

Abstract
We report on a white Afrikaans family from eastern South Africa with three members affected with North Sea progressive myoclonus epilepsy, resulting from a homozygous founder GOSR2 mutation (c.430G>T, p.Gly144Trp). The mutation was identified by exomic sequencing in a research study investigating childhood onset ataxias. All three subjects presented with ataxia, tremor, early gait difficulties, and myoclonic and generalized tonic clonic (GTC) epilepsy. Each patient underwent deep brain stimulation of the caudal Zona Incerta before coming to the attention of the authors. In each case there was a reduction in GTC seizures, and two patients exhibited a reduction in involuntary movements, as evaluated during long‐term follow‐up. In one case there was an improvement in gait and stance when assessed while the stimulation was on.
Keywords: deep brain stimulation, North Sea progressive myoclonus epilepsy, GOSR2
Progressive myoclonic epilepsies (PMEs) are genetic disorders, usually inherited in an autosomal recessive manner. They present with refractory epilepsy, including myoclonus and GTC seizures, cognitive decline, and other variable neurological features including ataxia.1 A form of PME resulting from a homozygosity for a founder missense mutation (c.430G>T, p.Gly144Trp) of the Golgi Qb‐SNARE gene (GOSR2), a Golgi vesicle transport gene, has been described. The mutation prevents the protein locating to the Golgi.2 This is now referred to as North Sea PME based on the common geographical origin of the patients and the presence of a founder haplotype.1, 2, 3, 4 The syndrome is characterized by early onset progressive ataxia, myoclonic epilepsy, and multiple seizure types. Cognition is preserved until late in the disease and scoliosis is a virtually consistent feature.3 Peripheral neuropathy, pes cavus, and mildly elevated Creatinine Kinase (CK) levels also occur.4
We describe three members of a white Afrikaans family from South Africa, each of whom was homozygous for the GOSR2 founder mutation. All had bilateral deep‐brain stimulators (DBS) of the caudal Zona Incerta (cZI) inserted before coming to the attention of the authors. We describe the effect of the DBS on their clinical phenotype.
Patients and Methods
The family originates from a small town in the Mpumalanga province of South Africa. Cases 2 and 3 are female siblings. Case 1 is a first cousin of cases 2 and 3, through his father and their mother (Fig. 1A). This family was entered into a research study investigating the genetic basis of childhood ataxias in Oxford, United Kingdom. Exome sequencing was performed using the SureSelect Human All Exon v5 kit (Agilent Technologies, Santa Clara, CA, USA) for exome capture and the Illumina HiSeq 2000 platform as described by Parolin Schenkenberg et al.5 Analysis was performed according to the Genome Analysis Toolkit's recommended work flow. Rare Minor allele frequency (MAF) < 0.01), putative functional variants were filtered according to a recessive model of inheritance. The homozygous mutation c.430G>T, p.Gly144Trp in GOSR2 was identified in cases 1 and 2. Sanger sequencing confirmed the homozygous mutation in the two probands, with all four parents being documented heterozygotes. DNA from case 3 was of poor quality and she was not tested.
Figure 1.
