

Abstract
CircRNAs have been identified play a key role in various different types of cancer. However, their role in lung adenocarcinoma remains unclear. In this study, we explored the specific circular transcriptome and characterized the circRNA expression profiles of five paired lung adenocarcinoma (LAC) tissues relative to adjacent normal tissues from LAC patients using next-generation sequencing (NGS). To illuminate circRNAs function, their gene targets were initially predicted before using, Kyoto Encyclopedia of Genes and Genomes (KEGG) and Gene Ontology (GO) analyses to further analyse the associated significant cell signalling pathways and functions. The potential interactions between circRNA-miRNA-mRNA were also investigated. Additionally, qRT-PCR assay, Western blot and Immunohistochemistry were performed to validate the differential expression of circRNA, microRNA and mRNA in the LAC group in comparison to the control group. Two-hundred-eighty-five dysregulated circular transcripts were found in LAC tissues, among which 102 and 183 were either up or down regulated, respectively. Our biological analysis suggested that the host genes of differentially expressed circRNAs targeted to cancer-related processes and mechanisms. The interaction maps of the circRNA-miRNA-target gene were constructed using Cytoscope. In further study, hsa_circ_0002360 was found to be the most significantly overexpressed circRNA in LAC tissues by interacting with miRNA and its corresponding mRNA. Our results showed that hsa_circ_0002360 was aberrantly and abundantly expressed and implicated in the development of LAC, suggesting a valuable therapeutic target for LAC treatment.
Keywords: Circular RNA, hsa_circ_0002360, lung adenocarcinoma (LAC), RNA-sequencing (RNA-Seq)
Introduction
Based on the 2018 cancer statistics, lung cancer is the second leading cancer in men, comprising 14% of the total new cancer cases and 26% of total cancer deaths. The mortality of lung cancer among women is higher than that for breast cancer, accounting for 25% of total cancer deaths in women. For all types of cancers combined, the 5-year relative survival rate for lung cancers that are typically diagnosed at a late stage is less than 5% [1]. According to histologic types and prognosis, non-small cell lung cancer (NSCLC) accounts for approximately 85% of all types of lung cancer. These are further subcategorized as adenocarcinoma (44%), squamous cell carcinoma (23%), large cell carcinoma (4%), and unspecified lung carcinoma (28%). Adenocarcinoma occurs mostly in the peripheral airways of the lung and is the most common subtype in non-smokers [2]. CircRNAs have been shown to be enriched and stable in exosomes [3], human cell-free saliva [4], and peripheral blood [5], which makes circRNAs a particularly promising candidate biomarker of diseases. This further suggests that the identification of new circRNAs biomarkers which are indicative to the efficacy of targeted therapies, will improve treatment and reduce unwanted side-effects in non-responsive individuals. The development of circRNA biomarkers that could act as diagnostic tools, therapeutic targets or predictors of outcome of disease is urgently needed and will improve the diagnosis and treatment of LAC.
CircRNA was first discovered in RNA viruses via electron microscopy in 1976 [6]. CircRNAs are produced from precursor mRNA (pre-mRNA) by back-splicing circularization, which is processed by the spliceosome and modulated by both cis and trans regulators, resulting in the exclusion of a circular RNA molecule with a 3’,5’-phosphodiester bond at the junction site [7]. However, due to its low abundance, few circRNAs with functional potential were initially identified, and their biogenesis and possible functions were poorly understood. This lead to the conclusion that the majority of circRNAs originate from failed alternative splicing and represent inert splicing by-products [8]. Next-generation RNA sequencing (RNA-seq) of nonpolyadenylated transcriptomes has recently shed new light on the cell type-specific and tissue-specific expression of circRNAs as well as on the regulation of their biogenesis. Abundant circular RNAs were produced by escaping from the debranching of intron lariats in human cells, which were more stable than their parental linear mRNAs, due to their circular structure [9]. Their special closed-loop structure ensures that circRNAs are highly conserved and stable relative to other noncoding RNAs, such as long noncoding RNAs and miRNAs (micro-RNAs). Therefore, circRNAs could be promising biomarkers for diagnosis of many diseases, including cancer. It has been shown that circRNAs can function at the transcriptional or post-transcriptional level by titrating miRNAs, regulating transcription and interfering with pre-mRNA splicing [10]. Recent studies show that circRNAs regulate multiple biological processes via a variety of mechanisms such as binding and preventing microRNAs as sponges to regulate the expression of relevant genes, regulating RNA Pol II transcription, and directing protein synthesis [7,10-13]. Growing evidence has shown an abundance of circRNAs in many species and their implication in important functions in the physiological and pathological processes of several diseases [12]. For instance, circRNAs play roles in neurological diseases and multiple types of cancers [14], and confirms the possibility as promising diagnostic biomarkers and therapeutic target for LAC.
Compared with microarray analysis, NGS is an open technology that can identify undiscovered nucleotide sequences. Here we aim to identify, characterize and compare all expressed circRNAs in LAC tissues and adjacent non-tumor tissues. Furthermore we aimed to understand the potential role of hsa_circ_0002360 in LAC. To discover other potential therapy targets for LAC, we comprehensively investigated the relationship of hsa_circ_0002360 with its potential miRNA-mRNA binding.
Materials and methods
Ethics statement
This project was approved and supervised by the Ethics Committee of the Affiliated People’s Hospital of Jiangsu University. All patients included in this study provided written informed consent before surgery.
Subjects and samples
The study included forty-three patients with LAC who underwent partial or radical surgery at the Department of thoracic surgery, The Affiliated People’s Hospital of Jiangsu University, China between January 2016 and January 2018. Ten subjects with LAC and normal controls (n=5 per group) participated in the RNA-sequencing program. We excluded patients with a history of other types of cancer and any preoperative chemotherapy or radiotherapy prior to this study. After surgery, the tumor tissue and the paired adjacent healthy normal tissue were immediately preserved in liquid nitrogen for RNA isolation. All tissue specimens were examined by histopathologically and confirmed for the presence of LAC. The pathological diagnosis was in accordance with the tumor-node-metastasis classification scheme for each stage (2017 Union for International Cancer Control, UICC) and histopathologic grade of LAC (WHO 2015).
