

Climate-warming brown carbon aerosols lose warming capacity during atmospheric transport.
Abstract
Light-absorbing organic aerosols, known as brown carbon (BrC), counteract the overall cooling effect of aerosols on Earth’s climate. The spatial and temporal dynamics of their light-absorbing properties are poorly constrained and unaccounted for in climate models, because of limited ambient observations. We combine carbon isotope forensics (δ13C) with measurements of light absorption in a conceptual aging model to constrain the loss of light absorptivity (i.e., bleaching) of water-soluble BrC (WS-BrC) aerosols in one of the world’s largest BrC emission regions—South Asia. On this regional scale, we find that atmospheric photochemical oxidation reduces the light absorption of WS-BrC by ~84% during transport over 6000 km in the Indo-Gangetic Plain, with an ambient first-order bleaching rate of 0.20 ± 0.05 day−1 during over-ocean transit across Bay of Bengal to an Indian Ocean receptor site. This study facilitates dynamic parameterization of WS-BrC absorption properties, thereby constraining BrC climate impact over South Asia.
INTRODUCTION
High loadings of carbonaceous aerosols affect the climate system over South Asia, yet both the magnitude and specific mechanisms remain poorly constrained (1, 2). The systematic underestimation of the absorption aerosol optical depth (AAOD) over South Asia by factors of 2 to 3 in climate models relative to observation-based estimates illustrates the currently large uncertainties (3). This uncertainty may be related to several different factors, including inaccurate estimates of source emissions of black carbon (BC) and light-absorbing organic carbon (OC) [also known as brown carbon (BrC)], atmospheric longevity of these light-absorbing aerosols, and uncertainties in optical properties of ambient and long-range transported BC and BrC. The BrC component is little studied outside laboratory experiments and immediate source regions (4–6), yet some estimates suggest that it can be a substantial component of the total aerosol absorption over areas such as South Asia (7). The light absorption measurements of BrC from liquid extracts are less prone to interferences from other absorbers such as BC (4, 8). This study seeks to quantify the dynamically changing light-absorbing properties of water-soluble BrC (WS-BrC) in the South Asian outflow and during its long-range over-ocean transport. We focus on the WS fraction of BrC (i.e., WS-BrC), as the WS fraction of total OC aerosol is commonly 50 to 80% over South Asia (8).
The WS-BrC aerosols may contribute to the AAOD offset, yet is little studied in source-to-receptor systems. BrC is generally characterized by a complex and dynamic mixture of organic compounds, originating from both primary sources and secondary formation (8). Hence, the chemical composition and concentration of BrC are variable across source regions and receptors. Modeling studies of BrC often involve a range of assumptions regarding their solubility, mixing state, and atmospheric processing. However, a scarcity of field-based observational constraints on BrC makes it challenging to test and improve models. Recent global modeling studies have suggested that the positive forcing induced by BrC absorption may be so large that it could offset the entire magnitude of the negative direct radiative forcing due to organic aerosol scattering (9, 10).
Light absorption of BrC is typically characterized by a power-law wavelength dependence, increasing from the visible to the ultraviolet (UV) ranges (11). The absorption and spectral dependence of BrC are believed to be affected by atmospheric processes and properties such as oxidation, exposure to sunlight, temperature variations, relative humidity, and pH, as well as source variability (12–16). However, BrC optical properties and lifetimes are presently not at all, or only poorly, considered in climate models (17). The extent to which atmospheric transport and processing could affect the BrC optical properties remains sparsely investigated and poorly understood, which, in turn, results in substantial uncertainties in predicting and mitigating their climate impact.
The South Asian Pollution Experiment 2016 (SAPOEX-16) addressed WS-BrC absorptive properties during long-range transport in the ambient South Asian atmosphere. South Asia is a region with high BrC emissions and aerosol loadings [Fig. 1; e.g., (18)]. The vast and densely populated Indo-Gangetic Plain (IGP) is a particularly high emission region. To this end, samples were collected simultaneously during the dry winter period in the high-intensity SAPOEX-16 campaign at a megacity source site in central IGP (Delhi), an IGP outflow site [Bangladesh Climate Observatory at Bhola (BCOB)], and a large-footprint Indian Ocean regional receptor site [Maldives Climate Observatory at Hanimaadhoo (MCOH)]. We combine the down-wind evolution of WS-BrC mass absorption cross section (MACWS-BrC) and absorption Ångström exponent (AAE) with the stable carbon isotope signature (δ13C) of WSOC, diagnosing extent of aging, and develop a conceptual aging model to elucidate the potential effects of atmospheric processing, and thereby provide observational constraints on the dynamic nature of ambient absorptive properties of WS-BrC over South Asia.
Fig. 1. Meteorology and general aerosol characteristics during the SAPOEX-16.
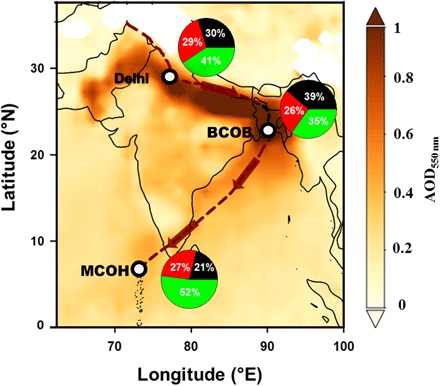
The average AOD at 550 nm during January to March 2016 over the South Asian region and sampling sites of Delhi, BCOB, and MCOH are shown. The trail (dashed line) is the Hybrid Single-Particle Lagrangian Integrated Trajectory (HYSPLIT) model–based mean air mass back trajectory (BT) of the IGP cluster showing the dominating air mass transport from Delhi to BCOB to MCOH (shown by arrows). The pie charts depict the particulate matter (PM) composition in terms of mean relative abundances of (i) TCA mass (black; i.e., TCA = OM + EC), (ii) anthropogenic WS inorganic species (green; i.e., WSISanth: Cl− + NO3− + SO42− + NH4+ + K+), and (iii) largely nonanthropogenic fraction [red; i.e., NAF: PMtot – (TCA + WSISanth)]. The AOD data were obtained from NASA Moderate Resolution Imaging Spectroradiometer (MODIS) (https://giovanni.gsfc.nasa.gov/giovanni/).
RESULTS
Meteorological setting and general aerosol composition during SAPOEX-16
The meteorology of South Asia is driven by the monsoon circulation. During the dry winter (November to March), winds are predominantly NE (referred to as “northeast/winter monsoon”), leading to the buildup of a thick haze of air pollution (AOD > 0.4) containing carbonaceous aerosols that spreads far out over the Bay of Bengal and northern Indian Ocean (1). Cluster analysis revealed the major air mass transport pathways reaching Delhi, BCOB, and MCOH during the SAPOEX-16. The major source regions/clusters were IGP for Delhi; IGP and northern Bay of Bengal (along the eastern coast of India) for BCOB; and IGP, central Arabian Sea (including southern India), eastern Arabian Sea (including western Indian margin), and southern Bay of Bengal for MCOH (see fig. S1 for air mass clusters and fig. S2 for relative cluster contribution).
