an experimental paradigm with clinical relevance as this benzodiazepine is still used for prolonged sedation in intensive care unit (ICU) settings (Spence, et al., 2018,Zhang, et al., 2017). Again, despite achieving a modest level of sedation, characterized primarily by mild ataxia, hippocampal tau phosphorylation was observed immediately upon cessation of the 24-hour midazolam infusion. Moreover, this effect was augmented by age suggesting that, following its chronic administration, the aged brain is more susceptible to midazolam-induced increases in hippocampal tau phosphorylation.
Surprisingly, we also observed pronounced hippocampal tau hyperphosphorylation in young non-transgenic mice, one week following the administration of a 24-hour midazolam infusion. The degree of tau phosphorylation at one week was somewhat different compared with the 24-hour time point, as it became more pronounced at AT8, while disappearing at PHF-1. This suggests that in the setting of chronic midazolam administration, hippocampal tau phosphorylation not only persists for up to 1 week, but also implies that the pattern of epitopes affected shifts over time. Hence, future studies are indicated to determine whether this phosphorylation persists at time points even more remote than 1 week following midazolam exposure. This persistence of tau phosphorylation following chronic administration of an anesthetic is not a unique finding, as Le Freche et al. observed a similar phenomenon following the chronic administration of the inhalational anesthetic sevoflurane (Le Freche, et al., 2012). However, this persistence of midazolam-induced tau phosphorylation is the first time this has been described with an intravenous sedative that has been clinically associated with cognitive decline.
One of the key functions of tau is to bind and stabilize microtubules. Tau hyperphosphorylation has been associated with the dissociation of tau from microtubules resulting in disruption of the microtubule network and the impairment of axonal transport (Feinstein and Wilson, 2005,Mandelkow, et al., 2003,Mi and Johnson, 2006). In hTau mice, we observed that the chronic administration of midazolam resulted in a decrease capacity of tau to bind to preassembled, taxol-stabilized microtubules. This suggests that one of the major functions of tau, which is also critical to normal axonal transport (Mandelkow, et al., 2003), is disrupted by midazolam. As fast axonal transport has been consistently demonstrated to be disrupted in several animal models of AD (Buxbaum, et al., 1998,Morfini, et al., 2002,Smith, et al., 2007), any anesthetic that can further cause microtubule dysregulation could theoretically accelerate this neurodegenerative disorder.
Interestingly, just as was the case following acute midazolam administration, we did not observe any increases in insoluble tau levels in the cortex of 6-month-old hTau mice. following a chronic infusion, despite the aforementioned disruption of tau function. While this is somewhat surprising, it could be related to the fact that the tau aggregation analyses were performed immediately upon cessation of midazolam infusion. Thus, although though the tau hyperphosphorylation was associated with an increase in tau levels in the free (unbound) fraction and a concurrent decrease in the bound fraction, it is conceivable that not enough time elapsed for the unbound tau to more fully aggregate. Moreover, hTau mice express human, non-mutant tau, which would be expected to aggregate slower than other mice expressing the proaggregant P301L mutation (Lewis, et al., 2000). Also, the hTau used in our studies were still relatively young, which could have contributed to the absence of an increase in insoluble tau levels.
Recent studies have demonstrated that both intravenous (Run, et al., 2009,Whittington, et al., 2015,Whittington, et al., 2011) and inhalational anesthetics (Le Freche, et al., 2012,Tao, et al., 2014) can induce tau hyperphosphorylation in the absence of hypothermia; however, clinically most of these classes of anesthetics have not been unequivocally associated with cognitive impairment in humans. In contrast, benzodiazepines are commonly used sedative and anxiolytic agents that have been clinically associated with an increased risk of short-term cognitive impairment (Chun, 2005,Curran, 1986,Ghoneim and Mewaldt, 1990,Hirshman, et al., 2003,Lister, 1985). Moreover, using a prospective population based study design with long-term follow-up, Billioti de Gage et al. recently demonstrated that ever use of benzodiazepines was associated a 50% increase in dementia risk within 15 years following the start of this pharmacological class of drugs (Billioti de Gage, et al., 2012), findings that, although controversial, have been further supported by a follow-up study (Billioti de Gage, et al., 2014). The current study demonstrates that acute and chronic midazolam exposure increases tau phosphorylation, which theoretically could contribute to development or exacerbation of dementia. However, this transient exposure appears to be insufficient to propagate higher degrees of pathology typically associated with tau hyperphosphorylation such as aggregation and the development of memory deficits.