Total RNA isolation and quality control (QC)
Total RNA was isolated from samples using TRIzol reagent (Life Technologies, CA, USA) following the manufacturer’s protocol. The integrity and amount of total RNA samples were measured using a Qubit 2.0 Fluorometer (Thermo Fisher Scientific), NanoDrop 2000 spectrophotometer (Thermo Scientific, USA), and 2200 Tape Station System (Agilent). The spectrophotometric absorbance of RNA samples at 230, 260 and 280 nm was from 1.8 and 2.1 (ratio A260/280), >1.8 (ratio A260/230) was accepted as the integrity criterion. RNA samples were stored at -80°C before use.
RNA library construction and circRNA sequencing analysis
Total RNA from five matched samples of LAC tissues and adjacent non-tumor tissues was treated with Epicenter Ribo-Zero rRNA Removal Kit (Illumina, CA, USA) and RNase R (Epicenter, CA, USA) to remove ribosomal and linear RNA respectively. The enriched RNA was broken into fragments, followed by cDNA synthesis and PCR amplification with terminal modification and linker prim. Then, the RNA-seq libraries were constructed using the TruSeq® Stranded Total RNA HT/LT Sample Prep Kit (Illumina, CA, USA) according to the manufacturer’s protocol. Libraries were assessed for quality and quantified using the Agilent 2100 system (Agilent Technologies, Santa Clara, CA, USA). Paired-end reads were harvested from the Illumina HiSeq X10 sequencer and were quality controlled by Q30. After 3’ adaptor trimming, N 3-containing sequences, the reads less than 50 bp or low-quality were removed using the Cutadapt software (version 1.9.3). The high quality trimmed reads were used subsequently to analyze circRNAs. Sequencing data were aligned to the Human Genome (NCBI xl_ref_Xenopus_laevis_v2) with Bowtie2 software [15], and Find_circ software (e.g., CIRI) [16] were utilized for circRNA identification. To compare differences in circRNA expression profiles between the LAC and adjacent nontumor tissue samples, we calculated the “fold change” (i.e., the ratio of the group averages) between the groups for each circRNA. A t-test was used to estimate the statistical significance of the differences. Significant differential expression of circRNA was defined by fold-change >2.0 or <0.5 with a P-value <0.05. The R software limma package (R version 3.3.1) was used to normalize the raw data and subsequent data processing. We selected the Data/Sort & Filter function in Microsoft Excel to filter the acquired data and rank the differently expressed circRNAs based on various parameters, such as fold-change and P-value. The sequencing procedures and analyses were performed by OE Biotech. (Shanghai, China).
Bioinformatic analysis
To further explore the potential function and signalling pathways of the differently expressed circRNAs in LAC, Gene ontology (GO) term analysis was performed based on a targetome using the DAVID gene annotation tool (http://david.abcc.ncifcrf.gov/) [17]. The -log10 (P-value) yielded an enrichment score representing the significance of GO term enrichment among differentially expressed genes. KEGG analysis was performed to determine the involvement of target genes in different biological pathways using KOBAS software (KEGG Orthology-Based Annotation System) [18]. In this context, the -log10 transformed data (P-value) yielded an enrichment score indicating the significance of pathway correlations.
miRNA prediction and circRNA-miRNA-mRNA network construction
Previous studies have verified that circRNAs contain corresponding miRNA binding sites and can function as miRNA sponges to regulate their potential functions. Therefore, we constructed a circRNA-miRNA co-expression network consisting of the top 300 circRNA-miRNAs with the lowest p-value with Cytoscape software version 3.2.1. To further elucidate the role of circRNAs in LAC, we chose the 10 most dysregulated circRNAs of which, five up-regulated and five down-regulated circRNAs. The different circRNAs and their host genes were screened out using HTseq [19] and DEGSeq [20]. The Arraystar target prediction software was applied to predict the miRNA targets of these circRNAs based on TargetScan and miRanda applications [21]. Then the differently expressed circRNAs were annotated in detail using the circRNA-miRNA interaction information and searched for miRNA response elements (MREs) for the differently expressed circRNAs identified from the sequence data. Cytoscape 3.2.1 was used to diagram the potential map of the circRNA-miRNA-mRNA interaction network in LAC.
Validation of the circRNA candidates using qRT-PCR
To further validate the data from RNA sequencing, the ten preceding significantly different circRNAs were selected and validated by real-time PCR in twenty LAC samples. Specific divergent primers for each circRNA were designed according to the sequence of the linear transcripts, synthesized by Generay Biotech (Generay, PRC), and all divergent primers are shown in Table S1. Total RNA was extracted as described above, digested using RNase R, and purified cDNA was synthesized using the HiScript II Q RT SuperMix for qPCR (Vazyme, Nanjing, China). Real-time PCR was performed on a LightCycler® 480 II Real-time PCR Instrument (Roche, Swiss) according to the manufacturer’s instructions for the QuantiFast® SYBR® Green PCR Kit (Qiagen, Germany). The real-time PCR program was initiated by denaturation at 95°C for 10 min, followed by 40 cycles of 95°C for 10 s and 60°C for 30 s. Expression of circRNA was normalized to glyceraldehyde-3-phosphate dehydrogenase (GAPDH, standard control) and calculated using the 2-ΔΔC method. All experiments were repeated three times for statistical analysis.
Detecting hsa-mir-3620-5p and PHF19 in LAC tissue
qRT-PCR was applied to detect hsa-mir-3620-5p and PHF19 levels in eighteen LAC tissues and adjacent non-tumor tissues. Western blot was performed according to established protocols [22], using anti-PHF19 monoclonal antibody (1:500, Abcam, Cambridge, MA, USA). An anti-α-tubulin (1:1000, Sigma, St. Louis, MO, USA) was used as a loading control. Furthermore, immunohistochemistry was carried out as described previously on tumor tissues and adjacent normal tissues using the appropriate primary (PHF19 (1:100)) and secondary (the HRP-conjugated anti-rabbit) antibodies.