The SAPOEX-16 campaign was strategically designed to intercept these high aerosol loadings, characteristic of this anthropogenically dominated aerosol regime of the winter-time South Asian outflow (as seen in Fig. 1). High PM2.5 loadings were observed across the IGP at Delhi (208 ± 57 μg m−3) and BCOB (103 ± 70 μg m−3) (table S1). The daily mean concentrations of elemental carbon (EC), OC, and WSOC varied by an order of magnitude between the IGP sites Delhi and BCOB versus at the receptor site MCOH (fig. S3). Taking organic matter (OM) to OC ratio to be 1.7 for the continental IGP sites (Delhi, BCOB) and 2.1 for the remote receptor site MCOH, based on an earlier study in the region (19), the concentration of total carbonaceous aerosols (TCAs) at Delhi (59 ± 17 μg m−3), BCOB (41 ± 33 μg m−3), and MCOH (5 ± 2 μg m−3) constituted about 30 ± 10%, 39 ± 14%, and 21 ± 7% of total PM mass, respectively.
The chemical composition of the carbonaceous aerosols changed both between sites and over time (Fig. 2). The OC to EC ratios during the SAPOEX-16 period varied from as high as 7.9 in Delhi to as low as 1.2 in MCOH and are affected both by emission source variability and by processing during long-range transport (19). The aging of the air parcel also affected the WS inorganic ion fractions. A clear dominance (>95%) of non–sea-salt sulfate (nss-SO42−) fraction to total sulfate was observed at MCOH (the nss-SO42− was as high as 65% of PM), suggesting large contributions of SO42− and SO2 (followed by oxidation of SO2 gas to SO42− during long-range transport) from diesel combustion and coal-fired power plants in India and Bangladesh (20). Similar aerosol compositions in the outflow from South Asia have been previously suggested to be related to aging of aerosols during long-range transport (21–23).
Fig. 2. Aerosol chemical composition during SAPOEX-16.
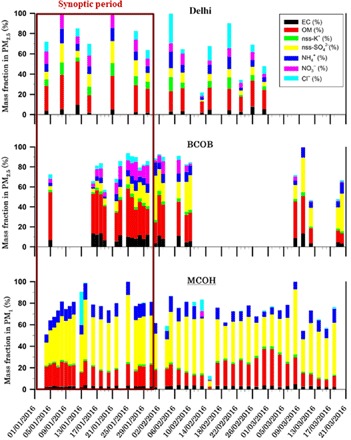
Fractional mass contribution of measured chemical species in PM2.5 over Delhi (n = 15) and BCOB (n = 24) and PM1 over MCOH (n = 43) during the SAPOEX-16 campaign. Synoptic period refers to the period when air masses from the IGP exiting Bay of Bengal passed over the northern region of Maldives (MCOH), as evidenced in the BTs (fig. S1). Relative air mass cluster contribution at each site revealed this period to be between 2 and 31 January 2016 (fig. S2). The mass fraction of the WS inorganic ions is corrected for contribution from sea salt.
Specific inorganic elemental ratios in aerosols may also be used as qualitative source tracers. The non–sea-salt potassium (nss-K+) to EC ratios ranging between 0.2 and 1.0 during SAPOEX-16 are indicative of the influence of biomass burning emissions during winter in the South Asian outflow. In addition, nss-SO42− to EC ratios varying between 2 and 14 also suggest a contribution from fossil fuel emissions (20). These ratios have been previously reported for near-source and receptor sites in South Asia during dry winter months (21–23). The WS-BrC source-receptor system study focused on the period when the air masses from the IGP exiting Bay of Bengal passed over the northern region of Maldives (MCOH), as evidenced by back trajectory (BT) analysis (fig. S1). The relative air mass cluster contribution at each site revealed this period to be between 2 January and 31 January 2016 (fig. S2). We refer to this period as the “synoptic period” (as seen in Fig. 2). January to February is a dry monsoon period for all these three sites. During this time, the air over IGP typically travels with westerly winds, in direction from Delhi to Bangladesh. A substantial fraction of the air masses entering the Bay of Bengal from Bangladesh then travel along India’s eastern shore and over the Maldives. Although this generalized pathway is by no means the only one passing over the Maldives during this time of the year, we use BT analysis to select the samples during which this occurs. Studies have suggested synoptic conditions over South Asia during winter [e.g., (1, 24)].
WSOC abundance and evolution of its stable carbon isotope signature
The WSOC is a potentially large but highly variable portion of TCAs. During SAPOEX-16, the fraction WSOC in the total OC showed a clear increase between source-to-receptor sites, with 26 ± 10% in Delhi, 39 ± 6% at BCOB, and 74 ± 13% at MCOH (Fig. 3A). This is consistent with ranges from 30% to more than 70% of total OC reported also earlier for South Asia (21–23, 25, 26). The δ13CWSOC signature also increased along the same Delhi-BCOB-MCOH source-to-receptor sequence, with only a minor variability at each site. On average, the lightest/depleted 13CWSOC component was observed at Delhi (−25.4 ± 1.0‰) followed by BCOB (−24.2 ± 0.6‰), with the heaviest/enriched 13CWSOC being observed at MCOH (−20.9 ± 0.6‰).
Fig. 3. Evolution of WS-BrC in the South Asian outflow.

(A) The fractional contribution of WSOC, water-insoluble OC (WIOC), and stable carbon isotope ratio of WSOC (δ13CWSOC) at Delhi, BCOB, and MCOH for the synoptic period of the SAPOEX-16 campaign when air masses were overwhelmingly from IGP and over the Bay of Bengal to MCOH (and thus similar air masses connecting the three stations, see figs. S1 and S2). (B) Absorption coefficient (babs) and mass absorption cross section of WS-BrC at 365 nm (MACWS-BrC 365) and AAE for 330 to 400 nm at Delhi, BCOB, and MCOH during the SAPOEX-16 campaign. Note the order of magnitude difference in scale for babs of MCOH compared to Delhi and BCOB.
Light absorption of WS-BrC in the South Asian outflow
WS-BrC is characterized by comparably high AAEs and thus predominantly absorbs at UV wavelengths. During SAPOEX-16, measurements of WS extracts of BrC revealed large differences in light absorption characteristics between source-to-receptor sites (Fig. 3B). The average mass absorption cross section measured at 365 nm (MACWS-BrC 365) at Delhi (2.5 ± 0.3 m2 g−1) and BCOB (1.4 ± 0.2 m2 g−1) was nearly an order of magnitude higher compared to at the receptor site MCOH (0.4 ± 0.1 m2 g−1). Hence, the MACWS-BrC 365 at MCOH decreased to only ~16% of the value in the IGP. The imaginary part of the refractive index at 365 nm (KWS-BrC) was derived using the measured MACWS-BrC 365 values (see note S5 and fig. S4). The KWS-BrC values during SAPOEX-16 period over South Asia were relatively higher than reported for other geographical regions (17). Corresponding to the MACWS-BrC 365, the wavelength dependence given as the AAE between 330 and 400 nm (AAE330–400nm) increased along the sequence Delhi-BCOB-MCOH. The variability of optical properties of WS-BrC is high over the IGP, e.g., Delhi, compared to MCOH. The dynamics of the local boundary layer with lower mixing heights and considerable diurnal mixing height variability (0.1 to 2.6 km), as well as the influence of advected aerosols from upwind crop-residue burning regions along with variable local emissions, strongly affect the aerosol concentrations and optical properties in nearer-source regions such as the IGP (26). The monotonic increase in AAE330–400nm and decrease in MACWS-BrC 365 between the source-to-receptor sites signify the evolving light absorption characteristics of WS-BrC during long-range transport.
