

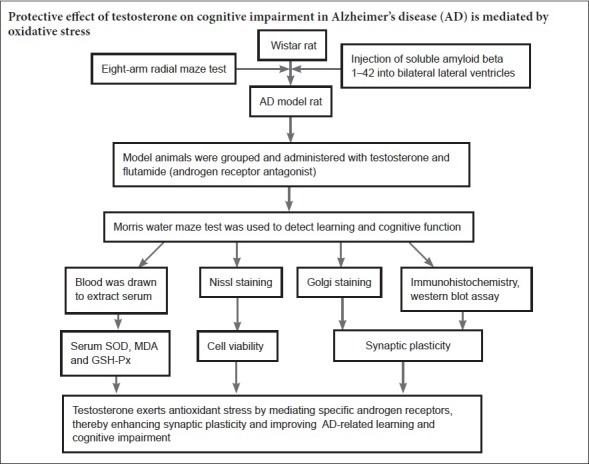
Keywords: nerve regeneration, Alzheimer's disease, testosterone, cognitive dysfunction, synaptic plasticity, free radicals, Morris water maze, androgen receptor, flutamide, postsynaptic density protein 95, amyloid beta 1–42, neurodegenerative change, neural regeneration
Abstract
Cognitive dysfunction in Alzheimer’s disease is strongly associated with a reduction in synaptic plasticity, which may be induced by oxidative stress. Testosterone is beneficial in learning and memory, although the underlying protective mechanism of testosterone on cognitive performance remains unclear. This study explored the protective mechanism of a subcutaneous injection of 0.75 mg testosterone on cognitive dysfunction induced by bilateral injections of amyloid beta 1–42 oligomers into the lateral ventricles of male rats. Morris water maze test results demonstrated that testosterone treatment remarkably reduced escape latency and path length in Alzheimer’s disease rat models. During probe trials, testosterone administration significantly elevated the percentage of time spent in the target quadrant and the number of platform crossings. However, flutamide, an androgen receptor antagonist, inhibited the protective effect of testosterone on cognitive performance in Alzheimer’s disease rat models. Nissl staining, immunohistochemistry, western blot assay, and enzyme-linked immunosorbent assay results showed that the number of intact hippocampal pyramidal cells, the dendritic spine density in the hippocampal CA1 region, the immune response and expression level of postsynaptic density protein 95 in the hippocampus, and the activities of superoxide dismutase and glutathione peroxidase were increased with testosterone treatment. In contrast, testosterone treatment reduced malondialdehyde levels. Flutamide inhibited the effects of testosterone on all of these indicators. Our data showed that the protective effect of testosterone on cognitive dysfunction in Alzheimer’s disease is mediated via androgen receptors to scavenge free radicals, thereby enhancing synaptic plasticity.
Chinese Library Classification No. R459.1; R749
Introduction
Alzheimer’s disease (AD), the most common dementia in the older adult population (Yang et al., 2016), is characterized pathologically by extracellular senile plaques composed of amyloid beta peptide (Aβ), as well as intracellular neurofibrillary tangles and synaptic loss (Selkoe, 2011; Jia et al., 2013; Cherry et al., 2014). AD is a neurodegenerative disorder that results in progressive cognitive dysfunction and neuronal death. It places a large burden on families of patients, and is an increasingly important public health problem worldwide due to an aging population.
Aβ oligomers are recognized as the primary neurotoxic agents in AD and are associated with cognitive impairment and neuronal loss, which, combined with impaired brain energy metabolism and oxidative stress, are implicated in cognitive decline in AD (Rege et al., 2015; Wang et al., 2016). Furthermore, the pathological accumulation of extracellular Aβ and intracellular hyperphosphorylated tau in the brain is hypothesized to promote inflammation, oxidative stress, and neuronal loss in this disease (Asih et al., 2017; Song et al., 2017). Synaptic impairment is an early event leading to cognitive dysfunction in AD; most oxidative stress localizes to the synapse, and synaptic loss is the basis of cognitive decline in AD (Wang et al., 2016). It has been reported that resveratrol is neuroprotective against Aβ-induced oxidative stress and memory loss (Wang et al., 2016).
It is known that a decline in serum testosterone levels is a risk factor for AD (Rosario et al., 2008), and that a reduction in serum testosterone levels is associated with pathogenesis in male patients with AD. Furthermore, Holland et al. (2011) reported that serum testosterone levels of male patients with AD were much lower than in healthy subjects, and that cognitive dysfunction was ameliorated following testosterone replacement therapy. Moreover, several studies have demonstrated a positive correlation between serum testosterone levels and cognitive performance (Jia et al., 2013, 2015). Studies have attested to the neuroprotective effects of sex hormones in animal models of AD, but clinical trial data remain controversial (Grimm et al., 2016).
Furthermore, sex hormones are considered to be associated with the regulation of synaptic plasticity (Mukai et al., 2010; Jia et al., 2015, 2016; Huo et al., 2016). Several studies have indicated that androgen reverses the reduction in hippocampal dendritic spine density and the enhancement of abnormal ultrastructure in excitatory synapses, as well as the expression of synaptic marker proteins following castration, suggesting that androgen exerts an improvement on synaptic plasticity (Li et al., 2013; Pan et al. 2016). Jia et al. (2015) reported that testosterone dramatically increases the expression levels of synaptophysin, which is a known biomarker of synaptogenesis that indicates the amount, density, and distribution of synapses. In addition, Jia et al. (2016) demonstrated that testosterone improves cognitive performance and synaptic plasticity in male AD mice by mediating specific androgen receptors, and that these protective effects of testosterone were blocked by flutamide, a specific androgen-receptor antagonist. Testosterone may improve cognitive dysfunction in AD patients, but the underlying mechanisms of androgenic action on cognitive dysfunction remain unclear.
Based on the previous observations, in this study we used an AD rat model, induced by bilaterally injecting Aβ1–42 oligomers into the lateral ventricles. Our aim was to determine whether the potential protective mechanisms attributed to testosterone’s effects on cognitive dysfunction are associated with synaptic plasticity and oxidative stress, and whether flutamide is effective in blocking these effects of testosterone.
Materials and Methods
Animals
Forty male 3-month-old Wistar rats (SCXK [Meng] 2016-0001) weighing 280–300 g were provided by the Laboratory Animal Center of Inner Mongolia University, China. The experimental procedures followed the United States National Institutes of Health Guide for the Care and Use of Laboratory Animals (NIH Publication No 85-23, revised 1985). All animal protocols used were approved by the Institutional Animal Care and Use Committee, Baotou Medical College, China (approval No. 2018-009).
AD modeling
Rats were anesthetized by intraperitoneal injection of 2% pentobarbital sodium (Yimin Pharmaceutical Co., Beijing, China). Guide cannulae were implanted in the bilateral ventricles and attached to the skull surface using jeweler’s screws and dental cement. Based on the atlas of the rat brain (Paxinos and Watson, 1986), stereotaxic coordinates were as follows: anterior–posterior, 0.8 mm from bregma; medial–lateral, ± 1.5 mm from the midline; and dorsal–ventral, 3.7 mm from the skull surface. After 7 days, soluble Aβ1–42 oligomers (St. Louis, MO, USA; CAT#A4559) were injected into the bilateral ventricles through the guide cannulae, using injection needles connected by polyethylene tubing to 5 μL Hamilton microsyringes. Five μL of Aβ1–42 (2 μg/μL) was stereotaxically injected into the bilateral ventricles at a rate of 1 μL/min. After 7 days of once-daily Aβ1–42 injections, the eight-arm maze test was conducted to evaluate the cognitive performance of AD model rats.