Statistical analysis
Statistical analysis was performed using GraphPad.Prism.v6.01 (GraphPad Software, CA, USA), R software version 3.2.1 (http://www.r-project.org/) and Microsoft Excel (Microsoft, DC, USA). Student’s t-test was used or comparisons between groups. A two-sided P-value of <0.05 was considered statistically significant.
Results
Clinical characteristics of the participants
Based on the histopathological examination results, ten fresh tissue samples of which five were LAC samples, and five were taken from adjacent normal tissue samples from the same patient were finally selected to perform RNA-sequencing in the study. The clinical data of each patient are shown in Table S2.
Differentially expressed circRNAs based on circRNA sequencing analysis
The characteristics of the circRNA expression profiles between LAC tissues and adjacent non-cancerous tissues from five patients were performed by high-throughput human circRNA sequencing. The distribution of the Back-spliced Junction Counts from all the datasets after normalization was visualized and it was shown that the distribution of log2 ratios were similar among the tested samples, except the fourth and fifth normal samples (Figure 1A). Most circRNAs of the samples were from the sense-overlapping portion (89.60%), and a small proportion of circRNAs were aligned to exonic (3.99%), intronic (0.95%), intergenic (4.51%), and antisence (0.95%) (Figure 1B). Moreover, to examine the genomic localization of the differentially expressed LAC circRNAs, the distribution of differentially expressed circRNAs along the human chromosomes were analyzed and the up-regulated and down-regulated circRNAs in each chromosome were displayed (Figure 1C). Cluster analysis segregated samples into different groups based on their expression levels and hypothetic relationships among the samples. The hierarchical clustering results showed statistically different circRNA expression profiling among 10 samples (Figure 2A). The Scatter-Plot and Volcano plot were produced to visualize the significantly different circRNAs (fold change >2.0 or <0.5, P value <0.05) between LAC tissues and non-cancerous tissues. The sequence data showed that 285 circRNAs were differently expressed in LAC tissues (fold-change >2.0 or <0.5, P-value <0.05). Among those circRNAs, 102 circRNAs were upregulated, and 183 were downregulated in LAC tissues (Figure 2B, 2C). The data indicates that circRNAs have a different expression pattern in LAC tissues compared with that in adjacent healthy lung tissues. These aberrantly expressed circRNAs may participate in LAC tumorigenesis and play an important role in tumor occurrence and development, providing plethora of unctional circRNAs in lung adenocarcinoma. Total detailed circRNA information is shown in Table S3.
Figure 1.

Expression profile of circRNAs from the LAC tissues and adjacent normal tissues. A. Box plots of Back-spliced Junction Counts in all samples were applied to visualize the normalized intensities of circRNAs for the two groups. B. Classifcation of the differentially expressed circRNAs based on genomic origin. C. The bar diagram shows the circRNA expressions on different chromosomes.
Figure 2.

Different expression pattern of circRNAs was detected by high-throughput sequencing in LAC tissues and paired adjacent non-cancerous tissues. A. Hierarchical clustering for analysis of differently expressed circRNA expression among samples. Each row represents one circRNA, and each column represents one sample. Red indicates higher expression level, green indicates lower expression level and the brightness of the color represents the degree of higher or lower gene expression. B. Scatter plot shows the difference in the expression of circRNAs between lung adenocarcinoma tisseus (Y-axis) versus the control (X-axis). The gray line in the middle represents no difference between groups, the upper and lower gray lines refer to 2.0-fold changes. The circRNAs above the upper and below the lower green lines indicate more than 2.0-fold differences between the two compared groups. C. Volcano plot of differentially expressed circRNAs in LAC. The vertical gray lines correspond to two-fold increased and decreased expression, and the horizontal gray line represents P=0.05. The green points and red points represent circRNAs that were differentially expressed with statistical significance.
The biological function of circRNA host genes
The functions of circRNAs are regulated by the host linear transcripts which from which they are transformed. The regulated linear transcripts of the different circRNAs were analyzed using the DAVID Bioinformatics Resources (http://david.abcc.ncifcrf.gov). GO (Gene Ontology) annotation and KEGG (Kyoto Encyclopedia of Genes and Genomes) pathway analyses were used to explore the potential functions of differentially expressed circRNAs between the two groups. Seven-hundred-nine and 1171 GO processes (P<0.05) were identified between the upregulated and downregulated circRNAs, respectively. Figure 3A depicts the top 10 dysregulated GO processes of each subgroup [biological process (BP), cell components (CC) and molecular function (MF)] based on the routine GO classification algorithms. The top three GO terms of the host genes of the upregulated circRNAs in LAC tissues compared with matched nontumor tissues were rich in the hypoxia-inducible factor-1alpha signalling pathway, regulation of the acetyl-CoA biosynthetic process from pyruvate, and intrinsic apoptotic signalling pathway in response to oxidative stress in biological process subgroups. The top three processes in the cellular component subgroups consisted of the following subgroups: mitochondrial pyruvate dehydrogenase complex, dynein complex, and mitochondrial matrix. Pyruvate dehydrogenase (acetyl-transferring) kinase activity, glycoprotein 6-alpha-L-fucosyltransferase activity, and histone binding were the top three processes in the molecular function subgroup. Next, the top 10 GO terms predicted by the downregulated circRNAs were identified as the same as those predicted for the upregulated circRNAs, which were based on the enrichment scores (Figure 3B). The top three processes of the transcripts of the downregulated circRNAs were rich in negative regulation of actin filament depolymerization, phospholipid biosynthetic process, and the G-protein coupled receptor signalling pathway, coupled to cyclic in the biological process subgroup, plasma membrane, cortical actin cytoskeleton, motile primary cilium in cellular component subgroup, and G-Protein coupled receptor activity, actin binding and growth factor binding in molecular function subgroup. Furthermore, the 80 and 109 KEGG pathways (P<0.05) were identified between the upregulated and downregulated circRNAs, respectively. Figure 4A shows the top 20 KEGG pathways in the differently expressed circRNAs-derived genes, which include central carbon metabolism in cancer (path: hsa05230), transcriptional misregulation in cancer (path: hsa05202) and the HIF-1 signalling pathway (path: hsa04066) of the upregulated circRNAs. The downregulated circRNAs were also enriched in cancer-related processes (Figure 4B), such as cell adhesion molecules (CAMs) (hsa04514), ECM-receptor interaction (hsa04512), and the Rap 1 signalling pathway (hsa04051). Thus, it indicates that the differently expressed circRNAs could promote tumorigenesis and progression through these pathways.
