

Mutations in the parkin gene (PRKN, PARK2) cause autosomal‐recessive early‐onset Parkinson's disease (PD). Reported parkin mutation frequencies vary across studies, but percentages up to 49% of familial and 20% of sporadic cases of early‐onset PD have been reported on.1, 2, 3, 4 It is difficult to clinically distinguish parkin mutations from other causes of PD. Clinical clues for mutations in the parkin gene are symmetrical onset, dystonia, hyperreflexia, slower disease progression, and a good response to levodopa.
In this report, we describe a 35‐year‐old Dutch man with “foot drop dystonia” resulting from a parkin mutation.
Case Description
The patient was referred to our neurology outpatient clinic with a gait disorder. He had progressive difficulties with walking for 6 years. He complained of walking bent over with his legs apart. While walking, it felt like his feet dropped to the floor. His complaints worsened during the day and during exercise. In the beginning, he only experienced this during strenuous effort, especially while playing football. He ran straddle‐legged on the lateral sides of his feet. He recovered in half an hour. At the end, playing football was not possible anymore. He also experienced trembling in his legs during walking and worsening with emotions and exhaustion. His girlfriend noticed a mumbled speech. He reported a restless sleep with flailing and kicking during sleep. Family history was negative for neurological or psychiatric disorders. There was no parental consanguinity. He has one healthy sister and no brothers or children.
Neurological examination on admission showed a monotonous voice, minimal hypomimia, mild general rigidity, and minimal bradykinesia during finger tapping. He walked with exaggerated lifting and subsequently dropping of the feet, slightly with the legs apart, which we classified as a dystonic gait (see Video 1).
Dopa‐responsive dystonia (DRD) was in our differential diagnosis, because of worsening during the day and the combination of foot dystonia with some bradykinesia.5 Importantly, mild parkinsonism may occur in DRD.6 Indeed, l‐dopa/carbidopa 100/25 mg three times daily appeared successful. Brain MRI showed no abnormalities. Cerebrospinal fluid (CSF) analysis showed normal concentrations of biopterin, neopterin, and homovanillic acid (however, during l‐dopa treatment). Genetic tests for mutations in the genes for guanosin triphosphate cyclohydrolase 1 (GCH1, DYT5), sepiapterin reductase (SPR), and tyrosin hydroxylase (TH) were negative. A dopamine transporter imaging with single‐photon emission computed tomography (DAT SPECT) scan showed bilateral and asymmetrical loss of striatal DAT binding with the right striatum showing the largest loss (Figure 1). Finally, multiplex litigation‐dependent probe amplification showed a compound heterozygous parkin mutation (i.e., deletion exons 3–6 and deletion exon 3).
Figure 1.
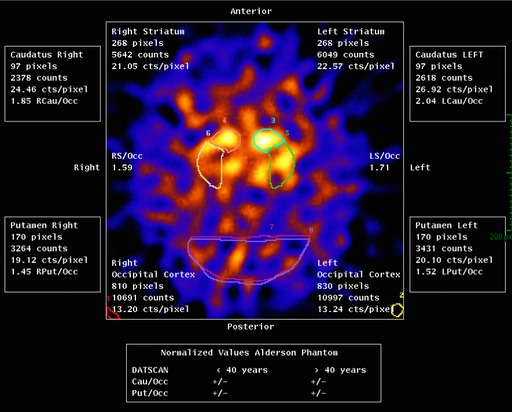
DAT SPECT scan showing bilateral asymmetrical loss of striatal DAT binding with the right striatum showing the largest loss.
To prevent early development of l‐dopa‐induced dyskinesia, which is more common in patients with parkin mutations,4 l‐dopa/carbidopa was replaced by pramipexole. A dose of pramipexole extended release 1.5 mg once‐daily appeared equally effective. One year later, pramipexole was increased. Four years later, he used 3.375 mg per day. His girlfriend then noticed altered behavior. He was indifferent, had little sleep, and suffered from binge eating and gambling. Impulse control disorder7 was diagnosed and pramipexole was replaced by l‐dopa/carbidopa again. Four months later, the patient was on a daily dose of 550/137.5 mg of l‐dopa/carbidopa. There was neither gambling nor binge eating anymore, but he felt that walking was worse than before. The latter could not be objectified during neurological examination.
At the last evaluation, 7 months after the pramipexole was replaced by l‐dopa/carbidopa, he still felt that walking was worse than before. He did not have other complaints after withdrawal of the pramipexole.
Discussion
DRD mostly presents in childhood or adolescence with dystonia and sometimes mild parkinsonism. Typically, there is diurnal fluctuation in which symptoms are improved by sleep. It is characterized by a successful and sustained response to a low dose of l‐dopa. It is mostly the result of a mutation in the GCH1 gene. If not, a mutation in the SPR or TH gene may be the cause, though these mutations lead to a complex (i.e., not purely dystonic) phenotype.6 Instead of mutation analyses, CSF investigation may be done. Normal CSF concentrations of neopterin, biopterin, and homovanillic acid preclude pathogenic mutations in the GCH1, SPR, and TH genes. In retrospect, there was no indication for these tests in our patient.
