

Early‐onset dystonia is not encountered very frequently in adult movement disorders and child neurology clinics. A significant proportion of early‐onset dystonia cases are estimated to be genetic, such as dopa‐responsive dystonias (DYT1 and DYT6); however, some cases may have a treatable inborn error of metabolism, such as organic acidurias, glucose transporter 1 deficiency, Wilson's disease, disorders of iron accumulation (e.g., PKAN), Niemann Pick C, and DYT5.1 Choosing the appropriate diagnostic test for these patients can be challenging and expensive. Next‐generation sequencing techniques, such as whole‐genome sequencing, whole‐exome sequencing, or sequencing panels targeting specific disease‐causing genes, have improved our diagnostic capabilities significantly and allow us to analyze massive number of genes simultaneously. Here, we report on two cases of congenital dystonia and central hypomyelination discovered by whole‐exome sequencing.
Case
A 21‐year‐old Italian male, conceived from a nonconsanguinous marriage, presented with poor head control and hypotonia at 2 months of age and strabismus and optic pallor at 4 months of age. Choreoathetotic/dyskinetic movements began at 6 months of age that progressed to generalized dystonia. The patient's brother had a similar presentation. Both had severe motor and language delay, sensorineural hearing loss, facial dysmorphism with a long face, and ocular hypotelorism, and neither developed the ability to walk. The older brother has partial hearing loss and did develop basic academic skills, has some language, and can do some reading. The younger brother is deaf and has not developed academic skills. The mother had similar facial features, but no neurological or cognitive problems. Both patients had minimal response to levodopa, pimozide, trihexyphenidyl, clonidine, baclofen, clonazepam, and tetrabenazine. Workup, including serum carnitine, acyl‐carnitine profile, very‐long‐chain fatty acids, creatine phosphokinase, arylsufatase, ß‐galactosidase, carbohydrate deficient transferrin, complete blood count, complete metabolic profile, liver function tests, uric acid, lactate, pyruvate, ammonia, alpha‐fetoprotein, immunoglobulins, inborn error screen, acanthocyte smear, biotinidase, thyroid studies, vitamin B12, folate, copper, ceruloplasmin, creatine, guanidinoacetate, urine and serum amino acids, urine organic acids, cerebrospinal fluid studies (protein, glucose, cells, immunoglobulins, lactate, pyruvate, oligoclonal bands, myelin basic protein, neurotransmitter metabolites, tetrahydrobiopterin, and neopterin), and electrophysiologic studies, was unremarkable. MRI of the brain performed on the older brother revealed delayed myelination, atrophy in posterior occipital lobes, hypoplasia of the superior cerebellar vermis, and thinning of the corpus callosum (Fig. 1). Genetic workup, including subtelomeric fluorescence in situ hybridization and screening for MERRF, MELAS, NARP, and Leigh's mutations, was negative. Nonrandom X‐inactivation in studies of maternal DNA suggested X‐linked transmission. Exome sequencing was performed on the entire family. Exons were captured with the Nimblegen SeqCap EZ Human Exome Library and sequenced with 2 × 100 base‐pair (bp) paired‐end reads on an Illumina HiSeq2000 (Illumina, Inc., San Diego, CA). Sequence was aligned to Hg19 using the Burrows‐Wheeler Aligner (0.5.9)13, and variants were called with the Genome Analysis Toolkit (v. 1.6). More than 97% of bases sequenced had a quality score >10 and variants with a quality score <10 were removed to avoid false positives. Variants that had an allele frequency of >1% in dbSNP, the 1000 Genomes Project, and ESP were filtered out, leaving 564 variants present in at least 1 of the 2 brothers. Assuming an autosomal‐recessive or X‐linked mode of inheritance, filtering for two shared variants in autosomes and one shared variant in X chromosome in the two affected brothers, both patients were found to have a 6‐bp deletion of c.261_266delGCTTCT, in exon 3 of BCAP31 (NM_001139457.1 MIM 300398), and the mother and sister were found to be carriers (Fig. 2). The 6‐bp deletion at Xq28 results in a BAP31 protein change of p.Leu87_Leu89delinsPhe (deletion of leucines 88 and 89 and leucine 87 changed to phenylalanine). Analysis with MutationTaster showed that the mutated leucine residues are highly conserved and the mutation would potentially impact multiple functional domains, including the TOPO domain of the protein. The impact of this mutation on the protein was clearly predicted to be deleterious in Provean, with a score of −16.7 (cut‐off score for deleterious change: −2.5). The deletion was confirmed by Sanger sequencing and is not in the Exome Aggregation Consortium (ExAC) database.