DISCUSSION
The SAPOEX-16 high-intensity campaign allowed a synoptic study of the source-receptor dynamics of WS-BrC in the outflow from IGP—one of the globally largest source region. The changing composition and behavior of WS-BrC were herewith possible to investigate by intercepting and characterizing the emissions from sources in the IGP (megacity Delhi) in a system with consistency in NE monsoon geographical region and after long-range over-ocean transport, with limited additional sources along the way. The surface concentrations of the aerosol components, e.g., WS-BrC, are highest in Delhi while lower at BCOB and lowest at MCOH. However, the regional AOD distribution (as seen in Fig. 1) shows a maximum around Bangladesh (BCOB) and not over Delhi. This is likely due to differences in vertical pollution gradients and in the cumulative boundary layer loading of pollution (1). Delhi has strong ground-based sources (and thus high ground-based concentrations), while its pollution has not yet had time to fully mix through the vertical column (reflected by the AOD). In contrast, the BCOB-downwind receptor observatory lacks strong local sources (and thus lower surface concentrations), yet the total vertically integrated pollution load that has accumulated and has time to mix vertically during transport through the IGP gives a high columnar AOD at BCOB. A clear evolution of both increasing carbon isotope signature (δ13CWSOC) and consistently trending optical properties of WS-BrC (decreasing MACWS-BrC 365 and increasing AAE) was revealed during the long-range transport from source-to-receptor sites in South Asia.
Isotope forensics of aerosol processing
The δ13C signature can shed light on both sources and atmospheric processing. Emissions from various sources cover a wide range of δ13C values, including marine-, fossil-, and land-based vegetation types. The δ13C signature is also influenced by kinetic isotope effects from atmospheric processes, such as oxidation, oligomerization, and condensation of semivolatile species onto particles (27–29). Studies suggest that aerosol photochemical aging (e.g., photodissociation and photooxidation) during formation and transport can lead to a net release of CO2, CO, or volatile organic compounds of isotopically lighter carbon, which leads to relatively enriched (i.e., increasing) δ13C values in the remaining aerosols (30–33). In contrast, aerosol carbon (aerosol-C) stemming from the formation of secondary organic aerosol (SOA) carries a net depleted δ13C signal as SOA-C is a reaction product, as opposed to a residual C pool (34, 35). At a source region like Delhi, the δ13C signature reflects a mixture of primary and secondary sources. However, aging will act on both of these two pools, inducing 13C enrichment (29). Hence, δ13C can be used as a diagnostic for aging of the carbonaceous aerosols during long-range transport (23, 29, 32).
The δ13CWSOC signatures observed at MCOH during SAPOEX-16 overlap with the isotopic range of marine-biogenic emissions. However, combining isotopic data with measurements of WSOC and inorganic ratios allow the exclusion of any significant contributions from marine-biogenic sources. The ratios of nss-SO42− to total SO42− and nss-K+ to total K+ were 98 ± 1% and 97 ± 4%, respectively, suggesting peripheral marine-biogenic contribution (fig. S5A). The limited effect of mixing of terrestrial and marine-biogenic sources on the light absorption of WS-BrC is further elaborated using a theoretical model in note S1 and shown in fig. S7. Furthermore, the size distribution data for MCOH demonstrate the absence of an Aitken mode (~50 nm), which would be expected for new particle formation from a marine-biogenic source and which instead centers around 100 nm (24), consistent with long-range transport of polluted air from the IGP (see aerosol size distribution at MCOH during SAPOEX-16 in fig. S5B). Last, this is also consistent with a contribution from hydroxyl functional groups of only ~10% in OM reported for an earlier winter campaign at MCOH, which is much lower than the ~50% expected for a marine-dominated aerosol regime (23). Together, this supports the notion of insignificant marine-biogenic contribution to the aerosols at MCOH and instead a predominance of long-range transport of the massive air pollution from the IGP. The enrichment in δ13CWSOC between source-to-receptor sites during SAPOEX-16 thus suggests a significant role of aerosol photochemical aging.
Evaluation of different mechanisms for observed optical dynamics
The absorptive properties of WS-BrC may be influenced by matrix effects, e.g., the pH. For South Asia, the variability of the pH is expected to vary only within a limited range due to buffering from the regional soil dust particles (36). The pH changes were assessed with a model and measurements, yielding very small variations across the three studied systems (note S6 and table S2). Furthermore, we investigated the effect on optical properties of the small pH differences between Delhi (6.14 ± 0.03) over BCOB (5.84 ± 0.16) to MCOH (5.76 ± 0.20). The slightly decreasing pH of WS extracts between Delhi-BCOB-MCOH was not comparable to the observed large changes in MAC and AAE. Last, the pH was adjusted for MCOH samples to the pH of BCOB (and further to the pH of Delhi), and there were no changes in MAC (<0.01 m2 g−1) or AAE (<0.03) due to these relatively small pH adjustments (fig. S6). Thus, we conclude that intersite pH variability does not affect the WS-BrC optical properties reported here.
Changes in aerosol size distribution during long-range transport may potentially affect the optical properties. However, aerosol WSOC and WS-BrC have been found to be predominantly in the fine mode for both fresh and aged aerosols (4). As seen in Fig. 4, in the present study, we have compared the MACs and AAEs of WS-BrC from two campaigns conducted at MCOH: the present 2016 SAPOEX campaign and the 2012 CARDEX campaign. Virtually no differences (both P values are <0.05) are observed among these results, despite the fact that PM1 samples were collected during SAPOEX-16 and PM2.5 samples were collected during CARDEX-12. Furthermore, the WS-BrC MACs and AAEs from the 2016 SAPOEX study from Delhi (PM2.5) are overlapping with our 2012 Delhi study, where total suspended particle filters were collected. Moreover, the average values of MACWS-BrC 365 (0.4 ± 0.1 m2 g−1) and AAE330–400nm (6.9 ± 0.5) in PM1 fraction at MCOH in the present study are also comparable with values of MAC365 (0.5 ± 0.2 m2 g−1) and AAE300–700nm (6.9 ± 1.9) reported in PM10 size fraction over the Bay of Bengal (21). Overall, within the framework of the present study, we find it unlikely that the changes in WS-BrC light absorption are driven by changes in the aerosol size distributions during long-range transport.
Fig. 4. Atmospheric dynamics of WS-BrC optical properties.