As with many pre-clinical anesthesia neurotoxicity studies, it would be certainly premature to extrapolate these findings to clinical practice at this juncture. First of all, midazolam is extremely valuable as a sedative and anxiolytic agent in the immediate perioperative period as well as in the patient population requiring post-operative sedation. Nevertheless, these pre-clinical findings further strengthen the notion that clinical studies should ultimately examine the impact of more prolonged benzodiazepine exposure on tau pathology in humans, especially given that this sedative class has been associated with a higher incidence of delirium in humans when compared with alpha-2 agonists (Riker, et al., 2009) as well as an increased risk of incident dementia (Billioti de Gage, et al., 2012).
In summary, acute midazolam administration in non-transgenic and transgenic mice results in hippocampal and cortical tau hyperphosphorylation that persists beyond recovery from its immediate sedative effects. This hippocampal tau phosphorylation pattern, following acute midazolam administration differs greatly between C57BL/6 mice and hTau mice. Furthermore, acute midazolam-induced hyperphosphorylation does not impact tau aggregation or spatial memory in hTau mice, suggesting that acute exposure is unlikely to accelerate neurofibrillary pathology. Chronic midazolam administration is associated with the persistence of tau phosphorylation and a decrease in the capacity of tau to bind to exogenous, preassembled microtubules. Hence, future studies should be directed at further establishing the pathological impact of a more prolonged or repeated exposure to this benzodiazepine.
Midazolam is not the most commonly used benzodiazepines. The most prescribed are aloprazam, diazepam, clonazepam, and lorazepam, but their effects on pathological markers of dementia are not yet understood. It is particularly a concern since a report has found that around 5.2% of US adults used benzodiazepines, with this percentage climbing to 8.7% in the elderly (Olfson, et al., 2015). Thus, future in vivo studies should be performed with other clinically used benzodiazepines to determine their comparative effects on tau pathology. The identification of such mechanisms may important for ultimately determining how benzodiazepine use increases the risk of incident dementia.
Highlights.
Benzodiazepines are linked to an increased risk of incident dementia in the elderly
The effect of benzodiazepines on tau pathology in vivo is unknown
Acute and chronic midazolam treatment in mice led to persistent tau hyperphosphorylation
Benzodiazepines might lead to dementia by increasing tau pathology
Verifications :
-
1)
The authors declare that there are no current or potential conflicts of interest including any financial, personal or other relationship with other people or organizations within three years of beginning the work submitted that could inappropriately influence their work.
-
2)
Supported by grant 2R01GM101698 (RAW) from the National Institutes of Health, and grants to Emmanuel Planel from the Fonds de Recherche en Santé du Québec (16205, 20048), the Natural Sciences and Engineering Research Council of Canada (354722), and the Alzheimer Society of Canada.
-
3)
The data contained in this manuscript have not been previously published, have not been submitted elsewhere and will not be submitted elsewhere while under consideration at Neurobiology of Aging.
-
4)
Animals were handled according to procedures approved by the Columbia University Animal Care and Use Committee approved the experimental protocol, and appropriate measures were taken to minimize pain and discomfort as per National Institutes of Health (NIH) guidelines.
-
5)
All authors have reviewed the contents of the manuscript being submitted, approve of its contents and validate the accuracy of the data.