Figure 3.

GO analysis for circRNA co-expression genes. A. Top 10 GO enriched biological process, celluar process, and molecular process of linear counterparts of up-regulated circRNAs in lung adenocarcinoma tissues. B. Top 10 GO enriched biological process, celluar process, and molecular process of linear counterparts of down-regulated circRNAs in lung adenocarcinoma tissues. The -log10 (p-value) yields an enrichment score representing the significance of GO term enrichment between differently expressed circRNAs.
Figure 4.

Top 20 KEGG pathways for circRNA co-expression genes between LAC tissues and adjacent non-tumor tissues. A. Top 20 KEGG pathways of up-regulated circRNAs in lung adenocarcinoma tissues. B. Top 20 KEGG pathways of down-regulated circRNAs in lung adenocarcinoma tissues. KEGG pathway analysis was performed to determine the involvement of linear transcripts in different biological pathways. The -log10 (p-value) yields an enrichment score indicating the significance of pathway correlations.
Predicting the interaction between circRNA and miRNA
Findings over the past several years have strongly supported a vital role for miRNA in the initiation and progression of human malignancies, indicating the potential to classify tumors with high accuracy and predict outcome [23]. CircRNA, as regulatory factors of miRNA, may affect the occurrence and development of cancers. Several studies have shown that some circRNAs are able to absorb cancer-associated miRNAs via microRNA response elements (MREs) to promote cancer pathogenesis [24]. To facilitate the investigation, we constructed the network to reveal the co-expression pattern of circRNA-miRNA based on TargetScan and miRanda. The constructed network map consisted of the top 300 relationships between circRNAs and miRNAs ranked by the p-value of their hypergeometric distribution. Our result shows that 141 miRNAs interact with 103 circRNAs among the top 300 relationships and may predict close connections with LAC (Figure 5). For example, the downregulation of hsa-miR-106b-3p leads to the upregulation of the target gene, CDKN1A, and plays a critical role in the cell cycle progression by arresting marine-treated cells in the G0/G1 phase [25]. The network shows that circRNA does not interact with miRNA in parallel, but a single miRNA could be targeted by several circRNAs, whereas a single circRNA could also target multiple miRNAs. For example, hsa-miR-1293 may bind circRNA_22795, circRNA_10620, circRNA_07376, circRNA_15924, and circRNA_11662, whereas circRNA_11662 was predicted to target hsa-miR-4437, hsa-miR-4312, hsa-miR-5004-5p, hsa-miR-1247-5p, and hsa-miR-4433a-5p. The co-expression network suggests that miRNA-mediated regulation between circRNAs and mRNAs is implicated in lung adenocarcinoma.
Figure 5.

The mapping network of circRNA-miRNA interactions in LAC based on sequence-pairing prediction. The interaction network of differentially expressed circRNAs (red elliptic nodes) and their complementary binding miRNAs (blue triangular nodes) was presented by the cytoscape software.
Prediction of interactions between circRNAs, miRNAs, and target genes
CircRNAs have been shown to sponge miRNAs, leading to the de-repression of miRNA targets and thereby act as ‘miRNA sponges’ within the cellular RNA network [26]. Two circRNA-miRNA-mRNA networks were constructed to reveal the interactions between circRNA, miRNA, and mRNA. We selected the ten most dysregulated circRNAs (five up-regulated and five down-regulated), and mapped them with the miRanda database to obtain the targeted miRNA of the circRNA. These miRNAs were then mapped to the database to confirm the matched mRNA, and all these matched mRNAs were intersected with our mRNA sequencing result. A total of five circRNAs, 29 miRNAs and 98 mRNAs were contained in the upregulated circRNA network (Figure 6A). The downregulated circRNA network consisted of 5 circRNAs, 93 miRNAs and 450 mRNAs (Figure 6B). In the upregulated circRNA network, hsa-mir-125a-3p, which was targeted by hsa_circ_0039161, was confirmed to have a negative relationship with the lymph node metastasis phase in non-small cell lung cancer (NSCLC) [27]. The hsa-mir-125a-3p expression trend in our data was consistent with other studies, which suggests that hsa_circ_0039161 might play complex roles in the spread and invasion of the LAC into other tissues. As shown in Figure 6B, hsa-mir-8485 had the most degrees and hsa_circ_0004062, hsa_circ_0004777, and hsa_circ_0004417 had the most interactions with miRNAs, indicating that they might play a key role in the down-regulated circRNA network.
Figure 6.

Interactions between circRNAs, miRNAs, and target genes. CircRNAs could regulate the expression level of mRNAs via absorbing with MREs of miRNAs. A. Network contained miRNAs down-regulation and both circRNAs and mRNAs up-regulation. B. Network contained miRNAS up-regulation and both circRNAs and mRNAs down-regulation. Triangular nodes, square nodes, and circular nodes, represent circRNAs, miRNAs and mRNAs respectively. Red color represents the up-regulated circRNAs and light blue color represents the down-regulated circRNAs.