Figure 1.
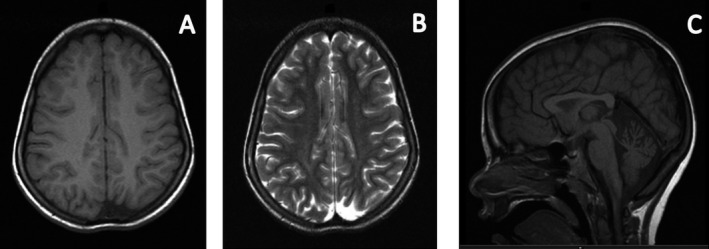
Axial T1W (A) and T2W (B) images showing focal atrophy and areas of dysmyelination in posterior occipital lobes. Sagittal T1W (C) showing hypoplasia of the superior cerebellar vermis and thinning of the corpus callosum.
Figure 2.
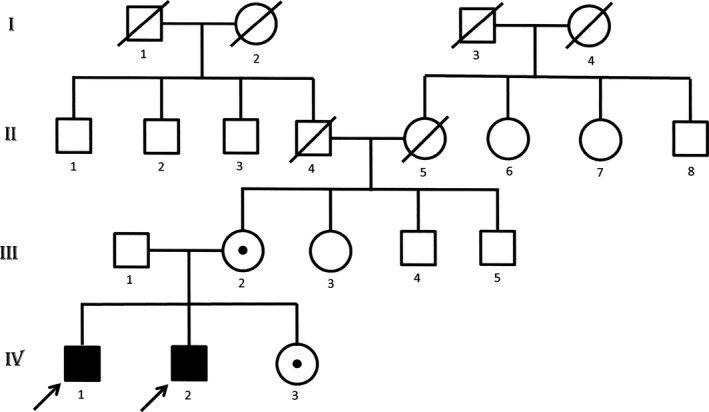
Family tree of the probands marked with arrows. The subjects who underwent exome sequencing were the 2 affected brothers (IV 1 and 2), sister (IV 3), and mother (III 2) who were found to be carriers. The father of the probands (III 1) was not exome sequenced, but did not have the deletion based on Sanger sequencing.
Discussion
The brothers described in this report were found to have a 6‐bp deletion in BCAP31 at Xq28, and the mother and patient's sister were carriers. BCAP31 encodes B‐cell‐receptor‐associated protein 31 (BAP31) an endoplasmic reticulum (ER) membrane protein abundant in neurons. It functions as a chaperone protein in ER‐associated degradation, programmed cell death, and export of ER proteins to the Golgi apparatus. Lack of the BAP31 protein function disrupts ER metabolism, causes disorganization of Golgi, affecting ER‐to‐Golgi crosstalk, and results in impaired protein trafficking. Three families with probands with strabismus, optic atrophy, deafness, congenital dystonia, and central hypomyelination2 were recently described to have truncating mutations in BCAP31. Only these seven cases have been described in the literature so far by Cacciagli et al. who identified three different mutations in the BCAP31 gene, but all of them caused a loss of function of the BAP31 protein. The deletion in the BCAP31 gene identified in the brothers in this report is expected to be in frame and thus the protein would still be produced, although with a predicted deleterious effect on protein function. These brothers may be somewhat less affected than the patients with truncating mutations based on some residual protein activity.
Another syndrome of deafness, dystonia, and optic atrophy to be considered in the differential diagnosis is Mohr‐Tranebjaerg syndrome, which is an X‐linked disease caused by mutations in the nuclear gene TIMM8A, encoding a protein involved in mitochondrial transport processes.3 Mitochondrial disorders, such as Leber's optic atrophy, can also be considered. The brothers in this report had dystonia, deafness, and optic atrophy, but did not have psychiatric symptoms or pyramidal signs that have been reported with Mohr‐Tranebjaerg syndrome, and workup ruled out mitochondrial disorders, such as Leber's optic atrophy. Other X‐linked disorders associated with dystonia include DYT3 (Lubag syndrome), ornitine transcarbamylase deficiency, Lesch Nyhan syndrome, and cerebral creatinine deficiency syndrome‐1, but workup was negative for these disorders. Brain MR findings of hypomyelination and thinning of corpus suggest consideration of causes such as the recently described POL3A mutation4 resulting in an autosomal‐recessive hypomyelination with leukodystrophy disease and TUBB4A gene mutations that underlie the hypomyelination with atrophy of the basal ganglia and cerebellum syndrome5; however, the imaging characteristics as well as the clinical picture of these brothers do not match these diagnoses.