Acknowledgments
Supported by grant 2R01GM101698 (RAW) from the National Institutes of Health, and grants to Emmanuel Planel from the Fonds de Recherche en Sante du Quebec (16205, 20048), the Natural Sciences and Engineering Research Council of Canada (354722), and the Alzheimer Society of Canada. The authors also thank Peter Davies, PhD (Professor, Departments of Pathology and Neuroscience, The Feinstein Institute for Medical Research, Manhasset, NY, USA) for the gift of the PHF-1, CP13, and TG5 antibodies and Dr. Peter Seubert (Neotope Biosciences, San Francisco, CA, USA) for the gift of the 12E8 antibody.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Disclosure statement
The authors declare no competing financial interest.
References
- Acil M, Basgul E, Celiker V, Karagoz AH, Demir B, Aypar U 2004. Perioperative effects of melatonin and midazolam premedication on sedation, orientation, anxiety scores and psychomotor performance. European journal of anaesthesiology 21(7), 553–7. [DOI] [PubMed] [Google Scholar]
- Andorfer C, Kress Y, Espinoza M, de Silva R, Tucker KL, Barde YA, Duff K, Davies P 2003. Hyperphosphorylation and aggregation of tau in mice expressing normal human tau isoforms. J Neurochem 86(3), 582–90. [DOI] [PubMed] [Google Scholar]
- Augustinack JC, Schneider A, Mandelkow EM, Hyman BT 2002. Specific tau phosphorylation sites correlate with severity of neuronal cytopathology in Alzheimer’s disease. Acta Neuropathol 103(1), 26–35. [DOI] [PubMed] [Google Scholar]
- Billioti de Gage S, Begaud B, Bazin F, Verdoux H, Dartigues JF, Peres K, Kurth T, Pariente A 2012. Benzodiazepine use and risk of dementia: prospective population based study. Bmj 345, e6231. doi: 10.1136/bmj.e6231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Billioti de Gage S, Moride Y, Ducruet T, Kurth T, Verdoux H, Tournier M, Pariente A, Begaud B 2014. Benzodiazepine use and risk of Alzheimer’s disease: case-control study. Bmj 349, g5205. doi: 10.1136/bmj.g5205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Binder LI, Frankfurter A, Rebhun LI 1985. The distribution of tau in the mammalian central nervous system. J Cell Biol 101(4), 1371–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brion JP, Passareiro H, Nunez J, Flament-Durand J 1985. Mise en évidence immunologique de la protéine tau au niveau des lésions de dégénérescence neurofibrillaire de la maladie d’Alzheimer. Arch Biol (Bruxelles) 95, 229–35. [Google Scholar]
- Buee L, Bussiere T, Buee-Scherrer V, Delacourte A, Hof PR 2000. Tau protein isoforms, phosphorylation and role in neurodegenerative disorders. Brain Res Brain Res Rev 33(1), 95–130. [DOI] [PubMed] [Google Scholar]
- Buffett-Jerrott SE, Stewart SH 2002. Cognitive and sedative effects of benzodiazepine use. Curr Pharm Des 8(1), 45–58. [DOI] [PubMed] [Google Scholar]
- Buxbaum JD, Thinakaran G, Koliatsos V, O’Callahan J, Slunt HH, Price DL, Sisodia SS 1998. Alzheimer amyloid protein precursor in the rat hippocampus: transport and processing through the perforant path. J Neurosci 18(23), 9629–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiba S, Nishiyama T, Yoshikawa M, Yamada Y 2009. The antinociceptive effects of midazolam on three different types of nociception in mice. J Pharmacol Sci 109(1), 71–7. [DOI] [PubMed] [Google Scholar]
- Chun MM 2005. Drug-induced amnesia impairs implicit relational memory. Trends in cognitive sciences 9(8), 355–7. doi: 10.1016/j.tics.2005.06.015. [DOI] [PubMed] [Google Scholar]