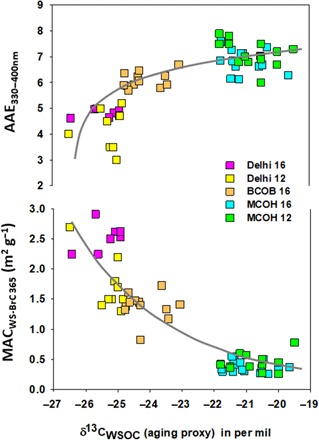
Measurements of the two–optical parameter mass absorption cross section at 365 nm (MACWS-BrC 365) and AAE for 330 to 400 nm (AAE330–400nm) of WS-BrC are shown. MACWS-BrC 365 (bottom panel) and AAE330–400nm (top panel) from SAPOEX-16 are shown with suffix “16,” and previous campaigns measured separately at Delhi (26) and MCOH (23) are shown with suffix “12” (studies performed in the year 2012). A first-order aging model fit (gray line) elucidates the interdependence of MACWS-BrC 365, AAE330–400nm, and atmospheric processing (i.e., bleaching) during source-to-receptor transport in the South Asian outflow (see note S3 for mathematical formulation). δ13CWSOC is used as proxy for aging. The goodness of fit for MACWS-BrC 365 was root mean square deviation (RMSD) of 0.34 and normalized RMSD (NRMSD) of 0.27, with the corresponding values for AAE330–400nm being RMSD of 0.80 and NRMSD of 0.13.
Both laboratory and field experiments have shown that the WS-BrC light absorption is highly dynamic, with photoenhancement through the formation of chromophores and photodissociation through oxidation (5, 13, 37). Comparisons of WS-BrC optical properties at near-source sites with remote sites suggest lower MACs and higher AAEs at the receptor sites (21–23, 26, 32, 33, 38). Possible explanations for this dynamic evolution of WS-BrC optical properties in the South Asian outflow are the following: (i) contrasting emissions and mixing of various WSOC sources, (ii) mixture of WS-BrC with newly formed nonabsorbing SOA-WSOC, (iii) conversion of BrC from water-insoluble OC (WIOC) to WS pool leading to addition of nonabsorbing WSOC, and (iv) photolysis of chromophores and atmospheric oxidation during long-range transport that can change the composition and thus optical properties of WS-BrC. Below, we evaluate several of these putatively important effects.
Several previous studies have reported very similar 14C-WSOC values between source and receptor sites over South Asia (23, 26, 32), suggesting limited input of new aerosol-C. Addition of nonabsorbing (colorless) SOA-WSOC could putatively cause a reduction in MACWS-BrC 365. However, the AAE should not be affected by this process. What we observe here is that AAE330–400nm increases by a factor of ~2 during transport from Delhi to MCOH. This is in line with previous observations of limited diurnal variability in OC/EC ratios at MCOH, suggesting the limited role of fresh biogenic-SOA formation (23). Thus, we conclude that simple dilution from SOA cannot explain the evolution of light absorption of WS-BrC. Another possible mechanism is the potential conversion of WIOC to WSOC during transport, as has been elaborated using a theoretical model in note S2 and fig. S8. This suggests that ~50% of the WSOC at MCOH originates from the oxidation of WIOC (increase in WSOC/OC ratio between the sites can be seen in Fig. 3A). Thus, this could potentially lead to a dilution. The MACWS-BrC 365 value at MCOH is ~0.4 m2 g−1, while it is ~2.5 m2 g−1 at Delhi. Under the simplified assumption that it is oxidation of WIOC resulting in colorless WSOC, we would expect that MACMCOH/MACDelhi is ~0.5. However, what is observed is ~0.16. Together, these considerations and observations support our hypothesis that the reduction of MACWS-BrC 365 during transport is not driven by the addition of colorless WSOC. Instead, there is evidence for sunlight disrupting the conjugated structure of chromophores in UV-visible (UV-vis) regions. It is known that the WSOC pool contains chromophores such as aromatic rings and nitro and phenolic groups (8). Hence, it is likely that photolysis may contribute to the changing properties of WSOC during long-range transport (14). Specifically, direct photolysis by sunlight may efficiently photobleach the overall WS-BrC by affecting a suite of different chromophores with inherently specific photolysis rates (15). In addition, it is also known that atmospheric aging processes like oxidation of electron acceptors lead to a reduction in light absorption (5, 37, 39, 40).
Connecting isotopic shifts and changing aerosol absorption properties
Atmospheric processing by means of oxidation would simultaneously cause an enrichment in the δ13C signature and a shift in the spectral dependency and absorptivity. This is precisely what was observed during SAPOEX-16 (Fig. 4). MACWS-BrC 365 and AAE330–400nm showed clear but opposing trends with respect to the increasing δ13CWSOC signature. The loss of absorption may be caused by direct photodestruction of the chromophores and/or from photochemical oxidation; we jointly refer to these two processes as bleaching. In the present study, we cannot distinguish which mechanism is dominant in South Asia.
During SAPOEX-16, we observe a monotonic increase in AAE330–400nm between source-to-receptor sites in the South Asian outflow. This change in AAE330–400nm correlates with a monotonic increase of the δ13C signature of WSOC and thereby aging. During transport, some of the chromophores in WS-BrC are more likely to be bleached, as they have different absorption wavelength dependencies and have different propensities for bleaching (14, 15). This will cause a shift in the absorption of the mixture and thereby the AAE. However, why this causes the AAE to increase (and not decrease)—blueshift—is currently not clear. We thus hypothesize that the fate of WS-BrC light absorption in the South Asian outflow is therefore likely related to atmospheric photochemical oxidation of its chromophoric moieties. This hypothesis is in line with the results of the aging model fit and observations (see note S3 for mathematical derivation) and is also consistent with indications from several independent chamber studies that have observed similar trends in the optical properties of WS-BrC during aging (5, 14–16, 37, 40). These findings further strengthen our postulation that atmospheric photochemical oxidation is an important driver for the observed loss of WS-BrC absorptivity in the South Asian atmosphere.
Bounding the light absorption of BrC in South Asian outflow
Modeling studies of BrC optical properties and direct radiative forcing (DRF) have found it challenging to fully evaluate model performances due to a lack of observational constraints (7, 9). To that end, the climate impact of BrC predicted from some models, when included at all, are associated with large uncertainties, including assumptions of constant values in BrC absorption properties and atmospheric longevity (17, 18). Here, we found in the ambient South Asian atmosphere that bleaching significantly affects WS-BrC. Our findings show that atmospheric photochemical oxidation decreases the refractive indices of WS-BrC by ~10-fold in the real atmosphere, which is much greater than previously found in chamber studies (16). To quantitatively estimate the bleaching effect of WS-BrC, the apparent first-order rate of bleaching of ambient WS-BrC was calculated on the basis of the source-to-receptor observations for the South Asian outflow (Eq. 1). The synoptic air mass transport time associated with the over-ocean (i.e., BCOB to MCOH) WS-BrC plume evolution in combination with measurements of MACWS-BrC 365 indicated that the ambient WS-BrC bleached with an atmospheric half-life of 3.6 ± 0.8 days (Fig. 5). A first-order bleaching rate constant of WS-BrC was thus estimated to be 0.20 ± 0.05 day−1. The rate of change for AAE330–400nm was 0.12 ± 0.07 day−1 for the South Asian outflow. The present finding of WS-BrC bleaching half-life for one of the world’s largest BrC emission regions, South Asia, is substantially higher than previously observed half-life of BrC (9 to 15 hours) from continental origin (6).