Validation of hsa_circ_0002360 expression
Ten circRNAs involved in the circ-RNA-miRNA-mRNA network were selected to confirm the RNA-Seq data in another twenty pairs of samples by qRT-PCR. Two circRNA primers were not successfully designed, and they were removed in the following experiment. The results revealed that hsa_circ_0000264, hsa_circ_0039161, hsa_circ_0002360, and hsa_circ_0006276 expression were upregulated, whereas hsa_circ_0004062, hsa_circ_0004417, hsa_circ_0001320, and hsa_circ_0004777 were downregulated (Figure 7A). In particular, hsa_circ_0002360 was the most enrich circular RNA among the remaining circRNAs. Additionally, the qRT-PCR data was highly consistent with the circRNA sequencing data (Figure 7B), confirming the reliability of our RNA-Seq results.
Figure 7.
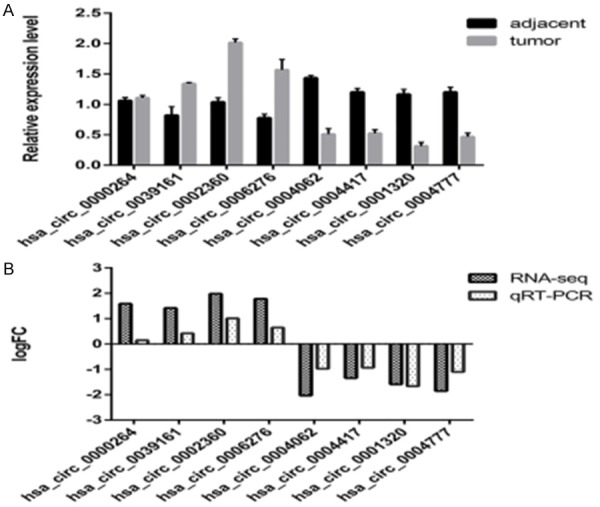
qRT-PCR validation of eight differentially expressed circRNAs. A. The relative expression level of circRNAs in 20 LAC tissues and paired adjacent nontumorous issues (P<0.05). B. Comparison of log2 fold changes (FCs) in circRNAs between RNA-Seq and qRT-PCR results.
hsa_circ_0002360 and PHF19 is up-regulated in LAC tissues while hsa-mir-3620-5p represents an opposite trend
Bioinformatic predictions suggest that hsa-mir-3620-5p may be one of the main target miRNAs of hsa_circ_0002360 and that PHF19 may be one of the target mRNAs of hsa-mir-3620-5p. To further verify the relationship, we detected the expression of hsa_circ_0002360, hsa-mir-3620-5p and its target in additional 18 cases of paired LAC tissues by real-time qRT-PCR or Western blot. The PCR results showed that the gene level of hsa_circ_0002360 and PHF19 were significantly higher in LAC tissues than adjacent normal tissues (P<0.05) (Figure 8A and 8C) while hsa-mir-3620-5p exhibited an opposite trend (P<0.05) (Figure 8B). Additionally, we showed the rich infiltration and expression of PHF19 in tumor tissues on the protein level (Figure 8D and 8E). Collectively, these data illustrates that there may be a possible regulatory pathway between hsa_circ_0002360, hsa-mir-3620-5p and PHF19.
Figure 8.
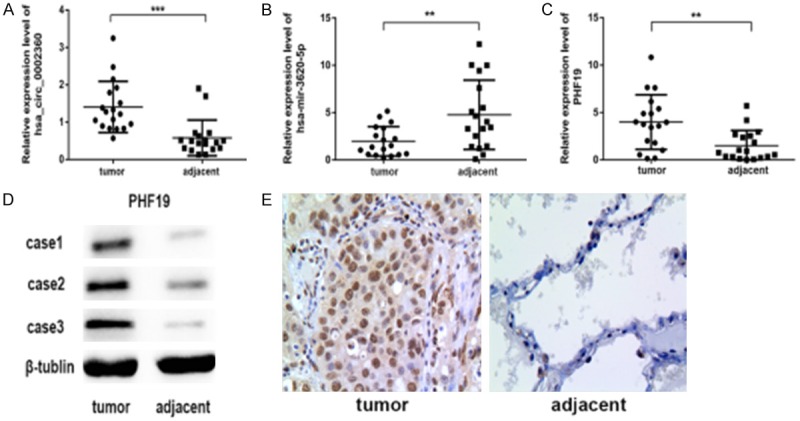
Validation for the expression of hsa_circ_0002360, hsa-mir-3620-5p and PHF19. A-C. qRT-PCR analysis shows the expression of hsa_circ_0002360, hsa-mir-3620-5p and PHF19 in the LAC tissues as compared to the normal controls. *P<0.05, n=3. D, E. Western blot and IHC analyses show the level of PHF19 protein in the LAC tissues as compared to control group. β-tublin acts as an internal control.
Discussion
To date, the main treatments of lung cancer are surgery, chemotherapy, and radiation therapy, and many types of studies have shown that combined chemotherapy significantly improves the survival rate of lung cancer patients over time [28]. However, owing to the aggressive nature of this cancer and the lack of effective diagnostic biomarkers to detect this disease, many patients respond poorly to chemotherapy. The successful response rates is only 30 to 40% [29], and the average 5-year survival rate is 17.4%, that can be as low as approximately 2% for those with stage IV (metastatic) disease at diagnosis. Since next-generation sequencing technology and bioinformatics analysis have rapidly developed and been widely used, the regulatory function of circRNAs in eukaryotic cells is beginning to draw attention. An increasing number of studies have shown that circRNAs participate in several different diseases, including neurological diseases [30], cardiovascular diseases [31] and multiple cancers for which they may serve as promising diagnostic biomarkers and potential therapeutic targets [14]. Currently, only a few circRNAs have been identified in association with lung cancer development, invasion, metastasis or patient prognosis including hsa_circ_0014130 [32], hsa_circ_0007385 [33] and circRNA_100876 [34]. However, the clinical values and roles of circRNAs in LAC remained not unclear and many circRNAs in LAC tissues need to be identified and characterised for their functional role.