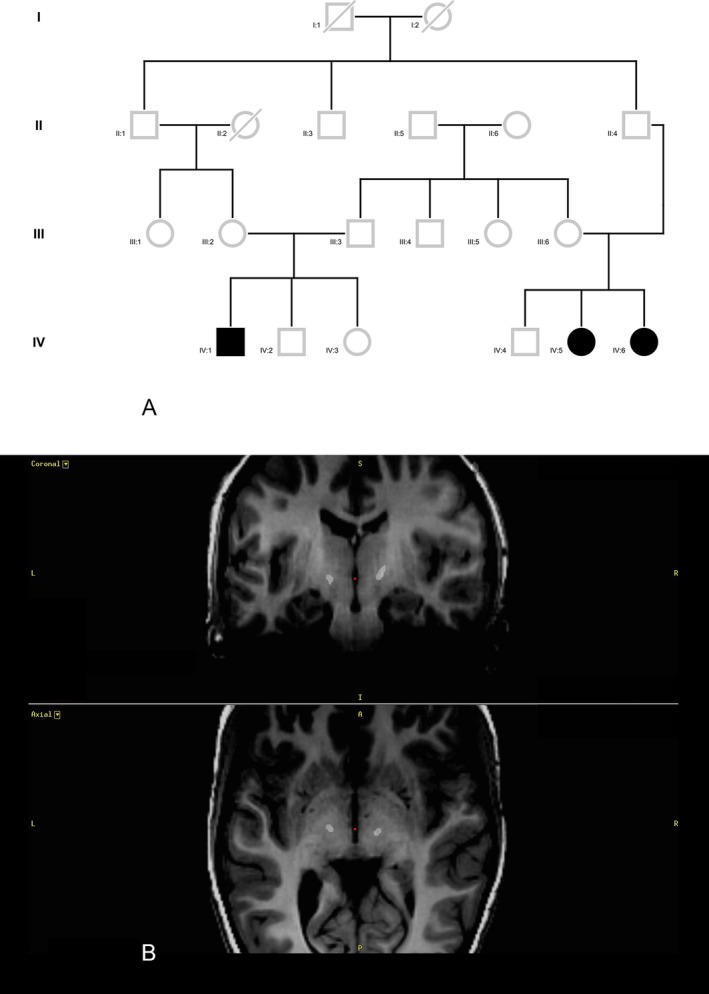
A: Pedigree: The subject in case 1 (IV:1) is a first cousin to the subjects in case 2 (IV:5) and case 3 (IV:6), once removed through his mother and their father. He is also a first cousin once removed through his mother's father and their father. In addition, there is a shared ancestor three generations previously. B: Case 1: Postoperative Stealth CT merged with preoperative MRI T1 sequences. The final position of the first contact of each electrode is shown in axial and coronal views.
Results
Case 1
Case 1 is a male, first seen at 10 years of age by D.G.A. He was born prematurely at 33 weeks and required 3 weeks of assisted ventilation. He had normal developmental milestones, walked at 13 months, and he did well academically and continues to do so. At 3 years of age he developed an upper limb tremor, and at 5 years he developed myoclonus. His first GTC seizure was at the age of 6 years and he was treated with carbamazepine. Despite treatment, his seizures worsened, as did his tremor and balance, and he also developed absence seizures. His family reported a GTC seizure every 2 to 3 months, absence seizures at a rate of 1 to 2 per week, and persistent myoclonus. Bilateral DBS surgery of the cZI (Fig. 1B) was performed at the age of 9 years for the progressively worsening “jerking.” The family reported that the DBS significantly improved his balance, and the myoclonus was reduced to a few episodes per day. Since the surgery, the family has not observed any GTC or absence seizures.
On examination at presentation he had no nystagmus and preserved smooth pursuit of eye movements with full range. He had frequent spontaneous myoclonus that was augmented by sound and touch. A symmetrical action tremor was found in all four limbs and he had dysdiadochokinesis. No reflexes could be elicited but he had normal sensation. He was able to walk ten steps in tandem but deviated more than three times. He did not have scoliosis or pes cavus. Nerve conduction studies and CK levels were normal. His carbamazepine was changed to sodium valproate, and the family reported that this alteration reduced the myoclonus to a few episodes per day.
After confirming the genetic diagnosis, at 15 years of age, the patient was admitted to an epilepsy‐monitoring unit in Johannesburg to determine if objective improvement from the DBS could be demonstrated. The Unified Myoclonus Rating Scale (UMRS) and video EEG were first performed while his DBS was on at 2.1 mA with a pulse width of 450 μs and rate of 130 Hz bilaterally.6 The stimulator was then switched off, he underwent EEG monitoring, and the UMRS was repeated after 24 hours. Section 4 of the UMRS showed a score of 27 (of 160) while the DBS was on, and worsened to 46 after the DBS had been off for 24 hours. Gait was profoundly affected with the stimulator off (Video 1, Clips 2 and 3).