Animal group assignment
Forty AD model rats were randomly divided into four groups (n = 10): AD, testosterone (CAS#58-22-0; Proteintech, Chicago, IL, USA), flutamide (CAT#13311-84-7; Proteintech), and flutamide + testosterone. Rats in the testosterone group were subcutaneously injected in the back daily, 1 hour before the Morris water maze test, with 0.75 mg testosterone that had been dissolved in 0.1 mL of sesame oil, for 2 days. Daily injections were also administered during the 6 days of Morris water maze testing, thus a total of eight injections were given. The administered dose of testosterone was as previously described by Jia et al. (2013). Rats in the flutamide group were injected once daily, 1 hour before Morris water maze testing, with 5 μg of flutamide over 5 minutes (1 μg/min) into the bilateral ventricles, for a total of six injections. In the flutamide + testosterone group, testosterone was subcutaneously injected while flutamide was administered into the bilateral ventricles to prevent any interaction of testosterone and flutamide at the same dosing site. Rats were pre-injected with 5 μg flutamide into each of the bilateral ventricles over a 1-minute period, and the needles were left in place for an additional 60 seconds to allow for diffusion of the solution away from the needle tip. All flutamide injections were given daily, 1 hour before testing, beginning 2 days prior to the Morris water maze test experiments. Rats in the AD group were given daily subcutaneous injections of 0.1 mL of sesame oil in the back, 1 hour before testing, beginning 2 days prior to the Morris water maze test experiments, for a total of eight injections.
Eight-arm radial maze test
The test apparatus was as described by Tadatsugu et al. (2008). The procedure was as follows: Rats were familiarized with the eight-arm radial maze (arm width: 12 cm, height: 50 cm, length: 60 cm, diameter of platform: 30 cm; Neuroscience, Inc., Osaka, Japan). Rats were acclimated daily for 2 days prior to training. On the first day, food pellets (Bio-Serv: A Holton Industries, Frenchtown, NJ, USA) were scattered over the maze surface. Four rats were simultaneously placed on the eight-arm radial maze and freely permitted to take pellets. The second day, one pellet was placed in each of eight arms, and one rat explored freely until all pellets were taken. All animals were trained with one trial per day after acclimation. Four arms were baited in each trial, and the test sequence was unchanged throughout the procedure. One rat was put on the central platform, which was closed off by a door, and after 30 seconds the door was opened. The rat made arm choices to obtain pellets until all four food pellets had been eaten or 10 minutes had elapsed. The number of entries into unbaited arms was scored as the total error, and rats were trained continually until reaching a level of, at most, one error/trial for five successive trials. The first entry into a never-baited arm was recorded as a reference memory error, while re-entry into an arm where the food pellet had already been eaten was recorded as a working memory error.
Morris water maze test
Morris water maze testing began on the third day of testosterone administration, as described previously (Jia et al., 2013). The water maze (WinFast PVR, Jiliang, Nanjing, China) consisted of a circular pool (60 cm in height, 180 cm in diameter) filled with opaque water (24 ± 1°C) to a depth of 35 cm. The circular pool was divided into four quadrants with four equidistant release points around the edge. A round escape platform (10 cm in diameter) was placed 1 cm under the water surface, in the center of the fourth quadrant. For the positioning navigation of the Morris water maze test, each rat received four trials per day for 5 consecutive days (maximal swimming time 60 seconds; 10 seconds on platform; inter-trial interval 30 minutes). The maze tests began at 13:00 each day. For each trial, the rat was placed in the water facing the pool wall at one of four start quadrant points. The time taken for the rat to find the hidden platform, defined as the escape latency, was recorded and used as a measure of spatial learning. The initial position was varied in a quasi-random fashion so that the animal started from each location once, and never started from the same place on any two consecutive trials. Once the platform was found, the rat was allowed to stay on the platform for 10 seconds. If the rat did not find the platform within 60 seconds, it was placed on the platform for 10 seconds, and the escape latency was recorded as 60 seconds. The escape latency, path length, and mean swim speed were measured in the navigation test. For the probe trials of the Morris water maze test, on day 6, the platform was removed, and the rat was allowed to swim freely for 120 seconds. The time spent in the target quadrant and the number of platform crossings was measured. When the maze test was finished, rats were taken out of the pool and dried with a towel to prevent hypothermia. The experimental protocol is provided in detail in Jia et al. (2013).
Preparation of tissue and histological staining
After the final day of the Morris water maze test, all rats were anesthetized with 2% pentobarbital sodium. The whole brain was removed from half of the rats in each group and stored for western blot assay and enzyme linked immunosorbent assay (ELISA). The remaining rats were perfused with saline before the whole brain was extracted. One side of the brain was fixed with 4% paraformaldehyde, dehydrated in alcohol, cleared in xylene, embedded in wax, and sectioned in the coronal plane into 5 μm slices for Nissl staining, as described by Huo et al. (2016). Five sections were selected at random from the hippocampal CA1 regions in each group to calculate the number of intact pyramidal cells within a 1 mm length of hippocampus.
The other half of the brain was processed for Golgi staining and immunohistochemistry, as described by Li et al. (2013), and was rapidly frozen and sliced into 100 μm coronal sections on a cryostat. Tissue was processed for Golgi–Cox staining using a Rapid Golgi Stain Kit (FD Neuro-Technologies, Inc., Ellicott City, MD, USA), as described by Li et al. (2013). Briefly, sections were mounted onto gelatin-coated slides, infiltrated in the solution mixture for 10 minutes, dehydrated in ethanol, cleared in xylene, and coverslipped using a resinous mounting medium. Representative neurons from three sections were selected at random and analyzed for each animal. Secondary and tertiary apical dendrites were selected for quantitative analysis, in which 10 μm segments were magnified at 1000× in digitized images using an optical microscope (Olympus, Tokyo, Japan). The density values of dendritic spines were expressed as the number of thorns per 10 μm of dendrite, as previously described (Jia et al., 2016).
Immunohistochemistry was performed as described by Sonmez et al. (2015). Sections were deparaffinized at 65°C overnight and treated with xylene for 30 minutes. Sections were rehydrated in a series of decreasing amounts of ethanol. After washing with distilled water and phosphate-buffered saline (PBS) for 10 minutes, sections were treated with 3% H2O2 solution for 10 minutes to inhibit endogenous peroxidase activity. Sections were incubated overnight at 4°C in primary rabbit polyclonal antibody against postsynaptic density protein 95 (PSD-95) (1:100; CAT#60291-1-1g; Proteintech). After washing three times with PBS, the secondary goat anti-rabbit antibody biotinylated IgG (CAT#AR1022; Boster, Wuhan, China) and streptavidin-peroxidase conjugate were added and incubated for 30 minutes at room temperature. Then the 3, 3′-diaminobenzidine tetrahydrochloride substrate was added to each section and observed after 2 minutes. Sections were washed with distilled water and covered with mounting media.