In this study, we first investigated the circRNA expression profiles in human LAC tissues by high-throughput circRNA sequencing. We found that circRNAs expression in LAC tissues (n=5) was significantly different from adjacent non-cancerous tissues (n=5) (Figure 2). Compared with non-cancerous tissues, our sequence data showed that a total of 102 circRNAs were significantly upregulated while 183 were downregulated in LAC tissues (Table S1). In the patterns, hsa_circ_0000264, hsa_circ_0039161, hsa_circ_0002360, hsa_circ_0006276, and hsa_circ_0009128 were significantly upregulated, while hsa_circ_0004062, hsa_circ_0004417, hsa_circ_0003176, hsa_circ_0001320, and hsa_circ_0004777 showed the opposite trend. We confirmed that their expression trend was strongly consistent with those of circRNA sequencing using qRT-PCR analysis. Especially, we found that the level of hsa_circ_0002360 was significantly higher in LAC tissues than adjacent normal tissues (Figure 7). The dysregulated circRNAs could be a novel biomarker for the diagnosis of lung adenocarcinoma or could be used as therapeutic targets. This will be investigated in larger patient cohorts in the near future.
Although the function of most circRNAs is still not completely understood, it is possible to estimate their biological process, cellular component, and molecular function by performing a functional enrichment analysis of their host genes. GO enrichment analysis indicated that the different LAC circular RNAs-derived genes were enriched for certain pathways associated with cancer, such as the apoptotic signalling pathway, HIF-1α signalling pathway, and protein kinase activity, which indicated the coding genes contributed to the pathogenesis of LAC (Figure 3). The KEGG pathway was also enriched in tumor samples, and several important pathways, closely associated with cancer, were identified including central carbon metabolism in cancer, transcriptional misregulation in cancers, the HIF-1α signalling pathway, adhesion junctions, cell adhesion molecules, and the rap1 signalling pathway (Figure 4). A previous study has shown that nicotine, which has a broad spectrum of tumor-promoting activities in lung cancer, interacts with α 5-nAChR, activates the ERK, and Akt signalling pathways, upregulates HIF-1α signalling, elevates VEGF, and thereby promotes LAC cell proliferation, angiogenesis, and invasion [22]. Rap1 is a member of the Ras family of monomeric G proteins that has received recognition for its role in tumorigenesis and cancer progression. In vivo and in vitro data both provide evidence that aberrant Rap1 activation contributes to tumor cell adhesion, migration and invasion [35]. The core components of the adherens junctions are classical cadherins and catenins. The loss or decrease in expression of E-cadherin usually correlates with tumor invasiveness, distant metastasis, and is considered unfavourable in the outcome of patient [36]. Cadherins, which are the key molecules that control cell-cell adhesion are often inactivated or functionally inhibited, resulting in disease development, and progression in cancer [37]. Thus, we propose that these differentially expressed circRNAs might participate in the tumorigenesis of LAC by regulating their parent genes.
CircRNA can act as a sponge and bind tumor-associated miRNAs or proteins to affect disease progression. For example, circRNA CiRS-7 contains many binding sites for miR-7, which enables it to act as a ceRNA of miR-7 and competitively inhibit its activity, therefore promoting the initiation and development of cancer [38]. The cir-ITCH promotes lung cancer cell proliferation by acting as a sponge for miR-7 and miR-214 [39]. To our knowledge, this is the first study that confirmed the increased expression of hsa_circ_0002360 in lung adenocarcinoma patients. Additionally, the potential function of hsa_circ_0002360 in LAC has also been analyzed. In our study, different circRNAs were confirmed to be relevant to LAC, and therefore we predicted the downstream miRNAs of significantly increased circRNAs, upstream miRNAs of decreased circRNAs and targeted mRNAs. Next we constructed the circRNA-miRNA-mRNA pathways based on bioinformatics analysis (Figures 5 and 6). Under the circRNA-miRNA-mRNA networks, we inferred hsa_circ_0002360/hsa-mir-3620-5p/PHF19 axis existed in the progression of LAC. Previous study has shown that PHF19 is increased in several types of cancer and promotes the tumor growth [40,41]. Furthermore, the tumorigenicity of PHF19 could be suppressed by miR-195-5p which has binding sites for it [42]. In our study we conducted Western Blot, qRT-PCR and IHC with LAC tissues and adjacent nontumor tissues (Figure 8). In agreement with previous study, the results indicate that PHF19 is dramatically increased in LAC patients. In addition, hsa-mir-3620-5p is significantly decreased. Loss or suppression of hsa-mir-3620-5p targeting PHF19 may cause aberrant overexpression of PHF19. Therefore, the upregulated hsa_circ_0002360 and the downregulated hsa-mir-3620-5p interaction might result in the upregulation of tumorigenic gene such as PHF19. The data indicates that hsa_circ_0002360 might initially inhibit the expression of hsa-mir-3620-5p, subsequently increasing the expression of the target genes, and finally participating in the pathogenesis of LAC. It is necessary to explore the regulatory mechanism of hsa_circ_0002360 in LAC in future study. Furthermore, a larger sample size is urgently needed to verify our findings and to avoid selection bias.
Conclusions
In conclusion, we detected differential circRNA expression profiles between LAC tissues and adjacent non-neoplastic tissues. Our results demonstrated that multiple circRNAs were aberrantly expressed in LAC tissues and that hsa_circ_0002360 might have the potential to participate in the LAC biogenetic process. Further studies are urgently needed to explore the therapeutic potential for the treatment of LAC including its biological complexity and clinical applicability in LAC.
Acknowledgements
The authors would like to thank to Zhijian Zhang, the Jiang Su University, China for excellent experimental assistance. Special thanks also to Xi Wei for the provision of immunohistochemistry techniques. This work was supported by Grants from the Project of Natural Science Foundation of Jiangsu Province, China (BK20151333) and National Natural Science Foundation of China (81672999).