While the DBS was on, the EEG showed intermittent generalized sharp and slow waves 3 to 4 Hz in 20% of the awake recording. These increased in stages 1 and 2 of sleep. Polyspikes were seen in approximately 10% of the awake recording accompanying the slow waves. Polyspike waves were also seen independently. These were often accompanied by a myoclonic jerk. There was also a diffuse electrical artifact seen from the stimulator. This artifact stopped when the stimulator was switched off, but the polyspike activity increased along with myoclonic jerks until it was seen in 50% of the awake recording by the following day. Just before switching the DBS back on, the patient experienced a GTC seizure from sleep. He had not had a GTC for more than 7 years. Since turning the stimulator back on, no further GTC seizures have been observed.
Both cases 2 and 3 have had their DBSs removed despite subjective improvement in seizure rate.
Case 2 is a female who presented at 30 years of age. She reported that she struggled with sports as early as 5 years and had jerking at 7 years of age. Her first GTC seizure was at the age of 8 years. She has been in a wheelchair since the age of 11 because of her balance problems. Cognition is normal and she completed secondary schooling. Case 3 is the younger sister of case 2 and presented to us at 27 years of age. She had struggled to walk, had frequent injuries, and was wheelchair bound before the age of 10 years. Myoclonus started at about 3 years and she had her first GTC seizure at 5 years of age. A scoliosis was diagnosed before puberty and required surgery at the age of 17. Both cases presented to our unit were being treated with carbamazepine. This was changed to sodium valproate at presentation, which reduced their myoclonus subjectively.
Both patients had DBS surgery to the cZI before presenting to us. Case 2 was implanted at 26 years and case 3 at 24 years of age. In case 2 there was a reported reduction in her GTC seizures from several per month to one every 3 to 4 months and there was improvement in upper limb tremor, and in her sister's case there was a subjectively reported improvement of the seizures from 5 to 7 per month to 3 to 5 per month but they did not notice the same benefit in tremor or jerking.
At the time of case 2's DBS battery replacement, the wound became septic and the DBS was removed. Her GTC seizures have increased to 1 per month. In case 3, dysphagia slowly developed, which improved when the battery ran flat. The patient elected to have the DBS removed. Her family reports that the myoclonus has changed to being persistent from being occasional, and the GTC seizures occur 7 times per month on average.
On examination without the DBS, the two sisters had supranuclear up‐gaze palsies and ocular pursuit was interrupted, with a decreased range horizontally (Video 2, Clips 1 and 4). They also showed severe spontaneous myoclonus that was augmented with sound and touch, and neither could sit unaided (Video 2, Clip 2). Case 3 also had a loss of pain and temperature sensation on her feet in a slipper distribution and was areflexic. She had a rigid spine, resulting from spinal surgery, and pes cavus (Video 2, Clip 3). Her CK level was elevated at 421 U/L. Nerve conduction studies showed axonopathic peroneal amplitudes of 1.46 mV proximally and 1.17 mV distally with a normal velocity. The sural nerves could not be stimulated.
Discussion
We have described three related cases of North Sea PME in one Afrikaans South African family. Afrikaans individuals have ancestry from a small group of German, Dutch, and Huguenot settlers who immigrated to the Cape Colony in the 1600s.7 This is consistent with the North Sea, predominantly Dutch, origin of the mutation. This family is the only one described in which family members have had DBS of the cZI. In all three cases there was a reported reduction in GTC seizure frequency and possibly a reduction in myoclonus. Using video EEG, in one case, we have observed a reduction in myoclonus.
The mechanism of action for DBS in reducing seizures is still debated. The disruption of rhythmic discharges between the thalamus and cortex where abnormal signals are replaced, also referred to as “neural hijacking,” has been suggested.8 The cZI is well placed to have distant effects from high‐frequency stimulation with its connections and adjacent connections to the cortex, cerebellum, thalamus, subthalamic nucleus, and other basal ganglia structures.9 This may explain why we noted improvement in our patients.