Western blot assay
Western blot assays were conducted as previously described by Jia et al. (2016). Proteins were transferred from gels to polyvinylidene difluoride membranes by electroblotting. The membranes were reacted with PSD-95 rabbit polyclonal antibody (1:2000; CAT#60291-1-1g; Proteintech) at room temperature for 12 hours, then incubated with secondary goat anti-rabbit polyclonal antibody at room temperature for 1 hour (1:2000; CAT#60291-1-1g; Proteintech). The intensities of the immunoreactive bands were quantified by Quantity One software (Bio-Rad, Hercules, CA, USA). The grayscale densitometric scanning ratio of PSD-95 to GAPDH (1:1000; CAT#23660-1-AP; Boster, Wuhan, China) was calculated. Half of the rats in each group were used for western blot assays, and hippocampal protein from each rat was used in three different experiments (n = 15).
ELISA
Brain tissues were homogenized and centrifuged, and serum samples were thawed to room temperature. An ELISA kit (CAT#10269-1-AP; Lianshuo, Shanghai, China) was used according to the manufacturer’s instructions to measure serum activity of superoxide dismutase (SOD) (CAT#10269-1-AP; Lianshuo) and glutathione peroxidase (GSH-Px) (CAT#10274-1-AP; Lianshuo), and malondialdehyde (MDA) (CAT#11306-1-AP; Lianshuo) content.
Statistical analysis
All data were analyzed using SPSS 17.0 software (SPSS, Chicago, IL, USA), and presented as the mean ± SD. Multiple comparisons were conducted to determine significant differences using a one-way analysis of variance followed by the least significant difference post hoc test. P < 0.05 was considered statistically significant.
Results
Effects of testosterone on cognitive dysfunction in AD ratmodels in the Morris water maze test
In the positioning navigation test, there were significant differences in cognitive performance in the Morris water maze test between AD and testosterone groups (P < 0.05). Administration of testosterone significantly decreased the escape latency and path length (all P < 0.05; Figure 1), while there were no significant differences between the AD, flutamide, and flutamide + testosterone groups. In addition, there were no significant differences between any groups in the mean swim speed (data not shown).
Figure 1.

Effects of testosterone on cognitive functions of AD rat models in positioning navigation of the Morris water maze test.
(A) Escape latency is given in seconds and (B) path length in meters. Data are expressed as the mean (n = 10; one-way analysis of variance followed by the least significant difference post hoc test). *P < 0.05, vs. AD group. AD: AD group; T: testosterone group; F: flutamide group; F + T: flutamide + testosterone group. AD: Alzheimer’s disease.
In probe trials, testosterone administration significantly increased the number of platform crossings and the percentage of time spent in the target quadrant (all P < 0.05; Figure 2). Flutamide administration alone did not notably influence the probe trial parameters, and when flutamide and testosterone were administered concurrently, the effects of testosterone on the probe trial parameters were inhibited. Thus, flutamide itself did not play a role in cognitive performance in AD model rats, but it could inhibit the effects of testosterone on these abilities in the Morris water maze test.
Figure 2.

Effects of testosterone on cognitive functions of AD rat models in probe trials of the Morris water maze test.
(A, B) Data are presented as (A) percentage time spent in the target quadrant and (B) number of platform crossings in the probe trials. Data are expressed as the mean ± SD (n = 10; one-way analysis of variance followed by the least significant difference post hoc test). *P < 0.05, vs. AD group. AD: AD group; T: testosterone group; F: flutamide group; F + T: flutamide + testosterone group. AD: Alzheimer’s disease.
Effects of testosterone on the number of intact pyramidal cells in the hippocampal CA1 region in AD rat models
There was a significant difference between the AD and testosterone groups in the number of intact pyramidal cells in the hippocampal CA1 region (P < 0.05). A significant elevation in the number of intact hippocampal pyramidal cells was observed in the testosterone group using Nissl staining (Table 1 and Figure 3). Administration of flutamide combined with testosterone (Figure 3D) resulted in a marked decrease in the number of intact hippocampal pyramidal cells compared with testosterone alone (Figure 3B), while no significant differences were found between the AD (Figure 3A) and flutamide groups (Figure 3C).
Table 1.
Number of intact pyramidal cells and dendritic spines from secondary and tertiary apical dendrites in the hippocampal CA1 region, and levels of MDA, SOD, and GSH-Px in different groups
| AD | T | F | F + T | |
|---|---|---|---|---|
| Number of intact pyramidal cells (Nissl) | 38.3±5.75 | 74.6±9.30* | 41.43±6.10 | 53.2±8.15 |
| Number of dendritic spines (Golgi) | 1.35±0.18 | 2.04±0.26* | 1.41±0.21 | 1.62±0.22 |
| SOD (IU/mL) | 175.36±13.45 | 230.22±20.06* | 181.70±16.43 | 194.06±15.28 |
| MDA (nM) | 3.10±0.39 | 1.92±0.32* | 3.09±0.41 | 2.89±0.30 |
| GSH-Px (nM) | 35.06±3.95 | 53.64±5.28* | 38.85±3.82 | 40.16±4.05 |
Data are expressed as the mean ± SD (n = 10; one-way analysis of variance followed by the least significant difference post hoc test). *P < 0.05, vs. AD group. AD: AD group; T: testosterone group; F: flutamide group; F + T: flutamide + testosterone group. AD: Alzheimer’s disease; SOD: superoxide dismutase; MDA: malondialdehyde; GSH-Px: glutathione peroxidase.
Figure 3.
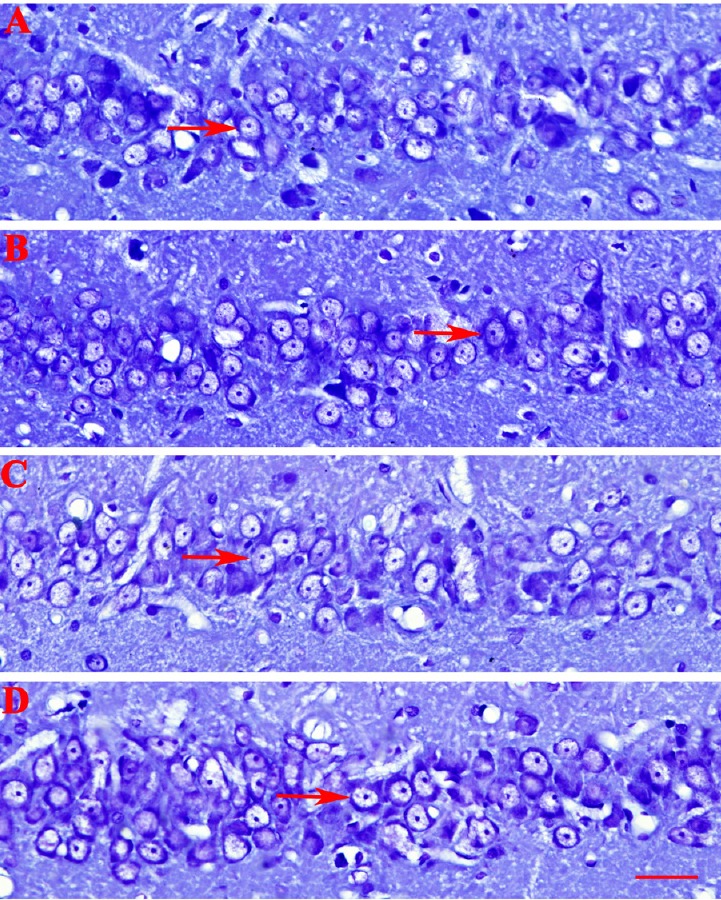
Nissl staining to investigate the number of intact pyramidal cells in different groups.