Disclosure of conflict of interest
None.
Supporting Information
Table S3
References
- 1.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2018. CA Cancer J Clin. 2018;68:7–30. doi: 10.3322/caac.21442. [DOI] [PubMed] [Google Scholar]
- 2.Langer CJ, Besse B, Gualberto A, Brambilla E, Soria JC. The evolving role of histology in the management of advanced non-small-cell lung cancer. J. Clin. Oncol. 2010;28:5311–5320. doi: 10.1200/JCO.2010.28.8126. [DOI] [PubMed] [Google Scholar]
- 3.Li Y, Zheng Q, Bao C, Li S, Guo W, Zhao J, Chen D, Gu J, He X, Huang S. Circular RNA is enriched and stable in exosomes: a promising biomarker for cancer diagnosis. Cell Res. 2015;25:981–984. doi: 10.1038/cr.2015.82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bahn JH, Zhang Q, Li F, Chan TM, Lin X, Kim Y, Wong DT, Xiao X. The landscape of MicroRNA, Piwi-interacting RNA, and circular RNA in human saliva. Clin Chem. 2015;61:221–230. doi: 10.1373/clinchem.2014.230433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Memczak S, Papavasileiou P, Peters O, Rajewsky N. Identification and characterization of circular RNAs as a new class of putative biomarkers in human blood. PLoS One. 2015;10:e0141214. doi: 10.1371/journal.pone.0141214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sanger HL, Klotz G, Riesner D, Gross HJ, Kleinschmidt AK. Viroids are single-stranded covalently closed circular RNA molecules existing as highly base-paired rod-like structures. Proc Natl Acad Sci U S A. 1976;73:3852–3856. doi: 10.1073/pnas.73.11.3852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jens M. Circular RNAs are a large class of animal RNAs with regulatory potency. Nature. 2013;495:333–338. doi: 10.1038/nature11928. [DOI] [PubMed] [Google Scholar]
- 8.Cocquerelle C, Mascrez B, Hétuin D, Bailleul B. Mis-splicing yields circular RNA molecules. FASEB J. 1993;7:155–160. doi: 10.1096/fasebj.7.1.7678559. [DOI] [PubMed] [Google Scholar]
- 9.Zhang Y, Zhang XO, Chen T, Xiang JF, Yin QF, Xing YH, Zhu S, Yang L, Chen LL. Circular intronic long noncoding RNAs. Mol Cell. 2013;51:792–806. doi: 10.1016/j.molcel.2013.08.017. [DOI] [PubMed] [Google Scholar]
- 10.Chen LL. The biogenesis and emerging roles of circular RNAs. Nat Rev Mol Cell Biol. 2016;17:205–211. doi: 10.1038/nrm.2015.32. [DOI] [PubMed] [Google Scholar]
- 11.Hansen TB, Jensen TI, Clausen BH, Bramsen JB, Finsen B, Damgaard CK, Kjems J. Natural RNA circles function as efficient microRNA sponges. Nature. 2013;495:384–388. doi: 10.1038/nature11993. [DOI] [PubMed] [Google Scholar]
- 12.Jens M. Circular RNAs are a large class of animal RNAs with regulatory potency. Nature. 2013;495:333–338. doi: 10.1038/nature11928. [DOI] [PubMed] [Google Scholar]
- 13.Wang Y, Wang Z. Efficient backsplicing produces translatable circular mRNAs. RNA. 2015;21:172–179. doi: 10.1261/rna.048272.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang Y, Mo Y, Gong Z, Yang X, Yang M, Zhang S, Xiong F, Xiang B, Zhou M, Liao Q. Circular RNAs in human cancer. Mol Cancer. 2017;16:25. doi: 10.1186/s12943-017-0598-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lee H, Lee KW, Lee T, Park D, Chung J, Lee C, Park WY, Son DS. Performance evaluation method for read mapping tool in clinical panel sequencing. Genes Genomics. 2018;40:189–197. doi: 10.1007/s13258-017-0621-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gao Y, Wang J, Zhao F. CIRI: an efficient and unbiased algorithm for de novo circular RNA identification. Genome Biol. 2015;16:4. doi: 10.1186/s13059-014-0571-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dennis G, Sherman BT, Hosack DA, Yang J, Gao WW, Lane HC, Lempicki RA. DAVID: database for annotation, visualization, and integrated discovery. Genome Biol. 2003;4:P3. [PubMed] [Google Scholar]
- 18.Goto S, Okuno Y, Hattori M, Nishioka T, Kanehisa M. LIGAND: database of chemical compounds and reactions in biological pathways. Nucleic Acids Res. 2002;30:402–404. doi: 10.1093/nar/30.1.402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Anders S, Pyl PT, Huber W. HTSeq--a Python framework to work with high-throughput sequencing data. Bioinformatics. 2015;31:166–169. doi: 10.1093/bioinformatics/btu638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang L, Feng Z, Wang X, Wang X, Zhang X. DEGseq: an R package for identifying differentially expressed genes from RNA-seq data. Bioinformatics. 2010;26:136–138. doi: 10.1093/bioinformatics/btp612. [DOI] [PubMed] [Google Scholar]
- 21.Shi Y, Shang J. Long noncoding RNA expression profiling using arraystar LncRNA microarrays. Methods Mol Biol. 2016;1402:43–61. doi: 10.1007/978-1-4939-3378-5_6. [DOI] [PubMed] [Google Scholar]
- 22.Ma X, Jia Y, Zu S, Li R, Jia Y, Zhao Y, Xiao D, Dang N, Wang Y. alpha5 Nicotinic acetylcholine receptor mediates nicotine-induced HIF-1alpha and VEGF expression in non-small cell lung cancer. Toxicol Appl Pharmacol. 2014;278:172–179. doi: 10.1016/j.taap.2014.04.023. [DOI] [PubMed] [Google Scholar]