- Curran HV 1986. Tranquillising memories: a review of the effects of benzodiazepines on human memory. Biological psychology 23(2), 179–213. [DOI] [PubMed] [Google Scholar]
- Drechsel DN, Hyman AA, Cobb MH, Kirschner MW 1992. Modulation of the dynamic instability of tubulin assembly by the microtubule-associated protein tau. Mol Biol Cell 3(10), 1141–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drubin D, Kobayashi S, Kellogg D, Kirschner M 1988. Regulation of microtubule protein levels during cellular morphogenesis in nerve growth factor-treated PC12 cells. J Cell Biol 106(5), 1583–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drubin DG, Feinstein SC, Shooter EM, Kirschner MW 1985. Nerve growth factorinduced neurite outgrowth in PC12 cells involves the coordinate induction of microtubule assembly and assembly-promoting factors. J Cell Biol 101(5 Pt 1), 1799–807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehlert FJ, Roeske WR, Gee KW, Yamamura HI 1983. An allosteric model for benzodiazepine receptor function. Biochemical pharmacology 32(16), 2375–83. [DOI] [PubMed] [Google Scholar]
- Feinstein SC, Wilson L 2005. Inability of tau to properly regulate neuronal microtubule dynamics: a loss-of-function mechanism by which tau might mediate neuronal cell death. Biochim Biophys Acta 1739(2-3), 268–79. [DOI] [PubMed] [Google Scholar]
- Ferrer I, Barrachina M, Puig B 2002. Glycogen synthase kinase-3 is associated with neuronal and glial hyperphosphorylated tau deposits in Alzheimer’s disease, Pick’s disease, progressive supranuclear palsy and corticobasal degeneration. Acta Neuropathol (Berl) 104(6), 583–91. [DOI] [PubMed] [Google Scholar]
- Ferrer I, Blanco R, Carmona M, Ribera R, Goutan E, Puig B, Rey MJ, Cardozo A, Vinals F, Ribalta T 2001. Phosphorylated map kinase (ERK1, ERK2) expression is associated with early tau deposition in neurones and glial cells, but not with increased nuclear DNA vulnerability and cell death, in Alzheimer disease, Pick’s disease, progressive supranuclear palsy and corticobasal degeneration. Brain Pathol 11(2), 144–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrer I, Gomez-Isla T, Puig B, Freixes M, Ribe E, Dalfo E, Avila J 2005. Current advances on different kinases involved in tau phosphorylation, and implications in Alzheimer’s disease and tauopathies. Curr Alzheimer Res 2(1), 3–18. [DOI] [PubMed] [Google Scholar]
- Ghoneim MM, Hinrichs JV, Mewaldt SP 1984a. Dose-response analysis of the behavioral effects of diazepam: I. Learning and memory. Psychopharmacology 82(4), 291–5. [DOI] [PubMed] [Google Scholar]
- Ghoneim MM, Mewaldt SP 1990. Benzodiazepines and human memory: a review. Anesthesiology 72(5), 926–38. [PubMed] [Google Scholar]
- Ghoneim MM, Mewaldt SP, Hinrichs JV 1984b. Dose-response analysis of the behavioral effects of diazepam: II. Psychomotor performance, cognition and mood. Psychopharmacology 82(4), 296–300. [DOI] [PubMed] [Google Scholar]
- Goedert M 1993. Tau protein and the neurofibrillary pathology of Alzheimer’s disease. Trends Neurosci 16(11), 460–5. [DOI] [PubMed] [Google Scholar]
- Goedert M, Jakes R, Crowther RA, Cohen P, Vanmechelen E, Vandermeeren M, Cras P 1994. Epitope mapping of monoclonal antibodies to the paired helical filaments of Alzheimer’s disease: identification of phosphorylation sites in tau protein. Biochem J 301 (Pt 3)(Pt 3), 871–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong CX, Lidsky T, Wegiel J, Zuck L, Grundke-Iqbal I, Iqbal K 2000. Phosphorylation of microtubule-associated protein tau is regulated by protein phosphatase 2A in mammalian brain. Implications for neurofibrillary degeneration in Alzheimer’s disease. J Biol Chem 275(8), 5535–44. [DOI] [PubMed] [Google Scholar]