Fig. 5. Bounding the light absorption of WS-BrC in the South Asian outflow.
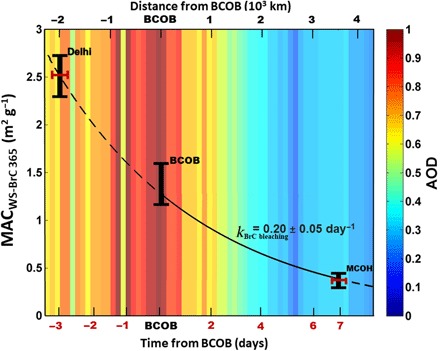
The decay/bleaching of the mass absorption cross section at 365 nm (MACWS-BrC 365) (vertical bars in black) of WS-BrC between source-to-receptor sites in the South Asian outflow is shown. The first-order rate of bleaching (k) is constrained (using Eq. 1) for the over-ocean transport of the BrC plume between BCOB and MCOH (solid line). The transport times of BrC plume evolution (horizontal bars in red) are calculated from BCOB as forward trajectories to MCOH and BTs to Delhi using the HYSPLIT model. The AOD at 550 nm is shown in combination with distance from BCOB. The AOD was obtained from NASA MODIS for the SAPOEX-16 campaign period.
Modeling studies have suggested that BrC may be an important atmospheric light-absorbing species in the South Asian region (9, 18). A lack of consensus on the DRF of BrC likely contributes to the model observation mismatch in AAOD estimates over South Asia. Up until now, the few climate model studies that have included the warming effects of BrC have treated the absorptivity (MAC) as a constant property (9, 17). However, to properly assess the climate impact of BrC, the dynamic nature must also be accounted for. Given the complexity of the BrC pool, it is expected to be very challenging to capture the loss of absorption resolved by an atmospheric chemistry module. Instead, the present systematic investigation of WS-BrC suggests that the loss of absorptivity—bleaching—may be parametrized as a generic atmospheric half-life term, quantified here as 3.6 ± 0.8 days. The implication of this finding is that the climate warming effects of WS-BrC are expected to be more regional in nature. Thus, the near-source warming effects may be large, but the effect is reduced comparably rapidly with atmospheric transport.
Although WSOC is a dominant part of aerosol-C over the regional scale (i.e., represented by MCOH in the present study), it should be noted that a substantial fraction of BrC may also be water-insoluble with other mechanisms, likely affecting its bleaching process (38). The fate of the water-insoluble BrC fraction remains to be investigated in more detail. We also stress that the present study deals with the aqueous BrC phase, and deriving the corresponding light absorption characteristics for the particle BrC phase requires correction factors (11).These factors can be greater than 1 when the light-absorbing compounds are enriched in accumulation mode aerosols. However, these factors are not well constrained, and it is unclear if the factors change during aerosol transport (4).
Together, this study highlights the significance of atmospheric processing effects on the optical properties of WS-BrC aerosols in the ambient South Asian atmosphere. The change in these optical properties with increasing aging time scales implies that current first-generation modeling-based estimates of DRF due to BrC light absorption, relying on set parameter values for BrC optical properties, may be inaccurate for South Asia. To evaluate accurately the climate effects of BrC, it is thus important to consider not only the lifetime of the aerosols themselves in modeling efforts but also the lifetimes of the intrinsic wavelength-dependent absorptive properties.
MATERIALS AND METHODS
Aerosol sampling
This study was part of the SAPOEX-16, a campaign conducted between 2 January 2016 and 20 March 2016, with sampling conducted at the Delhi observatory of the Indian Institute of Tropical Meteorology [28°35′N, 77°12′E ; 15 m above ground level (agl)], BCOB (22°17′N, 90°71′E ; 10 m agl) located on Bhola Island in the delta of Bay of Bengal about 300 km south of Dhaka (capital city of Bangladesh), and MCOH (6°78′N; 73°18′E; 1.5 m agl) located in the northern Indian Ocean. Detailed information about the observatory at MCOH and the Delhi operations can be found in (26) and (32), respectively.
At Delhi, PM2.5 aerosol samples (n = 30) were collected using an Air Pollution Monitoring 550 aerosol sampler (Envirotech Pvt. Ltd., India) equipped with U.S. Environmental Protection Agency standardized impactors, operated at 1 m3 hour−1 with a sampling duration of 12 hours (day and night). At BCOB, PM2.5 aerosol samples (n = 24) were collected using a high-volume sampler (model DH77, DIGITEL A.G., Switzerland) operated at 500 liters/min, with a sampling duration of 24 hours. At MCOH, PM1 aerosol samples (n = 43) were collected every 48 hours using a high-volume sampler (model DH77, DIGITEL A.G., Switzerland), operated at 500 liters/min. At MCOH, located at the northern tip of the island away from any local activities, wind-censored sampling was implemented during SAPOEX-16 to avoid any local island pollution. The sampler switched off automatically when wind speed was <1.2 m/s and when wind direction was southwest between 180° and 270°.
Aerosol samples were collected on precombusted (5 hours at 450°C) quartz fiber filters (Millipore, Billerica, MA). Filter blanks were collected approximately three times per month at each site during the campaign. Overall, 34 samples from the campaign were analyzed for WSOC concentrations, stable carbon isotope composition (δ13CWSOC), and optical properties (MACWS-BrC and AAE).
Concentration measurements of carbonaceous aerosols
The aerosol OC, EC, and total carbon (TC) concentrations were measured with a thermal-optical transmission analyzer (Sunset Laboratory, Tigard, OR, USA) using the National Institute for Occupational Safety and Health 5040 method. Potassium hydrogen phthalate was used as an external standard to verify the accuracy of the measurement of OC and TC during the analysis. These analyses gave an overall analytical uncertainty of <2.8% (1 SD, for n = 5).
The WSOC concentration was measured following earlier protocols [(33), with slight modification in (23)]: WSOC was extracted in water using ultrasonication, followed by centrifugation of the filter punches, and finally quantified in the filtered solutions using a high-temperature catalytic oxidation instrument (Shimadzu-TOC-VCPH analyzer; Shimadzu, Kyoto, Japan), following the nonpurgeable OC protocol. The presently used water-to-sample ratio was in the range of 0.10 to 0.24 cm3 m−3 (= volume extraction solvent/volume air sampled), which is well within the optimal water-to-sample range (>0.1 cm3 m−3), as suggested by theoretical considerations.
Concentration values of all measured parameters were blank-corrected by subtracting an average of the field blanks. The average relative SDs of triplicate analysis for TC and WSOC were respectively 3.1 and 7.0% at Delhi, 2.4 and 9.4% at BCOB, and 2.8 and 3.0% at MCOH. Detection limit for WSOC was 0.20 μg m−3 at Delhi, 0.16 μg m−3 at BCOB, and 0.14 μg m−3 at MCOH. EC was below detection limit in the blanks. The overall precision in the TC and WSOC concentrations and isotopic signatures was estimated, considering the analytical precision of concentration measurements (estimates from triplicate analysis), mass contributions from field blanks (estimates from several blanks), estimates from analysis of standard reference material NIST (National Institute of Standards and Technology) SRM (Standard Reference Material) 8785, and the precision of the isotope characterization (instrument precision) and the isotope signature of the field blanks.