Furthermore DBS of the cZI has been shown to improve seizures previously but never before in PME.10 In a small case series, DBS of the STN reduced seizures in other PMEs and was superior to DBS of the ventral intermediate nucleus of the thalamus.11
We also documented improvement of gait and stance with the stimulator turned on. This may be a result of the reduction in myoclonus but requires further investigation. Two of our cases had an up‐gaze palsy and case 1 did not. A vertical‐gaze palsy has been described in subthalamic and thalamic DBS.12 It is interesting to note that in our cases the only patient with normal up‐gaze was the patient with the DBS functioning, and in the two cases in which the DBS had been removed, the vertical‐gaze palsy is seen. The subject with the normal up‐gaze is also considerably younger than the other 2 patients.
These are interesting observations, and to our knowledge this is the first report of DBS for this condition. It is a very disabling condition for which any improvement could make a difference to the patient's level of functioning. Nevertheless, there are limitations, as these observations were largely retrospective and were with limited and unblinded assessments. Further research would be required and other potential targets considered to validate these incidental observations.
Author Roles
1. Research Project: A. Conception, B. Organization, C. Execution; D. Neurological input; 2. Genetic Analysis: A. Design, B. Execution, C. Review and Critique; 3. Manuscript: A. Writing of the first draft, B. Review and Critique.
D.G.A.: 1A, 1B, 1D, 3A
A.H.N.: 2C
K.A.F.: 2C
D.S.: 2C
J.M.: 2C
A.K.: 1A, 1B, 1C
Disclosures
Ethical Compliance Statement: We confirm that we have read the journal's position on issues involved in ethical publication and affirm that this work is consistent with those guidelines.
Funding Sources and Conflict of Interest: Funding to A.H.N. is acknowledged from Ataxia United Kingdom, Action Medical Research, The Henry Smith Charity, the Oxford Partnership Comprehensive Biomedical Research Centre funded by the Department of Health National Institute of Health Research (NIHR) Biomedical Research Centre Programme, and the Thames Valley Dementias and Neurodegenerative Diseases Research Network (DeNDRoN), United Kingdom. Funding to K.A.F. and D.S. is acknowledged from the Medical Research Council (United Kingdom) Computational Genomics Analysis and Training Programme (G1000902). The authors report no conflicts of interest.
Financial Disclosures for the previous 12 months: The authors report no sources of funding and no conflicts of interest.
Supporting information
Videos accompanying this article are available in the supporting information here.
Video 1. Clip one: case 1, eye movement. Normal range and smooth pursuit with no nystagmus. Clip two: case 1, tandem gait with DBS switched on. Considerable staggering, with difficulties in half turn. No support needed. Dystonic posturing of the right arm. Clip three: case 1, tandem gait with DBS switched off for 24 hours. Patient falls and requires support. Increase in myoclonus observed.
Video 2. Clip one: case 2, eye movements. Myoclonic movements of the body and head can be seen. Smooth pursuit is broken into saccades with full movement in the horizontal plane. There is a supranuclear up‐gaze palsy with normal down‐gaze. Clip two: case 2, myoclonus. Patient lying and cannot sit unaided. Generalized myoclonic movements of the entire body are seen. Action myoclonus is evident when patient attempts bilateral finger taps. Clip three: case 2, myoclonus and pes cavus. View of patient's lower limbs demonstrating pes cavus and myoclonus. Clip four: case 3, eye movements. Myoclonic movements of the face can be seen. Smooth pursuit is broken into saccades with full movement in the horizontal plane. There is a supranuclear up‐gaze palsy with normal down‐gaze.
Acknowledgments
We thank Dr. P. Slabbert and Dr. J. Vaidynathan, who operated on all three cases, for their kind assistance. We also thank Prof. M.A.J. de Koning‐Tijssen for her insight and guidance. Special thanks to the patients and their families for sharing their stories, giving their time, and teaching us all.
Relevant disclosures and conflicts of interest are listed at the end of this article.