- Greenberg SG, Davies P 1990. A preparation of Alzheimer paired helical filaments that displays distinct tau proteins by polyacrylamide gel electrophoresis. Proc Natl Acad Sci U S A 87(15), 5827–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grundke-Iqbal I, Iqbal K, Quinlan M, Tung YC, Zaidi MS, Wisniewski HM 1986a. Microtubule-associated protein tau. A component of Alzheimer paired helical filaments. J Biol Chem 261(13), 6084–9. [PubMed] [Google Scholar]
- Grundke-Iqbal I, Iqbal K, Tung YC, Quinlan M, Wisniewski HM, Binder LI 1986b. Abnormal phosphorylation of the microtubule-associated protein tau (tau) in Alzheimer cytoskeletal pathology. Proc Natl Acad Sci U S A 83(13), 4913–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirshman E, Fisher J, Henthorn T, Arndt J, Passannante A 2003. The effect of midazolam on conscious, controlled processing: evidence from the process-dissociation procedure. Memory & cognition 31(8), 1181–7. [DOI] [PubMed] [Google Scholar]
- Kosik KS, Joachim CL, Selkoe DJ 1986. Microtubule-associated protein tau (tau) is a major antigenic component of paired helical filaments in Alzheimer disease. Proc Natl Acad Sci U S A 83(11), 4044–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Freche H, Brouillette J, Fernandez-Gomez FJ, Patin P, Caillierez R, Zommer N, Sergeant N, Buee-Scherrer V, Lebuffe G, Blum D, Buee L 2012. Tau phosphorylation and sevoflurane anesthesia: an association to postoperative cognitive impairment. Anesthesiology 116(4), 779–87. doi: 10.1097/ALN.0b013e31824be8c7. [DOI] [PubMed] [Google Scholar]
- Lewis J, McGowan E, Rockwood J, Melrose H, Nacharaju P, Van Slegtenhorst M, Gwinn-Hardy K, Paul Murphy M, Baker M, Yu X, Duff K, Hardy J, Corral A, Lin WL, Yen SH, Dickson DW, Davies P, Hutton M 2000. Neurofibrillary tangles, amyotrophy and progressive motor disturbance in mice expressing mutant (P301L) tau protein. Nat Genet 25(4), 402–5. doi: 10.1038/78078. [DOI] [PubMed] [Google Scholar]
- Lister RG 1985. The amnesic action of benzodiazepines in man. Neuroscience and biobehavioral reviews 9(1), 87–94. [DOI] [PubMed] [Google Scholar]
- Maas T, Eidenmuller J, Brandt R 2000. Interaction of tau with the neural membrane cortex is regulated by phosphorylation at sites that are modified in paired helical filaments. J Biol Chem 275(21), 15733–40. [DOI] [PubMed] [Google Scholar]
- Mandelkow EM, Stamer K, Vogel R, Thies E, Mandelkow E 2003. Clogging of axons by tau, inhibition of axonal traffic and starvation of synapses. Neurobiol Aging 24(8), 1079–85. [DOI] [PubMed] [Google Scholar]
- Mi K, Johnson GV 2006. The role of tau phosphorylation in the pathogenesis of Alzheimer’s disease. Curr Alzheimer Res 3(5), 449–63. [DOI] [PubMed] [Google Scholar]
- Morfini G, Pigino G, Beffert U, Busciglio J, Brady ST 2002. Fast axonal transport misregulation and Alzheimer’s disease. Neuromolecular Med 2(2), 89–99. doi: 10.1385/NMM:2:2:089. [DOI] [PubMed] [Google Scholar]
- Nykanen NP, Kysenius K, Sakha P, Tammela P, Huttunen HJ 2012. Gamma-Aminobutyric acid type A (GABAA) receptor activation modulates tau phosphorylation. J Biol Chem 287(9), 6743–52. doi: 10.1074/jbc.M111.309385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Farrell PH 1975. High resolution two-dimensional electrophoresis of proteins. J Biol Chem 250(10), 4007–21. [PMC free article] [PubMed] [Google Scholar]