(A–D) Alzheimer’s disease, testosterone, flutamide, and flutamide + testosterone groups, respectively. Administration of testosterone elevated the number of intact hippocampal pyramidal cells. Administration of flutamide combined with testosterone resulted in a marked decrease in the number of intact hippocampal pyramidal cells compared with testosterone alone. The number of intact pyramidal cells (arrows; per 1 mm linear length of the same section of the hippocampal CA1 region) was counted under an optical microscope (original magnification, 100×; scale bar: 40 μm).
Effects of testosterone on dendritic spine density in the hippocampal CA1 region in AD rat models
Coronal sections in all groups were stained with Golgi–Cox solution. Neurons impregnated with Golgi–Cox solution clearly displayed dendritic spines (Figure 4). The secondary and tertiary apical dendrites in the hippocampal CA1 region were counted in digitized images (Table 1). The dendritic spine density in the AD group was 1.35 ± 0.18 thorns/μm. Treatment with testosterone produced a significant increase in dendritic spine density in the hippocampal CA1 region (2.04 ± 0.26 thorns/μm) compared with the AD group. Administration of flutamide alone did not markedly affect dendritic spine density (1.41 ± 0.21 thorns/μm). Concurrent treatment with testosterone and flutamide resulted in a significant decrease in dendritic spine density (1.62 ± 0.22 thorns/μm) compared with testosterone alone, and was not markedly different from spine density in the AD group.
Figure 4.
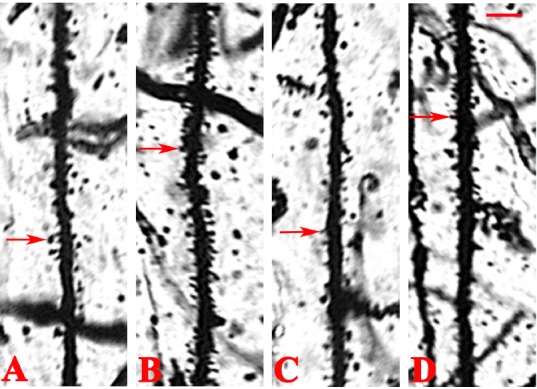
Golgi staining of dendritic spines from secondary and tertiary apical dendrites of the hippocampal CA1 region in different groups.
(A–D) Alzheimer’s disease, testosterone, flutamide, and flutamide + testosterone groups, respectively. Treatment with testosterone increased the dendritic spine density in the hippocampal CA1 region compared with the AD group. Concurrent treatment with testosterone and flutamide decreased dendritic spine density. Dendritic spines (arrows) in different groups were observed under an optical microscope (original magnification, 100×; scale bars: 40 μm).
Effects of testosterone on PSD-95 expression levels in AD rat models
Immunohistochemistry results demonstrated that testosterone significantly increased the number of PSD-95-positive cells compared with the AD group (P < 0.05; Figure 5). There were no obvious differences in immunostaining between the AD, flutamide, and flutamide + testosterone groups.
Figure 5.
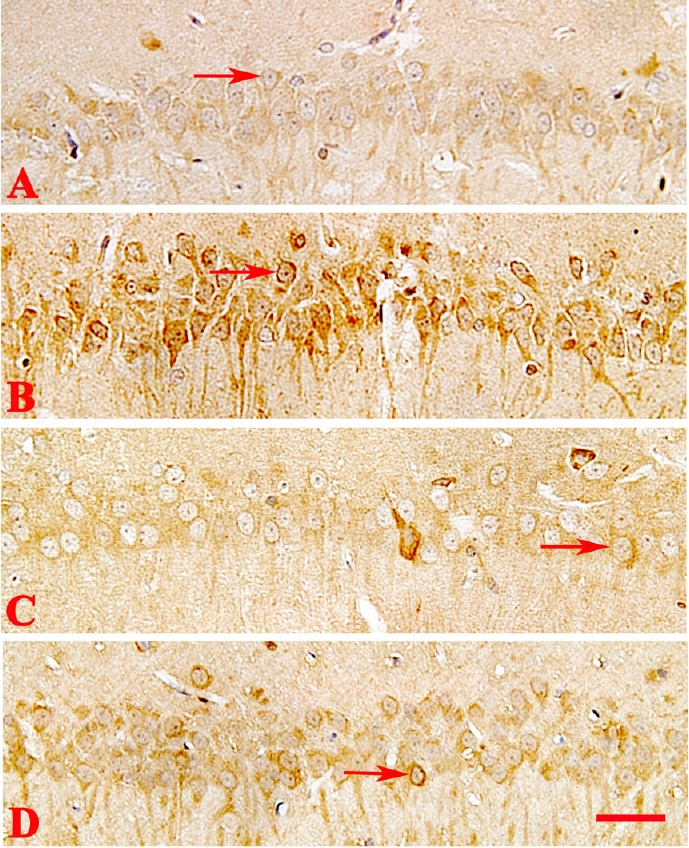
Immunohistochemical staining of postsynaptic density protein 95 (PSD-95) in the rat hippocampus of different groups.
(A–D) Alzheimer’s disease, testosterone, flutamide, and flutamide + testosterone groups, respectively. Testosterone increased the number of PSD-95-positive cells compared with the AD group. Flutamide inhibited the effect of testosterone on PSD-95-stained cells. PSD-95 expression (arrows) was observed using an optical microscope (original magnification, 100×; scale bar: 40 μm).
Western blot assay results showed that testosterone significantly increased PSD-95 expression levels compared with the AD group (P < 0.05; Figure 6). No significant differences in PSD-95 expression levels were detected between the AD, flutamide, and flutamide + testosterone groups. Thus, flutamide administration alone did not notably influence the expression levels of PSD-95. Moreover, when flutamide and testosterone were administered concurrently, the effect of testosterone on PSD-95 expression was inhibited.
Figure 6.