Furthermore, early‐onset PD may present with dystonia, in some cases as a DRD mimic. For this, nuclear imaging (e.g., DAT SPECT) may be helpful. These PD patients, as with our case, may show a mutation in the parkin gene.8
From his first presentation at the outpatient clinic, our patient had a persistent dystonic gait. However, anamnestically, he had claimed a paroxysmal exercise‐induced dystonia as his first symptom. Of interest, applying the recently launched diagnostic algorithm for paroxysmal exercise‐induced dyskinesias would have led to the same diagnosis as ours.9
Conclusion
We described a young man presenting with foot drop dystonia, mild parkinsonism, and fluctuations during the day, responding to l‐dopa. Therefore, dopa‐responsive dystonia was diagnosed. Genetic testing, however, showed a compound heterozygous parkin mutation. Clearly, parkin‐related early‐onset PD (also: parkin disease) should be kept in mind when analysing a dopa‐responsive dystonia patient.
Author Roles
(1) Clinical Project: A. Conception; B. Execution; (2) Clinical Assessment and Data; (3) Manuscript: A. Writing of the First Draft; B. Review and Critique.
L.J.S.: 3A, 3B
A.J.W.B.: 3B
A.G.M.: 1A, 1B, 2, 3B
Disclosures
Funding Sources and Conflicts of Interest: The authors report no sources of funding and no conflicts of interest.
Financial Disclosures for previous 12 months: The authors declare that there are disclosures to report.
Supporting information
A video accompanying this article is available in the supporting information here.
Video 1 Segment 1: Walking with exaggerated lifting and subsequently dropping of the feet, which we classified as a dystonic gait, also a reduced arm swing. Segment 2: Four years later, the patient talks about his symptoms. Segment 3: Observation of the gait. Segment 4: Neurological examination shows normal eye movements, some bradykinesia of the hands and feet, and normal muscle strength in the proximal and distal parts of the legs.
Relevant disclosures and conflicts of interest are listed at the end of this article.
References
- 1. Bonifati V. Autosomal recessive parkinsonism. Parkinsonism Relat Disord 2012;18(suppl 1):S4–S6. [DOI] [PubMed] [Google Scholar]
- 2. Lücking CB, Dürr A, Bonifati V, et al. Association between early‐onset Parkinson's disease and mutations in the parkin gene. N Engl J Med 2000;342:1560–1567. [DOI] [PubMed] [Google Scholar]
- 3. Kilarski LK, Pearson JP, Newsway V, et al. Systematic review and UK‐based study of PARK2 (parkin), PINK1, PARK7 (DJ‐1) and LRRK2 in early‐onset Parkinson's disease. Mov Disord 2012;27:1522–1529. [DOI] [PubMed] [Google Scholar]
- 4. Schrag A, Schott JM. Epidemiological, clinical, and genetic characteristics of early‐onset parkinsonism. Lancet Neurol 2006;5:355–363. [DOI] [PubMed] [Google Scholar]
- 5. Clot F, Grabli D, Cazeneuve C, et al. Exhaustive analysis of BH4 and dopamine biosynthesis genes in patients with Dopa‐responsive dystonia. Brain 2009;132:1753–1763. [DOI] [PubMed] [Google Scholar]
- 6. Tassin J, Dürr A, Bonnet A, et al. Levodopa‐responsive dystonia: GTP cyclohydrolase I or parkin mutations? Brain 2000;123:1113–1121. [DOI] [PubMed] [Google Scholar]
- 7. Sammler EM, Swingler RJ, Stuart A, Muqit M. Dopamine dysregulation syndrome in a patient with early onset Parkinsonism and Parkin gene mutations. Mov Disord 2009;24:2442–2443. [DOI] [PubMed] [Google Scholar]
- 8. Elia AE, Del Sorbo F, Romito LM, Barzaghi C, Garavaglia B, Albanese A. Isolated limb dystonia as presenting feature of Parkin disease. J Neurol Neurosurg Psychiatry 2014;85:827–828. [DOI] [PubMed] [Google Scholar]
- 9. Erro R, Stamelou M, Ganos C, et al. The clinical syndrome of paroxysmal exercise‐induced dystonia: diagnostic outcomes and an algorithm. Mov Disord Clin Pract 2014;1:57–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
A video accompanying this article is available in the supporting information here.
Video 1 Segment 1: Walking with exaggerated lifting and subsequently dropping of the feet, which we classified as a dystonic gait, also a reduced arm swing. Segment 2: Four years later, the patient talks about his symptoms. Segment 3: Observation of the gait. Segment 4: Neurological examination shows normal eye movements, some bradykinesia of the hands and feet, and normal muscle strength in the proximal and distal parts of the legs.