- Olfson M, King M, Schoenbaum M 2015. Benzodiazepine use in the United States. JAMA Psychiatry 72(2), 136–42. doi: 10.1001/jamapsychiatry.2014.1763. [DOI] [PubMed] [Google Scholar]
- Petry FR, Pelletier J, Bretteville A, Morin F, Calon F, Hebert SS, Whittington RA, Planel E 2014. Specificity of anti-tau antibodies when analyzing mice models of Alzheimer’s disease: problems and solutions. PLoS ONE 9(5), e94251. doi: 10.1371/journal.pone.0094251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Planel E, Bretteville A, Liu L, Virag L, Du AL, Yu WH, Dickson DW, Whittington RA, Duff KE 2009. Acceleration and persistence of neurofibrillary pathology in a mouse model of tauopathy following anesthesia. FASEB J 23(8), 2595–604. doi: 10.1096/fj.08-122424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Planel E, Krishnamurthy P, Miyasaka T, Liu L, Herman M, Kumar A, Bretteville A, Figueroa HY, Yu WH, Whittington RA, Davies P, Takashima A, Nixon RA, Duff KE 2008. Anesthesia-induced hyperphosphorylation detaches 3-repeat tau from microtubules without affecting their stability in vivo. J Neurosci 28(48), 12798–807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Planel E, Richter KEG, Nolan CE, Finley JE, Liu L, Wen Y, Krishnamurthy P, Herman M, Wang L, Schachter JB, Nelson RB, Lau L-F, Duff KE 2007. Anesthesia leads to tau hyperphosphorylation through inhibition of phosphatase activity by hypothermia. J Neurosci 27(12), 3090–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Planel E, Sun X, Takashima A 2002. Role of GSK-3 beta in Alzheimer’s disease pathology. Drug Development Research 56(3), 491–510. [Google Scholar]
- Rhoney DH, Murry KR 2003. National survey of the use of sedating drugs, neuromuscular blocking agents, and reversal agents in the intensive care unit. Journal of intensive care medicine 18(3), 139–45. doi: 10.1177/0885066603251200. [DOI] [PubMed] [Google Scholar]
- Riker RR, Shehabi Y, Bokesch PM, Ceraso D, Wisemandle W, Koura F, Whitten P, Margolis BD, Byrne DW, Ely EW, Rocha MG, Group SS 2009. Dexmedetomidine vs midazolam for sedation of critically ill patients: a randomized trial. JAMA 301(5), 489–99. doi: 10.1001/jama.2009.56. [DOI] [PubMed] [Google Scholar]
- Run X, Liang Z, Zhang L, Iqbal K, Grundke-Iqbal I, Gong CX 2009. Anesthesia induces phosphorylation of tau. J Alzheimers Dis 16(3), 619–26. doi: 10.3233/JAD-2009-1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santana Santos F, Wahlund LO, Varli F, Tadeu Velasco I, Eriksdotter Jonhagen M 2005. Incidence, clinical features and subtypes of delirium in elderly patients treated for hip fractures. Dement Geriatr Cogn Disord 20(4), 231–7. doi: 10.1159/000087311. [DOI] [PubMed] [Google Scholar]
- Shibasaki M, Masukawa D, Ishii K, Yamagishi Y, Mori T, Suzuki T 2013. Involvement of the K+-Cl- co-transporter KCC2 in the sensitization to morphine-induced hyperlocomotion under chronic treatment with zolpidem in the mesolimbic system. J Neurochem 125(5), 747–55. doi: 10.1111/jnc.12258. [DOI] [PubMed] [Google Scholar]
