
Figure 7.
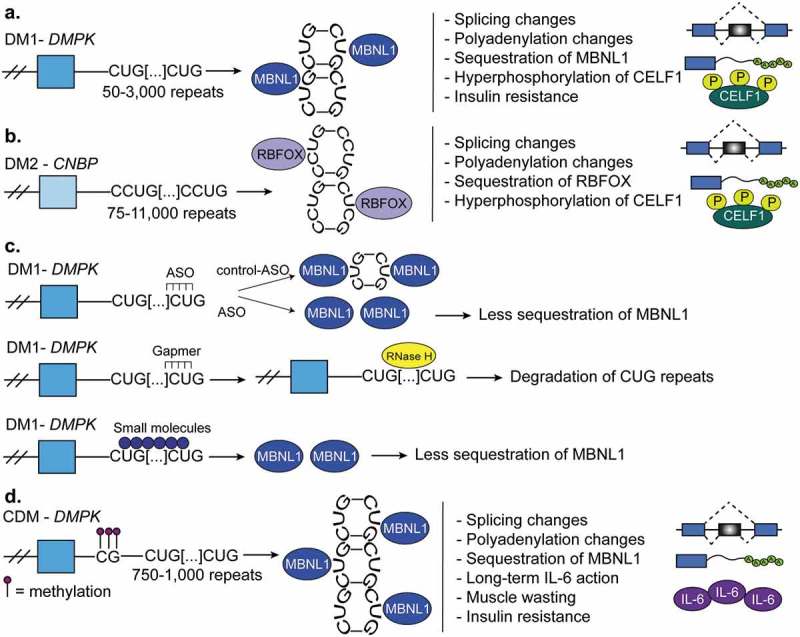
Myotonic dystrophy. (a). Myotonic dystrophy type-1 (DM1) is characterized by 50–3,000 CUG-repeats in the 3ʹUTR of DM1 protein kinase (DMPK) gene. The CUG-repeats cause RNA hairpins that sequester muscleblind like protein-1 (MBNL1) which leads to global changes in alternative splicing, polyadenylation, and mRNA stability. In DM1, the CUGBP Elav-like family member-1 (CELF1) is hyperphosphorylated and this leads to its gain-of-function. (b). Myotonic dystrophy type-2 (DM2) is characterized by 75–11,000 CCUG-repeats in the intron 1 of the CCHC-type zinc finger nucleic acid binding protein (CNBP, also known as ZNF9) gene. CCUG-hairpins sequester RNA binding fox-1 homolog proteins (RBFOX) which leads to extensive changes in alternative splicing and polyadenylation. CELF1 also becomes highly expressed and hyperphosphorylated in DM2. (c). Therapies for DM1. First, antisense oligonucleotides (ASOs) are used to block the transcription of portions CUG-repeats and in this manner MBNL sequestration is corrected. Second, gapmers that bind upstream of the CUG-repeats recruit RNAse-H which degrades the CUG-repeats and corrects the molecular defects of the disease. A third therapy is not RNA-based and instead uses various small molecules that block MBNL1 sequestration. This prevents the global misregulation of alternative splicing programs that occurs in DM1. (d). Congenital myotonic dystrophy (CDM) is characterized by 750–1,000 CUG-repeats at the 3ʹUTR of DMPK and hypermethylation of CpG islands upstream of the repeats. This results in sequestration of MBNL proteins as in DM1 and therefore, misregulation of alternative splicing, polyadenylation, and mRNA stability. Severe CDM results in muscle immaturity due to the improper activation of the cytokine, IL-6, an inhibitor of myoblast differentiation.