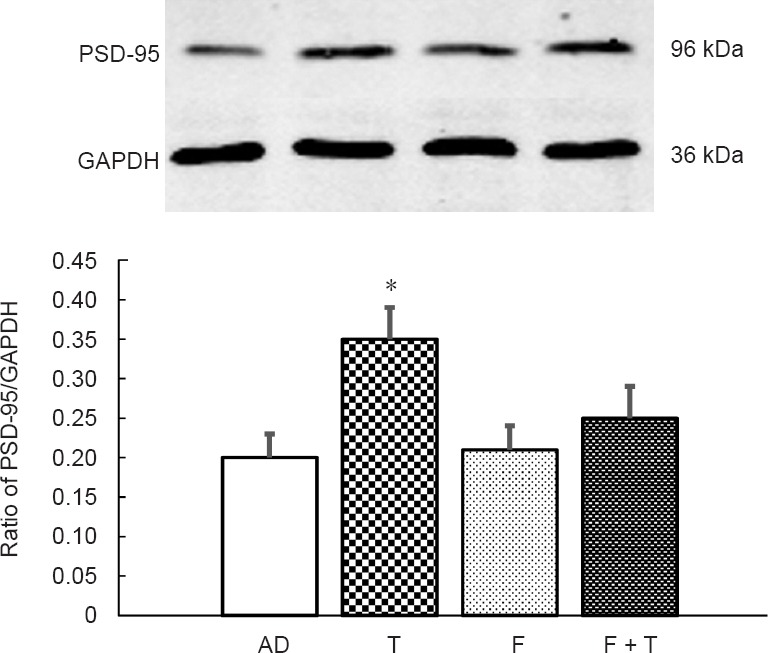
Western blot assay of relative PSD-95 protein expression levels in the rat hippocampus of different groups.
Compared with the AD group, testosterone significantly increased PSD-95 expression levels. When flutamide and testosterone were administered concurrently, the effect of testosterone on PSD-95 expression was inhibited. Expression levels are given as the ratio of PSD-95/GAPDH. Data are expressed as the mean ± SD (n = 15; one-way analysis of variance followed by the least significant difference post hoc test). The experiment was performed in triplicate. *P < 0.05, vs. AD group. AD: AD group; T: testosterone group; F: flutamide group; F + T: flutamide + testosterone group; AD: Alzheimer’s disease; GAPDH: glyceraldehyde phosphate dehydrogenase; PSD-95: postsynaptic density protein 95.
Effects of testosterone on SOD and GSH-Px activities and MDA content in AD rat serum
ELISA results showed that testosterone administration produced significant differences in SOD and GSH-Px activities, as well as MDA content (P < 0.05; Table 1). Testosterone treatment significantly reduced MDA content and increased SOD and GSH-Px activities compared with the AD group. There were no significant differences in MDA content and SOD and GSH-Px activity between the other three groups, indicating that flutamide alone did not obviously affect MDA content or SOD and GSH-Px activity. When flutamide and testosterone were administered concurrently, the effects of testosterone on the MDA content and SOD and GSH-Px activities were inhibited (Table 1).
Discussion
The mechanisms for the pathogenesis of AD are still unclear. Oxidative stress, either because of an overproduction of free radicals or a decrease in free radical scavenging ability, is recognized as a critical component of AD pathogenesis (Aliev et al., 2014). Mitochondrial damage induced by oxidative stress has been implicated as the primary cause of AD (Miwa and Saretzki, 2017). The oxidation or dysfunction of enzymes involved in energy metabolism can decrease glucose metabolism and cause a loss of adenosine triphosphate, which are key events that trigger the progressive neurodegeneration of AD (Tramutola et al., 2017). It has been reported that ammonia has toxic effects on brain metabolism, and there is confirmed ammonia accumulation in the brains of AD patients (Norenberg et al., 2009). Oxidative stress may be a crucial factor in the pathogenesis of ammonia neurotoxicity. The activation of N-methyl-D-aspartate receptors results in an activation of neuronal reactive oxygen species and nitric oxide formation, which disseminates oxidative stress in AD brains (Smith et al., 1997; Yang et al., 2011; Elena et al., 2014).
Furthermore, many researchers believe that Aβ aggregation is the initial cause of oxidative stress. In our study, the bilateral injection of Aβ1–42 oligomers into the ventricles increased the expression levels of oxidative stress biomarkers and induced cognitive dysfunction. Aβ oligomers, the early aggregates of Aβ peptides, have been suggested to cause synaptic plasticity dysfunction in the brains of early AD patients, and an increase in Aβ oligomers induces oxidative stress. Hence, to prevent and treat AD, it may be useful to decrease Aβ accumulation, thereby lowering oxidative stress levels (Ganjei, 2010). Our past results showed that testosterone notably reduces Aβ accumulation (Huo et al., 2016), but whether testosterone decreases Aβ accumulation by inhibiting oxidative stress remains unclear.
There is increasing evidence to suggest that the pathophysiology of AD is associated with inflammatory responses and oxidative stress mediated by microglia, which produce neurotoxic factors such as proinflammatory cytokines and nitric oxide, leading to neuronal degeneration (Chung et al., 2015). Oxidative stress, inflammation, disturbance of intracellular Ca2+ homeostasis, free radical generation in the brain, and excitotoxicity can all lead to neuronal cell death. It can be inferred from several studies that excitotoxicity, free radical generation, and altered synaptic function caused by oxidative stress are associated with AD pathology (Jing et al., 2016; Pradip et al., 2016).
Oxidative stress plays a key role in the pathogenesis of AD. Oxidative damage of macromolecules, involving protein carbonyl content formation, lipid peroxidation, and DNA oxidation, has been reported in AD patients (Rodrigo et al., 2017). Oxygen free radicals can cause cell injury not only through the peroxidation of polyunsaturated fatty acids in biofilms, but also through the metabolites of lipid peroxide (such as MDA); therefore, MDA content reflects the severity of cell injury by free radicals. As an important scavenger of oxygen free radicals, SOD disproportionates the superoxide anion radicals of metabolism and generates hydrogen peroxide, which is catalyzed into water by catalase and GSH-Px, thus protecting organs from free radical attack. The level of GSH-PX activity therefore indirectly reflects the scavenging ability of oxygen free radicals. If the superoxide anion concentration is too high, SOD in the body will be reduced due to excessive consumption, which damages neurons, causing neuronal metabolic disorders, degeneration, and necrosis, leading to cognitive deficits. Moreover, free radicals and unsaturated fatty acids are oxidized and decomposed into various metabolites by lipid peroxidation reactions (Yin et al, 2011). Thus, lipid peroxidation products such as MDA, 4-hydroxynonenal, and acrolein levels are considered to be biological indicators of oxidative stress (Liu et al., 2015).
A previous study (Aybek et al., 2007) showed that, in patients with late onset AD, serum MDA levels are markedly higher than in healthy controls. High MDA levels were proposed to be an essential factor in the pathogenesis and neuronal damage of AD. Our data showed that testosterone significantly reduced MDA content, and increased SOD and GSH-Px activities. Flutamide inhibited all testosterone-mediated effects, indicating that the protective effect of testosterone on cognitive dysfunction was mediated via androgen receptors to scavenge free radicals.
Several studies have indicated a positive correlation between improved cognitive performance and elevated serum testosterone levels (Driscoll et al., 2005; Jia et al., 2013, 2015). It is also been shown that a reduction in serum testosterone levels is a risk factor for AD, while cognitive performance improves after testosterone replacement therapy (Holland et al., 2011; Rosario et al., 2011).