- Sigel E, Baur R 1988. Allosteric modulation by benzodiazepine receptor ligands of the GABAA receptor channel expressed in Xenopus oocytes. J Neurosci 8(1), 289–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith KD, Kallhoff V, Zheng H, Pautler RG 2007. In vivo axonal transport rates decrease in a mouse model of Alzheimer’s disease. Neuroimage 35(4), 1401–8. doi: 10.1016/j.neuroimage.2007.01.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith PK, Krohn RI, Hermanson GT, Mallia AK, Gartner FH, Provenzano MD, Fujimoto EK, Goeke NM, Olson BJ, Klenk DC 1985. Measurement of protein using bicinchoninic acid. Anal Biochem 150(1), 76–85. [DOI] [PubMed] [Google Scholar]
- Spence J, Belley-Cote E, Devereaux PJ, Whitlock R, Um K, McClure G, Lamy A, LeManach Y, Connolly S, Syed S 2018. Benzodiazepine administration during adult cardiac surgery: a survey of current practice among Canadian anesthesiologists working in academic centres. Can J Anaesth 65(3), 263–71. doi: 10.1007/s12630-017-1047-1. [DOI] [PubMed] [Google Scholar]
- Tao G, Zhang J, Zhang L, Dong Y, Yu B, Crosby G, Culley DJ, Zhang Y, Xie Z 2014. Sevoflurane induces tau phosphorylation and glycogen synthase kinase 3beta activation in young mice. Anesthesiology 121(3), 510–27. doi: 10.1097/ALN.0000000000000278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian Q, Wang J 2002. Role of serine/threonine protein phosphatase in Alzheimer’s disease. Neurosignals 11(5), 262–9. doi: 10.1159/000067425. [DOI] [PubMed] [Google Scholar]
- Timic T, Joksimovic S, Milic M, Divljakovic J, Batinic B, Savic MM 2013. Midazolam impairs acquisition and retrieval, but not consolidation of reference memory in the Morris water maze. Behav Brain Res 241, 198–205. doi: 10.1016/j.bbr.2012.12.014. [DOI] [PubMed] [Google Scholar]
- Valentim AM, Olsson IA, Antunes LM 2013a. The anaesthetic combination of ketamine/midazolam does not alter the acquisition of spatial and motor tasks in adult mice. Lab Anim 47(1), 19–25. doi: 10.1258/la.2012.011179. [DOI] [PubMed] [Google Scholar]
- Valentim AM, Ribeiro PO, Olsson IA, Antunes LM 2013b. The memory stages of a spatial Y-maze task are not affected by a low dose of ketamine/midazolam. Eur J Pharmacol 712(1-3), 39–47. doi: 10.1016/j.ejphar.2013.04.027. [DOI] [PubMed] [Google Scholar]
- Weingarten MD, Lockwood AH, Hwo SY, Kirschner MW 1975. A protein factor essential for microtubule assembly. Proc Natl Acad Sci U S A 72(5), 1858–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whittington RA, Virag L, Gratuze M, Petry FR, Noel A, Poitras I, Truchetti G, Marcouiller F, Papon MA, El Khoury N, Wong K, Bretteville A, Morin F, Planel E 2015. Dexmedetomidine increases tau phosphorylation under normothermic conditions in vivo and in vitro. Neurobiol Aging 36(8), 2414–28. doi: 10.1016/j.neurobiolaging.2015.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whittington RA, Virag L, Marcouiller F, Papon MA, El Khoury NB, Julien C, Morin F, Emala CW, Planel E 2011. Propofol directly increases tau phosphorylation. PLoS ONE 6(1), e16648. doi: 10.1371/journal.pone.0016648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood JG, Mirra SS, Pollock NJ, Binder LI 1986. Neurofibrillary tangles of Alzheimer disease share antigenic determinants with the axonal microtubule-associated protein tau (tau) [published erratum appears in Proc Natl Acad Sci U S A 1986 Dec;83(24):9773]. Proc Natl Acad Sci U S A 83(11), 4040–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, Chen K, Ni H, Zhang X, Fan H 2017. Sedation of mechanically ventilated adults in intensive care unit: a network meta-analysis. Sci Rep 7, 44979. doi: 10.1038/srep44979. [DOI] [PMC free article] [PubMed] [Google Scholar]