- 23.Garzon R, Fabbri M, Cimmino A, Calin GA, Croce CM. MicroRNA expression and function in cancer. Trends Mol Med. 2006;12:580–587. doi: 10.1016/j.molmed.2006.10.006. [DOI] [PubMed] [Google Scholar]
- 24.Li J, Yang J, Zhou P, Le Y, Zhou C, Wang S, Xu D, Lin HK, Gong Z. Circular RNAs in cancer: novel insights into origins, properties, functions and implications. Am J Cancer Res. 2015;5:472–480. [PMC free article] [PubMed] [Google Scholar]
- 25.Tetik AV, Düzgün Z, Erdem C, Kaymaz BT, Eroglu Z, Çetintas VB. Matrine induced G0/G1 arrest and apoptosis in human acute T-cell lymphoblastic leukemia (T-ALL) cells. Bosn J Basic Med Sci. 2018;18:141–149. doi: 10.17305/bjbms.2017.2457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tay Y, Rinn J, Pandolfi PP. The multilayered complexity of ceRNA crosstalk and competition. Nature. 2014;505:344–352. doi: 10.1038/nature12986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jiang L, Huang Q, Zhang S, Zhang Q, Chang J, Qiu X, Wang E. Hsa-miR-125a-3p and hsa-miR-125a-5p are downregulated in non-small cell lung cancer and have inverse effects on invasion and migration of lung cancer cells. BMC Cancer. 2010;10:318–318. doi: 10.1186/1471-2407-10-318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bozcuk H, Artac M, Ozdogan M, Savas B. Does maintenance/consolidation chemotherapy have a role in the management of small cell lung cancer (SCLC)? A metaanalysis of the published controlled trials. Cancer. 2005;104:2650–2657. doi: 10.1002/cncr.21540. [DOI] [PubMed] [Google Scholar]
- 29.Tian Y, Wang Z, Liu X, Duan J, Feng G, Yin Y, Gu J, Chen Z, Gao S, Bai H, Wan R, Jiang J, Liu J, Zhang C, Wang D, Han J, Zhang X, Cai L, He J, Wang J. Prediction of chemotherapeutic efficacy in non-small cell lung cancer by serum metabolomic profiling. Clin Cancer Res. 2018;24:2100–2109. doi: 10.1158/1078-0432.CCR-17-2855. [DOI] [PubMed] [Google Scholar]
- 30.Chen BJ, Mills JD, Takenaka K, Bliim N, Halliday GM, Janitz M. Characterization of circular RNAs landscape in multiple system atrophy brain. J Neurom. 2016;139:485–496. doi: 10.1111/jnc.13752. [DOI] [PubMed] [Google Scholar]
- 31.Fan X, Weng X, Zhao Y, Chen W, Gan T, Xu D. Circular RNAs in cardiovascular disease: an overview. Biomed Res Int. 2017;2017:5135781. doi: 10.1155/2017/5135781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhang S, Zeng X, Ding T, Guo L, Li Y, Ou S, Yuan H. Microarray profile of circular RNAs identifies hsa_circ_0014130 as a new circular RNA biomarker in non-small cell lung cancer. Sci Rep. 2018;8:2878. doi: 10.1038/s41598-018-21300-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jiang MM, Mai ZT, Wan SZ, Chi YM, Zhang X, Sun BH, Di QG. Microarray profiles reveal that circular RNA hsa_circ_0007385 functions as an oncogene in non-small cell lung cancer tumorigenesis. J Cancer Res Clin Oncol. 2018;144:667–674. doi: 10.1007/s00432-017-2576-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yao JT, Zhao SH, Liu QP, Lv MQ, Zhou DX, Liao ZJ, Nan KJ. Over-expression of CircRNA_100876 in non-small cell lung cancer and its prognostic value. Pathol Res Pract. 2017;213:453–456. doi: 10.1016/j.prp.2017.02.011. [DOI] [PubMed] [Google Scholar]
- 35.Bailey CL, Kelly P, Casey PJ. Activation of Rap1 promotes prostate cancer metastasis. Cancer Res. 2009;69:4962–4968. doi: 10.1158/0008-5472.CAN-08-4269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Vasioukhin V. Adherens junctions and cancer. Subcell Biochem. 2012;60:379–414. doi: 10.1007/978-94-007-4186-7_16. [DOI] [PubMed] [Google Scholar]
- 37.Bruner HC, Derksen PWB. Loss of e-cadherin-dependent cell-cell adhesion and the development and progression of cancer. Cold Spring Harb Perspect Biol. 2018;10 doi: 10.1101/cshperspect.a029330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Peng L, Yuan XQ, Li GC. The emerging landscape of circular RNA ciRS-7 in cancer (review) Oncol Rep. 2015;33:2669–2674. doi: 10.3892/or.2015.3904. [DOI] [PubMed] [Google Scholar]
- 39.Li W, Lin Z, Kai F, Cheng ZX, Sun QC, Wang JJ. Circular RNA-ITCH suppresses lung cancer proliferation via inhibiting the Wnt/β-catenin pathway. Biomed Res Int. 2016;2016:1579490. doi: 10.1155/2016/1579490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Shuwen W, Gavin PR, Jiyue Z. A novel human homologue of Drosophila polycomblike gene is up-regulated in multiple cancers. Gene. 2004;343:69–78. doi: 10.1016/j.gene.2004.09.006. [DOI] [PubMed] [Google Scholar]
- 41.Li G, Warden C, Zou Z, Neman J, Krueger JS, Jain A, Jandial R, Chen M. Altered expression of polycomb group genes in glioblastoma multiforme. PLoS One. 2013;8:e80970. doi: 10.1371/journal.pone.0080970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Xu H, Hu YW, Zhao JY, Hu XM, Li SF, Wang YC, Gao JJ, Sha YH, Kang CM, Lin L. MicroRNA-195-5p acts as an anti-oncogene by targeting PHF19 in hepatocellular carcinoma. Oncol Rep. 2015;34:175–182. doi: 10.3892/or.2015.3957. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.