As the neurobiological basis of cognitive function, synapses are involved in cognitive performance (Liu et al. 2008), and morphological and functional abnormalities in synapses can cause cognitive dysfunction. Sex hormones are known to be associated with synaptic plasticity (Mukai et al. 2010). Several researchers have reported that androgen reverses the reduction in hippocampal dendritic spine density that occurs in castrated rats, demonstrating that androgens have a beneficial effect on synaptic plasticity (Leranth et al., 2003; Li et al., 2013). Moreover, Jia et al. (2015) reported that testosterone increases the expression levels of synaptophysin, which is considered a molecular biomarker of synaptogenesis. In addition, Phan et al. (2015) found that elevated estrogen levels induce new synapses and dendrites in brain regions associated with cognitive function.
The postsynaptic structural protein PSD-95 can serve as a marker of cognitive performance, as it is strongly associated with synaptic plasticity and mediates protein–protein interactions in the synaptic membrane that are critical for synaptic transmission (Kim and Sheng, 2004). In our study, testosterone restored cognitive dysfunction and increased dendritic spine density in the hippocampal CA1 region in AD model rats, as well as increasing expression levels of PSD-95. These results demonstrate that testosterone improves synaptic plasticity and ameliorates cognitive dysfunction in AD model rats, which is consistent with previous studies (Candemir et al., 2013; Jia et al., 2013, 2015, 2016; Huo et al., 2016).
Androgens are beneficial in improving neuronal differentiation and maintaining morphology (Leranth et al., 2004; Marron et al., 2005). Furthermore, androgens have been reported to elevate the viability of neurons when the brain undergoes toxic invasion or in the presence of apoptosis (Nunez et al. 2000). These observations are consistent with previous results by Spritzer and Galea (2007), which demonstrated enhanced survival of hippocampal neurons following the administration of testosterone or dihydrotestosterone, but not estrogen, indicating a specific androgenic action.
Testosterone metabolizes dihydrotestosterone and estradiol in vivo, which bind to androgen receptors and estrogen receptors in hippocampal and cortical regions, playing separate roles (Edinger and Frye, 2004; Spritzer et al., 2011). These results suggest that androgens and estrogens may have direct effects on these brain regions that are related to cognitive function (Kerr et al., 1995; Li et al., 1997; Kritzer, 2004). Additionally, dihydrotestosterone, but not estradiol, could restore memory retention in a water maze test in male mice (Benice and Raber, 2009), and Osborne et al. (2009) reported that male rats implanted with 3α-diol, a metabolite of dihydrotestosterone that binds to estrogen receptors, have reduced escape latency in the Morris water maze test.
Our data indicated that testosterone-mediated effects on cognitive function involve interactions between specific androgen receptors and androgens. In this study, the specific androgen-receptor antagonist, flutamide, inhibited testosterone-mediated improvement in cognitive dysfunction in AD model rats, reaffirming the involvement of androgen receptors in the observed responses. These findings indicate that the effects of testosterone on cognitive performance and PSD-95 expression levels are mediated via specific androgen receptors. Taken together, testosterone improved cognitive dysfunction in AD model rats by increasing the number of hippocampal neurons and enhancing synaptic plasticity.
Additional file: Open peer review report 1 (80.8KB, pdf) .
Footnotes
Conflicts of interest: The authors declare that there is no duality of interest associated with this manuscript.
Financial support: This study was supported by the Natural Science Foundation of Inner Mongolia Autonomous Region of China, No. 2017LH0301 (to JXJ), 2016MS08108 (to ZJY); the Science and Technology Planning Project of Inner Mongolia Autonomous Region of China, No. 201602069 (to ZJY); the PhD Scientific Research Fund of Baotou Medical College of China, No. BSJJ201606 (to JXJ); the “Dengfeng Project” Scientific Research Fund of Baotou Medical College of China, No. BYJJ-DF 201703 (to JXJ). Funders had no involvement in the study design; data collection, analysis, and interpretation; paper writing; or decision to submit the paper for publication.
Institutional review board statement: The procedures were approved by Institutional Animal Care and Use Committee, Baotou Medical College, China (approval No. 2018-009).
Copyright license agreement: The Copyright License Agreement has been signed by all authors before publication.
Data sharing statement: Datasets analyzed during the current study are available from the corresponding author on reasonable request.
Plagiarism check: Checked twice by iThenticate.
Peer review: Externally peer reviewed.
Open peer reviewer: Joaquin Martí-clúa, Universidad Autonoma de Barcelona, Spain.
P-Reviewer: Martí-clúa J; C-Editor: Zhao M; S-Editor: Yu J, Wang J, Li CH; L-Editor: Gardner B, Hindle A, Qiu Y, Song LP; T-Editor: Liu XL
Funding: This study was supported by the Natural Science Foundation of Inner Mongolia Autonomous Region of China, No. 2017LH0301 (to JXJ), 2016MS08108 (to ZJY); the Science and Technology Planning Project of Inner Mongolia Autonomous Region of China, No. 201602069 (to ZJY); the PhD Scientific Research Fund of Baotou Medical College of China, No. BSJJ201606 (to JXJ); the “Dengfeng Project” Scientific Research Fund of Baotou Medical College of China, No. BYJJ-DF 201703 (to JXJ).