Carbon isotope analysis of WSOC
The analytical method for the isolation of WSOC to determine δ13C builds on the method described previously (23, 33). Briefly, a filter area corresponding to at least 120 μg of WSOC was extracted in 10 ml of Milli-Q water. The WSOC extracts were freeze-dried and then successively redissolved in 150 μl of 1 M hydrochloric acid to decarbonate the samples. The samples were then transferred into precombusted (450°C for 5 hours) silver capsules and finally evaporated in the oven at 60°C. The dried WSOC samples were then ready for isotope analyses.
Measurements of the stable carbon isotope composition of WSOC (δ13C) were performed at the Stable Isotope Facility at Stockholm University (Stockholm, Sweden). The instrumental method used sample combustion with a Carlo Erba NC 2500 analyzer connected to a Finnigan MAT Delta V mass spectrometer via a split interface to reduce the gas volume.
Absorption measurements
The light extinction of water extracts (WSOC samples) was measured in the wavelength range of 190 to 1100 nm using a Hitachi U2010 UV-vis absorption spectrophotometer. For comparison with previous studies (23, 26), MACWS-BrC was computed for 365 nm and AAE was fitted within the range of 330 to 400 nm (see note S4 for mathematical derivation of MACWS-BrC 365 and AAE).
Ion analysis
WS inorganic constituents (Na+, NH4+, K+, Ca2+, Cl−, NO3−, and SO42−) were measured on a Dionex Aquion ion-chromatography (IC) system (Thermo Finnigan LLC). Water extracts from each quartz fiber filter were injected into the IC operating at 1.0 ml min−1. The Dionex system contains a guard column and an anion-cation separator column with a strong basic exchange resin and a suppressor column (AERS500/CERS 500) also with strong acid ion exchange resin. The anion eluent consisted of sodium carbonate (Na2CO3) and sodium bicarbonate (NaHCO3) solutions (1 M), and the cation eluent consisted of methanesulfonic acid solution (20 mM) prepared in double-distilled water. The instrument was calibrated using commercial Merck standards (~1000 parts per million). The quality assurance of the analytical data (within 5%) was ascertained by preparing standard stock solutions from analytical grade reagents and salts. The concentrations in the aerosol sample were suitably corrected for contribution from the blank filters.
Conceptual aging model for WS-BrC degradation in the South Asian outflow
We formulated an aging model based on the assumption that bleaching is the main driver of the time evolution of WS-BrC light absorption. Furthermore, as the time evolution of the δ13CWSOC signature is also affected by atmospheric processing, we used this parameter as a proxy for aging. A first-order decay of the light absorption was then expressed as a function of the δ13CWSOC signature (mathematical derivations in note S3). Within this model, we then obtained interrelations between the three parameters MACWS-BrC, δ13CWSOC, and AAE, which were fitted with the measurement-derived parameters from SAPOEX-16, as well as previous single-site campaigns from (23, 26), performed separately at Delhi and MCOH, respectively.
Calculation of the rate of WS-BrC bleaching during atmospheric transport
For the South Asian outflow, the rate of WS-BrC bleaching was calculated as
| (1) |
where k (day−1) is the apparent first-order WS-BrC bleaching rate constant and t (day) is the mean synoptic air mass transport time associated with the BrC plume evolution for the over-ocean transport of the outflow from the IGP from BCOB to MCOH.
The over-ocean transport has limited contribution from anthropogenic sources, and thus, the rate of WS-BrC bleaching was estimated for the plumes that were found to be transported between BCOB and MCOH. Air mass transport times were based on the BTs from the Hybrid Single-Particle Lagrangian Integrated Trajectory (HYSPLIT) model. For the month of January, all BTs between BCOB and MCOH were computed (details in the next section). The BTs also demonstrated that the various investigated plumes were downwind from the IGP only. On the basis of the time for transport of the plumes from BCOB to MCOH, the mean synoptic transport time was estimated to investigate possible effects of bleaching on WS-BrC evolution. The half-life was estimated as [Ln (2)/k].
Air mass BTs and cluster analysis
The sampling sites Delhi, BCOB, and MCOH encountered air masses from various geographical regions. Nine-day air mass BTs were generated for MCOH at an arrival height of 100 m, computed every 3 hours, using the National Oceanic and Atmospheric Administration HYSPLIT model (version 4) in such a way that air masses span across the preceding sites as well. Five-day and 3-day BTs were also generated for BCOB and Delhi, respectively. We assessed the relative contribution of each cluster for every sample collected at Delhi, BCOB, and MCOH and focused on “synoptic” IGP air mass cluster for the present BrC study. Overall, 7 samples from Delhi, 13 samples from BCOB, and 19 samples from MCOH collected during this period were such synoptic samples.
Supplementary Material
Acknowledgments
We would like to thank the Maldives Meteorological Services (MMS) and the government of the Republic of the Maldives for joint operation of MCOH, and especially technicians at MCOH for support with instruments. We would also like to thank the University of Dhaka in Bangladesh for supporting the BCOB. This study also benefitted from the research environment provided by the Bolin Centre for Climate Research at the Stockholm University. Funding: We acknowledge financial support from the Swedish Research Council for Sustainable Development (FORMAS contract no. 942-2015-1061), the Swedish Research Council (VR contract no. 348-2014-4244; 2017-01601), and Center on Excellence in Atmospheric Science funded by the Finnish Academy of Sciences Excellence (grant no. 307331). Author contributions: Ö.G., A.A., S.B., H.H., S.S., E.A., J.B., A.S., S.T., and Z.H. designed the SAPOEX-16 campaign. Samples were collected by S.D., S.B., K.B., and J.K. at MCOH, by A.S. at BCOB, and by D.S.B. and S.T. at Delhi. Ö.G. and A.A. conceived the study. S.D. analyzed the samples. S.D., Ö.G., and A.A. wrote the paper and produced the figures, with input from all coauthors. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the corresponding author (Ö.G.). The data will also be available in the Bolin Center Database (http://bolin.su.se/data/Dasari-2018).
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/5/1/eaau8066/DC1
Note S1. A theoretical model for ruling out marine-biogenic source contribution.
Note S2. A theoretical model for the degradation of WSOC and WIOC in the South Asian outflow.
Note S3. A conceptual aging model for the joint time and wavelength dependence of MACWS-BrC.
Note S4. Absorption measurements of MACWS-BrC and AAE.
Note S5. Estimating the imaginary part of the refractive index of WS-BrC.
Note S6. Testing a putative effect of pH on WS-BrC optical properties over the South Asian region.
Fig. S1. Air mass clusters during SAPOEX-16.
Fig. S2. Fractional contribution of air mass clusters during SAPOEX-16.
Fig. S3. Concentrations of EC, OC, and WSOC during SAPOEX-16.
Fig. S4. Imaginary part of the refractive index (KWS-BrC) at 365 nm.
Fig. S5. Asserting the peripheral contribution of marine-biogenic sources at MCOH during SAPOEX-16.
Fig. S6. Testing a putative effect of pH on WS-BrC optical properties.