This article reports 2 brothers in the same family affected by a recently diagnosed X‐linked disorder characterized by dystonia, deafness, and central hypomyelination. These patients had undergone extensive and expensive evaluations for a full battery of inborn errors and other metabolic diseases, mitochondrial mutations, and chromosome aberrations, yet these tests failed to yield a result, whereas exome sequencing provided a diagnosis with a single test. Although there are certainly limitations inherent in exome sequencing, this report highlights the value of exome sequencing as a new clinical tool to help physicians obtain diagnoses more efficiently and in a less expensive manner for patients and their families with unusual neurological symptoms.
Author Roles
(1) Research Project: A. Conception, B. Organization, C. Execution; (2) Statistical Analysis: A. Design, B. Execution, C. Review and Critique; (3) Manuscript Preparation: A. Writing of the First Draft, B. Review and Critique.
P.V.: 1B, 3A
E.B.‐K.: 1A, 1C, 3B
R.M.: 1C, 3B
S.D.: 1C
D.A.H.: 3B
Disclosures
Funding Sources and Conflicts of Interest: The authors report no sources of funding and no conflicts of interest.
Financial Disclosures for previous 12 months: P.V. received the Clinical Research Training Fellowship grant in Ataxia sponsored by the American Brain Foundation and the National Ataxia Foundation. D.A.H. received grant support from the National Institutes of Health (NIH; R01NS082416), the National Fragile X Foundation, the Anti‐Aging Foundation, and Pfizer. E.B.‐K. received Grant support from NIH, Centers for Disease Control and Prevention, Merck Foundation, and the Hope for Hayley Foundation and also received clinical trial support from Novartis, Roche, Alcobra, and Neuren.
Supporting information
A video accompanying this article is available in the supporting information here.
Video 1. The first part of the video demonstrates the older patient with congenital dystonia, facial dysmorphism with long narrow face, deep‐set eyes, prominent forehead, mid‐facial hypoplasia, and is able to speak a few words, follow some commands, and push his wheelchair. The second part of the video demonstrates the younger patient with severe motor and language delay, congenital dystonia, and facial dysmorphism and able to switch a light from on to off. The red skin change on his face is secondary to him sleeping on that side for a prolonged period of time.
Acknowledgment
The authors thank Casey M. Rand at Ann & Robert H. Lurie Children's Hospital of Chicago for running the protein prediction programs.
Relevant disclosures and conflicts of interest are listed at the end of this article.
References
- 1. van Egmond ME, Kuiper A, Eggink H, et al. Dystonia in children and adolescents: a systematic review and a new diagnostic algorithm. J Neurol Neurosurg Psychiatry 2015;88:774–781. [DOI] [PubMed] [Google Scholar]
- 2. Cacciagli P, Sutera‐Sardo J, Borges‐Correia A, et al. Mutations in BCAP31 cause a severe X‐linked phenotype with deafness, dystonia, and central hypomyelination and disorganize the Golgi apparatus. Am J Hum Genet 2013;93:579–586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Kojovic M, Pareés I, Lampreia T, et al. The syndrome of deafness‐dystonia: clinical and genetic heterogeneity. Mov Disord 2013;28:795–803. [DOI] [PubMed] [Google Scholar]
- 4. Khalifa M, Naffaa L. Exome sequencing reveals a novel WDR45 frameshift mutation and inherited POLR3A heterozygous variants in a female with a complex phenotype and mixed brain MRI findings. Eur J Med Genet 2015;58:381–386. [DOI] [PubMed] [Google Scholar]
- 5. Balint B, Bhatia KP. Isolated and combined dystonia syndromes—an update on new genes and their phenotypes. Eur J Neurol 2015;22:610–617. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
A video accompanying this article is available in the supporting information here.
Video 1. The first part of the video demonstrates the older patient with congenital dystonia, facial dysmorphism with long narrow face, deep‐set eyes, prominent forehead, mid‐facial hypoplasia, and is able to speak a few words, follow some commands, and push his wheelchair. The second part of the video demonstrates the younger patient with severe motor and language delay, congenital dystonia, and facial dysmorphism and able to switch a light from on to off. The red skin change on his face is secondary to him sleeping on that side for a prolonged period of time.