References
- Aliev G, Priyadarshini M, Reddy VP, Grieg NH, Kaminsky Y, Cacabelos R, Md Ashraf G, Jabir NR, Kamal MA, Nikolenko VN, Zamyatnin AA, Benberin VV, Bachurin SO. Oxidative stress mediated mitochondrial and vascular lesions as markers in the pathogenesis of Alzheimer disease. Curr Med Chem. 2014;21:2208–2217. doi: 10.2174/0929867321666131227161303. [DOI] [PubMed] [Google Scholar]
- Asih PR, Tegg ML, Sohrabi H, Carruthers M, Gandy SE, Saad F, Verdile G, Ittner LM. Martins RN multiple mechanisms linking type 2 diabetes and Alzheimer’s disease: testosterone as a modifier. J Alzheimers Dis. 2017;59:445–466. doi: 10.3233/JAD-161259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aybek H, Ercan F, Aslan D, Sahiner T. Determination of malondialdehyde, reduced glutathione levels and APOE4 allele frequency in late-onset Alzheimer’s disease in Denizli, Turkey. Clin Biochem. 2007;40:172–176. doi: 10.1016/j.clinbiochem.2006.09.005. [DOI] [PubMed] [Google Scholar]
- Benice TS, Raber J. Dihydrotestosterone modulates spatial working-memory performance in male mice. J Neurochem. 2009;110:902–911. doi: 10.1111/j.1471-4159.2009.06183.x. [DOI] [PubMed] [Google Scholar]
- Candemir M, Semiz S, Yonguc GN, Ozdemir MB, Abban-Mete G, Adiguzel E. Effect of testosterone propionate on hippocampal pyramidal neuron number in female rats. Singapore Med J. 2013;54:315–320. doi: 10.11622/smedj.2013124. [DOI] [PubMed] [Google Scholar]
- Cherry JD, Olschowka JA, Banion MK. Neuroinflammation and M2 microglia: the good, the bad, and the inflamed. J Neuroinflamm. 2014;11:1–15. doi: 10.1186/1742-2094-11-98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung WS, Welsh CA, Barres BA, Stevens B. “Do glia drive synaptic and cognitive impairment in disease. ” Nat Neurosci. 2015;18:1539–1545. doi: 10.1038/nn.4142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Driscoll I, Hamilton DA, Yeo RA, Brooks WM, Sutherland RJ. Virtual navigation in humans: the impact of age, sex, and hormones on place learning. Horm Behav. 2005;47:326–335. doi: 10.1016/j.yhbeh.2004.11.013. [DOI] [PubMed] [Google Scholar]
- Edinger KL, Frye CA. Testosterone’s analgesic, anxiolytic, and cognitive enhancing effects may be due in part to actions of its 5a-reduced metabolites in the hippocampus. Behav Neurosci. 2004;118:1352–1364. doi: 10.1037/0735-7044.118.6.1352. [DOI] [PubMed] [Google Scholar]
- Elena AK, Iliya NS, Lyudmila AT, Reddy VP, Aliev G, Kaminsky YG. Pathogenesis of Alzheimer disease: role of oxidative stress, amyloid-β peptides, systemic ammonia and erythrocyte energy metabolism. CNS Neurol Disord Drug Targets. 2014;13:112–119. doi: 10.2174/18715273113126660130. [DOI] [PubMed] [Google Scholar]
- Ganjei JK. Targeting amyloid precursor protein secretases: Alzheimer’s disease and beyond. Drug News Perspect. 2010;23:573–584. doi: 10.1358/dnp.2010.23.9.1507297. [DOI] [PubMed] [Google Scholar]
- Grimm A, Mensah-Nyagan AG, Eckert A. Alzheimer, mitochondria and gender. Neurosci Biobehav Rev. 2016;67:89–101. doi: 10.1016/j.neubiorev.2016.04.012. [DOI] [PubMed] [Google Scholar]
- Holland J, Bandelow S, Hogervorst E. Testosterone levels and cognition in elderly men: a review. Maturitas. 2011;69:322–337. doi: 10.1016/j.maturitas.2011.05.012. [DOI] [PubMed] [Google Scholar]
- Huo DS, Sun JF, Zhang BF, Yan XS, Wang H, Jia JX, Yang ZJ. Protective effects of testosterone on cognitive dysfunction in Alzheimer’s disease model rats induced by oligomeric beta amyloid peptide 1-42. J Toxicol Environ Health A. 2016;79:856–863. doi: 10.1080/15287394.2016.1193114. [DOI] [PubMed] [Google Scholar]
- Jia JX, Cui CL, Yan XS, Zhang BF, Song W, Huo DS, Wang H, Yang ZJ. Effects of testosterone on synaptic plasticity mediated by androgen receptors in male SAMP8 mice. J Toxicol Environ Health A. 2016;79:849–855. doi: 10.1080/15287394.2016.1193113. [DOI] [PubMed] [Google Scholar]
- Jia JX, Cui CL, Song W, Yan XS, Huo DS, Wang H, Yang ZJ. Effects of testosterone treatment on synaptic plasticity and behavior in senescence accelerated mice. J Toxicol Environ Health A. 2015;78:1311–1320. doi: 10.1080/15287394.2015.1085839. [DOI] [PubMed] [Google Scholar]
- Jia JX, Kang L, Li S, Geng D, Fan P, Wang L, Cui HX. Amelioratory effects of testosterone treatment on cognitive performance deficits induced by soluble Aβ1–42 oligomers injected into the hippocampus. Horm Behav. 2013;64:477–486. doi: 10.1016/j.yhbeh.2013.08.002. [DOI] [PubMed] [Google Scholar]
- Jing W, Michael FJ, Yu FX. Glia and TRPM2 channels in plasticity of central nervous system and Alzheimer’s diseases. Neural Plast. 2016;7:1–7. doi: 10.1155/2016/1680905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerr JE, Allore RJ, Beck SG, Handa RJ. Distribution of hormonal regulation of androgen receptor (AR) and AR messenger ribonucleic acid in the rat hippocampus. Endocrinology. 1995;136:3213–3221. doi: 10.1210/endo.136.8.7628354. [DOI] [PubMed] [Google Scholar]
- Kim E, Sheng M. PDZ domain proteins of synapses. Nat Rev Neurosci. 2004;5:771–781. doi: 10.1038/nrn1517. [DOI] [PubMed] [Google Scholar]
- Kritzer M. The distribution of immunoreactivity for intracellular androgen receptors in the cerebral cortex of hormonally intact adult male and female rats: localization in pyramidal neurons making corticocortical connections. Cereb Cortex. 2004;14:268–280. doi: 10.1093/cercor/bhg127. [DOI] [PubMed] [Google Scholar]
- Leranth C, Petnehazy O, MacLusky NJ. Gonadal hormones affect spine synaptic density in the CA1 hippocampal subfield of male rats. J Neurosci. 2003;23:1588–1592. doi: 10.1523/JNEUROSCI.23-05-01588.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leranth C, Hajszan T, MacLusky NJ. Androgens increase spine synapse density in the CA1 hippocampal subfield of ovariectomized female rats. J Neurosci. 2004;24:495–499. doi: 10.1523/JNEUROSCI.4516-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S, Kang L, Zhang C, Xie GS, Li N, Zhang Y, Du J, Cui HX. Effects of dihydrotestosterone on synaptic plasticity of hippocampus in male SAMP8 mice. Exp Gerontol. 2013;48:778–785. doi: 10.1016/j.exger.2013.04.014. [DOI] [PubMed] [Google Scholar]
- Li X, Schwartz PE, Rissman EF. Distribution of estrogen receptor-β-like immunoreactivity in rat forebrain. Neuroendocrinology. 1997;66:63–67. doi: 10.1159/000127221. [DOI] [PubMed] [Google Scholar]
- Liu L, Orozco IJ, Planel E, Wen Y, Bretteville A, Krishnamurthy P, Wang L, Herman M, Figueroa H, Yu WH, Arancio O, Duff K. A transgenic rat that develops Alzheimer’s disease-like amyloid pathology, deficits in synaptic plasticity and cognitive impairment. Neurobiol Dis. 2008;31:46–57. doi: 10.1016/j.nbd.2008.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L, Li J, Zhao F, Wang H, Qu Y, Mu D. Nitric oxide synthase in hypoxic or ischemic brain injury. Rev Neurosci. 2015;26:105–117. doi: 10.1515/revneuro-2014-0041. [DOI] [PubMed] [Google Scholar]