Fig. S7. Constraining the mixing of WSOC sources on WS-BrC light absorption in the South Asian outflow.
Fig. S8. Degradation of WSOC and WIOC during long-range transport in the South Asian outflow.
Table S1. Concentrations (mean ± SD) and element mass ratios of carbonaceous species during SAPOEX-16.
Table S2. pH of aerosol WSOC extracts during SAPOEX-16.
REFERENCES AND NOTES
- 1.Lawrence M. G., Lelieveld J., Atmospheric pollutant outflow from southern Asia: A review. Atmos. Chem. Phys. 10, 11017–11096 (2010). [Google Scholar]
- 2.Bond T. C., Doherty S. J., Fahey D. W., Forster P. M., Berntsen T., DeAngelo B. J., Flanner M. G., Ghan S., Kärcher B., Koch D., Kinne S., Kondo Y., Quinn P. K., Sarofim M. C., Schultz M. G., Schulz M., Venkataraman C., Zhang H., Zhang S., Bellouin N., Guttikunda S. K., Hopke P. K., Jacobson M. Z., Kaiser J. W., Klimont Z., Lohmann U., Schwarz J. P., Shindell D., Storelvmo T., Warren S. G., Zender C. S., Bounding the role of black carbon in the climate system: A scientific assessment. J. Geophys. Res. Atmos. 118, 5380–5552 (2013). [Google Scholar]
- 3.Gustafsson Ö., Ramanathan V., Convergence on climate warming by black carbon aerosols. Proc. Natl. Acad. Sci. U.S.A. 113, 4243–4245 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Liu J., Bergin M., Guo H., King L. E., Kotra N., Edgerton E., Weber R. J., Size-resolved measurements of brown carbon in water and methanol extracts and estimates of their contribution to ambient fine-particle light absorption. Atmos. Chem. Phys. 13, 12389–12404 (2013). [Google Scholar]
- 5.Wong J. P. S., Nenes A., Weber R. J., Changes in light absorptivity of molecular weight separated brown carbon due to photolytic aging. Environ. Sci. Technol. 51, 8414–8421 (2017). [DOI] [PubMed] [Google Scholar]
- 6.Forrister H., Liu J., Scheuer E., Dibb J., Ziemba L., Thornhill K. L., Anderson B., Diskin G., Perring A. E., Schwarz J. P., Jost P. C., Day D. A., Palm B. B., Jimenez J. L., Nenes A., Weber R. J., Evolution of brown carbon in wildfire plumes. Geophys. Res. Lett. 42, 4623–4630 (2015). [Google Scholar]
- 7.Chung C. E., Ramanathan V., Decremer D., Observationally constrained estimates of carbonaceous aerosol radiative forcing. Proc. Natl. Acad. Sci. U.S.A. 109, 11624–11629 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Laskin A., Laskin J., Nizkorodov S. A., Chemistry of atmospheric brown carbon. Chem. Rev. 115, 4335–4382 (2015). [DOI] [PubMed] [Google Scholar]
- 9.Feng Y., Ramanathan V., Kotamarthi V. R., Brown carbon: A significant atmospheric absorber of solar radiation? Atmos. Chem. Phys. 13, 8607–8621 (2013). [Google Scholar]
- 10.Zhang Y., Forrister H., Liu J., Dibb J., Anderson B., Schwarz J. P., Perring A. E., Jimenez J. L., Campuzano-Jost P., Wang Y., Nenes A., Weber R. J., Top-of-atmosphere radiative forcing affected by brown carbon in the upper troposphere. Nat. Geosci. 10, 486–489 (2017). [Google Scholar]
- 11.Sun H., Biedermann L., Bond T. C., Color of brown carbon: A model for ultraviolet and visible light absorption by organic carbon aerosol. Geophys. Res. Lett. 34, L17813 (2007). [Google Scholar]
- 12.Nguyen T. B., Lee P. B., Updyke K. M., Bones D. L., Laskin J., Laskin A., Nizkorodov S. A., Formation of nitrogen- and sulfur-containing light-absorbing compounds accelerated by evaporation of water from secondary organic aerosols. J. Geophys. Res. Atmos. 117, D01207 (2012). [Google Scholar]
- 13.Lambe A. T., Cappa C. D., Massoli P., Onasch T. B., Forestieri S. D., Martin A. T., Cummings M. J., Croasdale D. R., Brune W. H., Worsnop D. R., Davidovits P., Relationship between oxidation level and optical properties of secondary organic aerosol. Environ. Sci. Technol. 47, 6349–6357 (2013). [DOI] [PubMed] [Google Scholar]
- 14.Lee H. J., Aiona P. K., Laskin A., Laskin J., Nizkorodov S. A., Effect of solar radiation on the optical properties and molecular composition of laboratory proxies of atmospheric brown carbon. Environ. Sci. Technol. 48, 10217–10226 (2014). [DOI] [PubMed] [Google Scholar]
- 15.Romonosky D. E., Ali N. N., Saiduddin M. N., Wu M., Lee H. J., Aiona P. K., Nizkorodov S. A., Effective absorption cross sections and photolysis rates of anthropogenic and biogenic secondary organic aerosols. Atmos. Environ. 130, 172–179 (2016). [Google Scholar]
- 16.Sengupta D., Samburova V., Bhattarai C., Kirillova E., Mazzoleni L., Iaukea-Lum M., Watts A., Moosmüller H., Khlystov A., Light absorption by polar and non-polar aerosol compounds from laboratory biomass combustion. Atmos. Chem. Phys. 18, 10849–10867 (2018). [Google Scholar]
- 17.Wang X., Heald C. L., Ridley D. A., Schwarz J. P., Spackman J. R., Perring A. E., Coe H., Liu D., Clarke A. D., Exploiting simultaneous observational constraints on mass and absorption to estimate the global direct radiative forcing of black carbon and brown carbon. Atmos. Chem. Phys. 14, 10989–11010 (2014). [Google Scholar]
- 18.Jo D. S., Park R. J., Lee S., Kim S.-W., Zhang X., A global simulation of brown carbon: Implications for photochemistry and direct radiative effect. Atmos. Chem. Phys. 16, 3413–3432 (2016). [Google Scholar]
- 19.Bikkina S., Sarin M., PM2.5, EC and OC in atmospheric outflow from the Indo-Gangetic Plain: Temporal variability and aerosol organic carbon-to-organic mass conversion factor. Sci. Total Environ. 487, 196–205 (2014). [DOI] [PubMed] [Google Scholar]
- 20.Ramana M. V., Ramanathan V., Feng Y., Yoon S.-C., Kim S.-W., Carmichael G. R., Schauer J. J., Warming influenced by the ratio of black carbon to sulphate and the black-carbon source. Nat. Geosci. 3, 542–545 (2010). [Google Scholar]
- 21.Bikkina S., Sarin M. M., Light-absorbing organic aerosols (brown carbon) over the tropical Indian Ocean: Impact of biomass burning emissions. Environ. Res. Lett. 8, 044042 (2013). [Google Scholar]
- 22.Bikkina S., Sarin M. M., Brown carbon in atmospheric outflow from the Indo-Gangetic Plain: Mass absorption efficiency and temporal variability. Atmos. Environ. 89, 835–843 (2014). [Google Scholar]