References
- 1. Malek N, Stewart W, Greene J. The progressive myoclonic epilepsies. Pract Neurol 2015;15:164–171. doi: 10.1136/practneurol-2014-000994. [DOI] [PubMed] [Google Scholar]
- 2. Corbett MA, Schwake M, Bahlo M, et al. A mutation in the Golgi Qb‐SNARE gene GOSR2 causes progressive myoclonus epilepsy with early ataxia. Am J Hum Genet 2011;88:657–663. doi: 10.1016/j.ajhg.2011.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Boisse Lomax L, Bayly MA, Hjalgrim H, et al. “North Sea” progressive myoclonus epilepsy: phenotype of subjects with GOSR2 mutation. Brain 2013;136:1146–1154. doi: 10.1093/brain/awt021. [DOI] [PubMed] [Google Scholar]
- 4. van Egmond ME, Verschuuren‐Bemelmans CC, Nibbeling EA, et al. Ramsay Hunt syndrome: clinical characterization of progressive myoclonus ataxia caused by GOSR2 mutation. Mov Disord 2014;29:139–143. doi: 10.1002/mds.25704. [DOI] [PubMed] [Google Scholar]
- 5. Parolin Schnekenberg R, Perkins EM, Miller JW, et al. De novo point mutations in patients diagnosed with ataxic cerebral palsy. Brain 2015;138:1817–1832. doi: 10.1093/brain/awv117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Frucht SJ, Leurgans SE, Hallet M, Fahn S. The unified myoclonus rating scale. Adv Neurol 2002;89:361–376. [PubMed] [Google Scholar]
- 7. Nurse GT, Weiner JS, Jenkins T. The Peoples of Southern Africa and Their Affinities. Oxford: Oxford University Press; 1985. [Google Scholar]
- 8. Laxpati NG, Kasoff WS, Gross RE. Deep brain stimulation for the treatment of epilepsy: circuits, targets, and trials. Neurotherapeutics 2014;11:508–526. doi: 10.1007/s13311-014-0279-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Mitrofanis J. Some certainty for the “zone of uncertainty”? exploring the function of the zona incerta. Neuroscience 2005;130:1–15. doi: 10.1016/j.neuroscience.2004.08.017. [DOI] [PubMed] [Google Scholar]
- 10. Franzini A, Messina G, Marras C, et al. Deep brain stimulation of two unconventional targets in refractory non‐resectable epilepsy. Stereotact Funct Neurosurg 2008;86:373–381. doi: 10.1159/000175800. [DOI] [PubMed] [Google Scholar]
- 11. Wille C, Steinhoff BJ, Altenmüller D‐M, et al. Chronic high‐frequency deep‐brain stimulation in progressive myoclonic epilepsy in adulthood—report of five cases. Epilepsia 2011;52:489–496. doi: 10.1111/j.1528-1167.2010.02884.x. [DOI] [PubMed] [Google Scholar]
- 12. Fleury V, Spielberger S, Wolf E, et al. Vertical supranuclear gaze palsy induced by deep brain stimulation: report of two cases. Parkinsonism Relat Disord 2014;20:1295–1297. doi: 10.1016/j.parkreldis.2014.07.007. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Videos accompanying this article are available in the supporting information here.
Video 1. Clip one: case 1, eye movement. Normal range and smooth pursuit with no nystagmus. Clip two: case 1, tandem gait with DBS switched on. Considerable staggering, with difficulties in half turn. No support needed. Dystonic posturing of the right arm. Clip three: case 1, tandem gait with DBS switched off for 24 hours. Patient falls and requires support. Increase in myoclonus observed.
Video 2. Clip one: case 2, eye movements. Myoclonic movements of the body and head can be seen. Smooth pursuit is broken into saccades with full movement in the horizontal plane. There is a supranuclear up‐gaze palsy with normal down‐gaze. Clip two: case 2, myoclonus. Patient lying and cannot sit unaided. Generalized myoclonic movements of the entire body are seen. Action myoclonus is evident when patient attempts bilateral finger taps. Clip three: case 2, myoclonus and pes cavus. View of patient's lower limbs demonstrating pes cavus and myoclonus. Clip four: case 3, eye movements. Myoclonic movements of the face can be seen. Smooth pursuit is broken into saccades with full movement in the horizontal plane. There is a supranuclear up‐gaze palsy with normal down‐gaze.