- Marron TU, Guerini V, Rusmini P, Sau D, Brevini TA, Martini L, Poletti A. Androgen-induced neurite outgrowth is mediated by neuritin in motor neurones. J Neurochem. 2005;92:10–20. doi: 10.1111/j.1471-4159.2004.02836.x. [DOI] [PubMed] [Google Scholar]
- Miwa S, Saretzki G. Telomerase and mTOR in the brain: the mitochondria connection. Neural Regen Res. 2017;12:358–361. doi: 10.4103/1673-5374.202922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukai H, Kimoto T, Hojo Y, Kawato S, Murakami G, Higo S, Hatanaka Y, Ogiue-Ikeda M. Modulation of synaptic plasticity by brain estrogen in the hippocampus. Biochim Biophys Acta. 2010;1800:1030–1044. doi: 10.1016/j.bbagen.2009.11.002. [DOI] [PubMed] [Google Scholar]
- Norenberg MD, Rama Rao KV, Jayakumar AR. Signaling factors in the mechanism of ammonia neurotoxicity. Metab Brain Dis. 2009;24:103–117. doi: 10.1007/s11011-008-9113-6. [DOI] [PubMed] [Google Scholar]
- Nunez JL, Jurgens HA, Juraska JM. Androgens reduce cell death in the developing rat visual cortex. Brain Res Dev Brain Res. 2000;125:83–88. doi: 10.1016/s0165-3806(00)00126-7. [DOI] [PubMed] [Google Scholar]
- Osborne DM, Edinger KL, Frye CA. Chronic administration of androgens with actions at estrogen receptor beta have anti-anxiety and cognitive-enhancing effects in male rats. Age. 2009;31:191–198. doi: 10.1007/s11357-009-9114-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan WS, Han S, Kang L, Li S, Du J, Cui HX. Effects of dihydrotestosterone on synaptic plasticity of the hippocampus in mild cognitive impairment male SAMP8 mice. Exp Ther Med. 2016;12:1455–1463. doi: 10.3892/etm.2016.3470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paxinos G, Watson C. The rat brain in stereotaxic coordinates. 2nd edition. Orlando, FL, USA: Academic Press; 1986. [Google Scholar]
- Phan A, Suschkov S, Molinaro L. Rapid increases in immature synapses parallel estrogen-induced hippocampal learning enhancements. Proc Natl Acad Sci U S A. 2015;29:16018–16023. doi: 10.1073/pnas.1522150112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pradip KK, Anuradha K, Shivika R, Supriya S, Santoshkumar T, Chandishwar N, Neetu T. Mechanism of oxidative stress and synapse dysfunction in the pathogenesis of Alzheimer’s disease: understanding the therapeutics strategies. Mol Neurobiol. 2016;53:648–661. doi: 10.1007/s12035-014-9053-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rege SD, Geetha T, Broderick TL, Babu JR. Resveratrol protects β amyloid-induced oxidative damage and memory associated proteins in H19-7 hippocampal neuronal cells. Curr Alzheimer Res. 2015;12:147–156. doi: 10.2174/1567205012666150204130009. [DOI] [PubMed] [Google Scholar]
- Rodrigo GR, Mauricio NM, Karina VS, Daniel AS, Laura MM. Involvement of astrocytes in Alzheimer’s disease from a neuroinflammatory and oxidative stress perspective. Front Mol Neurosci. 2017;10:1–20. doi: 10.3389/fnmol.2017.00427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosario ER, Chang L, Head EH, Stanczyk FZ, Pike CJ. Brain levels of sex steroid hormones in men and women during normal aging and in Alzheimer’s disease. Neurobiol Aging. 2011;32:604–613. doi: 10.1016/j.neurobiolaging.2009.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosario ER, Pike CJ. Androgen regulation of β-amyloid protein and the risk of Alzheimer’s disease. Brain Res Rev. 2008;57:444–453. doi: 10.1016/j.brainresrev.2007.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selkoe DJ. Alzheimer’s disease. Cold Spring Harb Perspect Biol. 2011;3:1–16. doi: 10.1101/cshperspect.a004457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith CC, Smith LA, Bredemann TM, McMahon LL. 17β Estradiol recruits GluN2B-containing NMDARs and ERK during Induction of long-term potentiation at temporoammonic-CA1 synapses. Hippocampus. 2016;26:110–117. doi: 10.1002/hipo.22495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith MA, Richey Harris PL, Sayre L M, Joseph S, Beckman, George Perry. Widespread peroxynitrite mediated damage in Alzheimer’s disease. J Neurosci. 1997;17:2653–2657. doi: 10.1523/JNEUROSCI.17-08-02653.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song L, Li X, Bai XX, Gao J, Wang CY. Calycosin improves cognitive function in a transgenic mouse model of Alzheimer’s disease by activating the protein kinase C pathway. Neural Regen Res. 2017;12:1870–1876. doi: 10.4103/1673-5374.219049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonmez A, Sayın O, Gurgen SG, Calisir M. Neuroprotective effects of MK-801 against traumatic brain injury in immature rats. Neurosci Lett. 2015;597:137–142. doi: 10.1016/j.neulet.2015.05.001. [DOI] [PubMed] [Google Scholar]
- Spritzer MD, Daviau ED, Coneeny MK, Engelman SM, Prince WT, Rodriguez-Wisdom KN. Effects of testosterone on spatial learning and memory in adult male rats. Horm Behav. 2011;59:484–496. doi: 10.1016/j.yhbeh.2011.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spritzer MD, Galea LA. Testosterone and dihydrotestosterone, but not estradiol, enhance survival of new hippocampal neurons in adult male rats. Neurobiology. 2007;67:1321–1333. doi: 10.1002/dneu.20457. [DOI] [PubMed] [Google Scholar]
- Tadatsugu T, Azusa M, Kenichi S, Chiaki K. Participation of the cholinergic system in ameliorating effect of vasopressin fragment by Group I metabotropic glutamate receptor blockade. J Pharmacol Sci. 2008;108:335–340. doi: 10.1254/jphs.08146fp. [DOI] [PubMed] [Google Scholar]
- Tramutola A, Lanzillotta C, Perluigi M, Butterfield DA. Oxidative stress, protein modification and Alzheimer disease. Brain Res Bull. 2017;133:88–96. doi: 10.1016/j.brainresbull.2016.06.005. [DOI] [PubMed] [Google Scholar]
- Wang CY, Wang ZY, Xie JW, Wang T, Wang X, Xu Y, Cai JH. Dl-3-n-butylphthalide-induced upregulation of antioxidant defense is involved in the enhancement of cross talk between CREB and Nrf2 in an Alzheimer’s disease mouse model. Neurobiol Aging. 2016;38:32–46. doi: 10.1016/j.neurobiolaging.2015.10.024. [DOI] [PubMed] [Google Scholar]
- Wang X, Hu X, Yang Y, Takata T, Sakurai T. Nicotinamide mononucleotide protects against β-amyloid oligomer-induced cognitive impairment and neuronal death. Brain Res. 2016;15(1643):1–9. doi: 10.1016/j.brainres.2016.04.060. [DOI] [PubMed] [Google Scholar]
- Yang GF, Ji JG, Peng LW, He SZ. Oxidative proteins in human brains separated and identified by proteomics technology. Zhongguo Zuzhi Gongcheng Yanjiu. 2011;15:1990–1993. [Google Scholar]
- Yin H, Xu L, Porter NA. Free radical lipid peroxidation: mechanisms and analysis. Chem Rev. 2011;111:5944–5972. doi: 10.1021/cr200084z. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Guo JH, Yan HJ, et al. Bone marrow mesenchymal stem cell transplantation improves behavior performance of senile dementia rats. Zhongguo Zuzhi Gongcheng Yanjiu. 2016;20:5371–5377. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.