- 23.Bosch C., Andersson A., Kirillova E. N., Budhavant K., Tiwari S., Praveen P. S., Russell L. M., Beres N. D., Ramanathan V., Gustafsson Ö., Source diagnostic dual-isotope composition and optical properties of water-soluble organic carbon and elemental carbon in the South Asian outflow intercepted over the Indian Ocean. J. Geophys. Res. Atmos. 119, 11743–11759 (2014). [Google Scholar]
- 24.Nair V. S., Moorthy K. K., Babu S. S., Influence of continental outflow and ocean biogeochemistry on the distribution of fine and ultrafine particles in the marine atmospheric boundary layer over Arabian Sea and Bay of Bengal. J. Geophys. Res. Atmos. 118, 7321–7331 (2013). [Google Scholar]
- 25.Ram K., Sarin M. M., Tripathi S. N., Temporal trends in atmospheric PM2.5, PM10, elemental carbon, organic carbon, water-soluble organic carbon, and optical properties: Impact of biomass burning emissions in the Indo-Gangetic Plain. Environ. Sci. Technol. 46, 686–695 (2012). [DOI] [PubMed] [Google Scholar]
- 26.Kirillova E. N., Andersson A., Tiwari S., Srivastava A. K., Bisht D. S., Gustafsson Ö., Water-soluble organic carbon aerosols during a full New Delhi winter: Isotope-based source apportionment and optical properties. J. Geophys. Res. Atmos. 119, 3476–3485 (2014). [Google Scholar]
- 27.Miyazaki Y., Kawamura K., Jung J., Furutani H., Uematsu M., Latitudinal distributions of organic nitrogen and organic carbon in marine aerosols over the western North Pacific. Atmos. Chem. Phys. 11, 3037–3049 (2011). [Google Scholar]
- 28.Agnihotri R., Mandal T. K., Karapurkar S. G., Naja M., Gadi R., Ahammmed Y. N., Kumar A., Saud T., Saxena M., Stable carbon and nitrogen isotopic composition of bulk aerosols over India and northern Indian Ocean. Atmos. Environ. 45, 2828–2835 (2011). [Google Scholar]
- 29.Aggarwal S. G., Kawamura K., Molecular distributions and stable carbon isotopic compositions of dicarboxylic acids and related compounds in aerosols from Sapporo, Japan: Implications for photochemical aging during long-range atmospheric transport. J. Geophys. Res. Atmos. 113, D14301 (2008). [Google Scholar]
- 30.Pavuluri C. M., Kawamura K., Evidence for 13-carbon enrichment in oxalic acid via iron catalyzed photolysis in aqueous phase. Geophys. Res. Lett. 39, L03802 (2012). [Google Scholar]
- 31.Pavuluri C. M., Kawamura K., Enrichment of 13C in diacids and related compounds during photochemical processing of aqueous aerosols: New proxy for organic aerosols aging. Sci. Rep. 6, 36467 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kirillova E. N., Andersson A., Sheesley R. J., Kruså M., Praveen P. S., Budhavant K., Safai P. D., Rao P. S. P., Gustafsson Ö., 13C- and 14C-based study of sources and atmospheric processing of water-soluble organic carbon (WSOC) in South Asian aerosols. J. Geophys. Res. Atmos. 118, 614–626 (2013). [Google Scholar]
- 33.Kirillova E. N., Sheesley R. J., Andersson A., Gustafsson Ö., Natural abundance 13C and 14C analysis of water-soluble organic carbon in atmospheric aerosols. Anal. Chem. 82, 7973–7978 (2010). [DOI] [PubMed] [Google Scholar]
- 34.Fisseha R., Spahn H., Wegener R., Hohaus T., Brasse G., Wissel H., Tillmann R., Wahner A., Koppmann R., Kiendler-Scharr A., Stable carbon isotope composition of secondary organic aerosol from β-pinene oxidation. J. Geophys. Res. 114, D02304 (2009). [Google Scholar]
- 35.Miyazaki Y., Fu P. Q., Kawamura K., Mizoguchi Y., Yamanoi K., Seasonal variations of stable carbon isotopic composition and biogenic tracer compounds of water-soluble organic aerosols in a deciduous forest. Atmos. Chem. Phys. 12, 1367–1376 (2012). [Google Scholar]
- 36.Srinivas B., Sarin M. M., Rengarajan R., Atmospheric transport of mineral dust from the Indo-Gangetic Plain: Temporal variability, acid processing, and iron solubility. Geochem. Geophys. Geosyst. 15, 3226–3243 (2014). [Google Scholar]
- 37.Zhong M., Jang M., Dynamic light absorption of biomass-burning organic carbon photochemically aged under natural sunlight. Atmos. Chem. Phys. 14, 1517–1525 (2014). [Google Scholar]
- 38.Zhang X., Lin Y.-H., Surratt J. D., Weber R. J., Sources, composition and absorption Ångström exponent of light-absorbing organic components in aerosol extracts from the Los Angeles Basin. Environ. Sci. Technol. 47, 3685–3693 (2013). [DOI] [PubMed] [Google Scholar]
- 39.Adler G., Flores J. M., Riziq A. A., Borrmann S., Rudich Y., Chemical, physical, and optical evolution of biomass burning aerosols: A case study. Atmos. Chem. Phys. 11, 1491–1503 (2011). [Google Scholar]
- 40.Liu J., Lin P., Laskin A., Laskin J., Kathmann S. M., Wise M., Caylor R., Imholt F., Selimovic V., Shilling J. E., Optical properties and aging of light-absorbing secondary organic aerosol. Atmos. Chem. Phys. 16, 12815–12827 (2016). [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/5/1/eaau8066/DC1
Note S1. A theoretical model for ruling out marine-biogenic source contribution.
Note S2. A theoretical model for the degradation of WSOC and WIOC in the South Asian outflow.
Note S3. A conceptual aging model for the joint time and wavelength dependence of MACWS-BrC.
Note S4. Absorption measurements of MACWS-BrC and AAE.
Note S5. Estimating the imaginary part of the refractive index of WS-BrC.
Note S6. Testing a putative effect of pH on WS-BrC optical properties over the South Asian region.
Fig. S1. Air mass clusters during SAPOEX-16.
Fig. S2. Fractional contribution of air mass clusters during SAPOEX-16.
Fig. S3. Concentrations of EC, OC, and WSOC during SAPOEX-16.
Fig. S4. Imaginary part of the refractive index (KWS-BrC) at 365 nm.
Fig. S5. Asserting the peripheral contribution of marine-biogenic sources at MCOH during SAPOEX-16.
Fig. S6. Testing a putative effect of pH on WS-BrC optical properties.
Fig. S7. Constraining the mixing of WSOC sources on WS-BrC light absorption in the South Asian outflow.
Fig. S8. Degradation of WSOC and WIOC during long-range transport in the South Asian outflow.
Table S1. Concentrations (mean ± SD) and element mass ratios of carbonaceous species during SAPOEX-16.
Table S2. pH of aerosol WSOC extracts during SAPOEX-16.