

We developed an advanced clearing method with superior fluorescence-preserving capability for 3D imaging of whole organs.
Abstract
Various optical clearing methods have emerged as powerful tools for deep biological imaging. Organic solvent–based clearing methods, such as three-dimensional imaging of solvent-cleared organs (3DISCO), present the advantages of high clearing efficiency and size reduction for panoptic imaging of large samples such as whole organs and even whole bodies. However, 3DISCO results in a rapid quenching of endogenous fluorescence, which has impeded its application. Here, we propose an advanced method named FDISCO to overcome this limitation. FDISCO can effectively preserve the fluorescence of various fluorescent probes and can achieve a long storage time of months while retaining potent clearing capability. We used FDISCO for high-resolution imaging and reconstruction of neuronal and vascular networks. Moreover, FDISCO is compatible with labeling by multiple viruses and enables fine visualization of neurons with weak fluorescence labeling in the whole brain. FDISCO represents an effective alternative to the three-dimensional mapping of whole organs and can be extensively used in biomedical studies.
INTRODUCTION
High-resolution mapping of three-dimensional (3D) structures in intact tissues is indispensable in many biological studies. The conventional method of histological sectioning followed by the imaging of individual sections is commonly used and rather valuable (1, 2), but this process is often labor intensive and is prone to errors due to the loss or deformation of slices. Although recently developed automated serial sectioning and imaging techniques enable high-throughput data acquisition (3–6), these methods inevitably lead to sample destruction, which is the major limitation of mechanical sectioning.
In contrast to mechanical sectioning, multiple optical imaging techniques provide fast and simple choices for the 3D reconstruction of tissue structures by optical sectioning (7). However, for most of these techniques, including single-photon, two-photon, and light sheet fluorescence microscopy (LSFM), imaging depth is limited by the strong light scattering of biological tissues (8). Recently, tissue optical clearing techniques have emerged to reduce the scattering and improve the light penetration depth by introducing various chemical agents and tools (8–12). A number of clearing methods have been developed (8, 9, 13, 14), including aqueous-based clearing methods, such as CLARITY (15), PACT-PARS (16), CUBIC (17, 18), ScaleS (19), OPTIClear (20), and Ce3D (21), and organic solvent-based clearing methods, such as BABB (22), 3DISCO (23, 24), iDISCO (25), uDISCO (26), FluoClearBABB (27), Ethanol-ECi (28), and PEGASOS (29). Each kind of method has its own advantages: The former can preserve protein-based fluorescence, while the latter can provide a favorable speed of clearing (30). These methods provide essential tools for obtaining high-resolution 3D images of intact tissues and for elucidating many biological events (14).
Organic solvent–based clearing protocols, such as 3DISCO, can achieve the highest level of tissue transparency and shrinkage (8, 13), thereby facilitating the imaging of large samples, such as the brain, the spinal cord, lung tumors, mammary glands, immune organs, and embryos (23, 25). However, 3DISCO results in a rapid decline in endogenous fluorescence signals during the tissue clearing and storage procedure (with a half-life of approximately 1 to 2 days), which has impeded the application of 3DISCO, especially for large samples such as the whole brain (23, 26). To address this issue, Becker et al. have tried embedding specimens in a transparent solid resin (31). This method enabled long-term archiving but required a complex embedding procedure and incurred a risk of low imaging quality associated with reduced transparency. Renier et al. described iDISCO to visualize fluorescent reporters by immunolabeling (25). In practice, it has remained very challenging to achieve whole-mount and homogeneous staining of large volumes because of enormous molecular barriers. The recently developed uDISCO was able to maintain endogenous fluorescence signals for several months longer than 3DISCO by introducing new dehydrating and refractive index matching agents combined with antioxidants (26). However, uDISCO requires more processing steps with more chemicals and a longer incubation time, especially for myelin-rich tissues such as the spinal cord (26), and is not compatible with tdTomato experimentally.
In this work, we developed an advanced optical clearing method based on 3DISCO, named FDISCO (DISCO with superior fluorescence-preserving capability), by temperature and pH condition adjustments. FDISCO can achieve a high level of fluorescence preservation of various probes, such as fluorescent proteins (FPs) and chemical fluorescent tracers, with a short processing time while maintaining potent tissue-clearing capability. FDISCO allows 3D imaging of neuronal and vascular structures in various large samples, including the intact brain, kidney, and muscle, in combination with LSFM. Using FDISCO, we detected weakly labeled neurons with virus in the whole brain and analyzed the spatial distributions of cells projecting to the virus-injected region. FDISCO provides a novel and efficient method for morphological analysis of anatomical structures.
RESULTS
Development of FDISCO by temperature and pH adjustments
As mentioned above, 3DISCO is a powerful clearing protocol for imaging large volumes using tetrahydrofuran (THF) for tissue dehydration and dibenzyl ether (DBE) for refractive index matching, but 3DISCO rapidly quenches endogenously expressed FPs during the clearing procedure and has a very short storage time (23). To overcome this problem, we searched for an optimized protocol that can better preserve fluorescence. Inspired by the effect of temperature and pH on green fluorescent protein (GFP) stability reported in previous studies (32–36), we started by investigating the effects of the temperature and pH of the clearing agents (i.e., THF or DBE) on the fluorescence of enhanced GFP (EGFP).
First, we investigated the influence of the temperature and pH conditions of THF on fluorescence preservation by an in vitro assay of recombinant EGFP expressed in Escherichia coli. We dissolved EGFP in pH-adjusted (pH 9.0) or pH-unadjusted THF solutions under different temperature conditions. The experimental groups included 4°C/pH 9.0, 4°C, 25°C (used in the original 3DISCO), and 37°C. For each group, we recorded the fluorescence intensity of the EGFP/THF solutions over time (0, 6, 18, and 24 hours) and measured the emission and absorption spectra of the EGFP/THF solutions at 24 hours. The 4°C/pH 9.0 group demonstrated the best EGFP fluorescence preservation, which indicated that the EGFP fluorescence was quenched more slowly at lower temperatures and that the alkaline pH also contributed to fluorescence preservation (Fig. 1, A to C). The emission spectra of the different groups had the same characteristic peak at 509 nm [the original emission peak (32)], which indicated that the fluorescence characteristics of the retained EGFP were not disturbed by THF under the various temperature and pH conditions (Fig. 1C). The absorption spectra of EGFP incubated in THF solutions showed a fluorescence absorption band at 488 nm and a nonfluorescence absorption band at 383 nm, indicating a natural and denatured EGFP (Fig. 1, D and E) (36–38). As the temperature decreased, the fluorescence absorption band increased, and the nonfluorescence band decreased. In addition, the pH-adjusted group (4°C/pH 9.0) showed a higher peak value at 488 nm than the pH-unadjusted group (4°C) (Fig. 1D). These results indicated that less EGFP was denatured by THF under the lower temperature and alkaline pH condition.
Fig. 1. Development of FDISCO by temperature and pH adjustments.
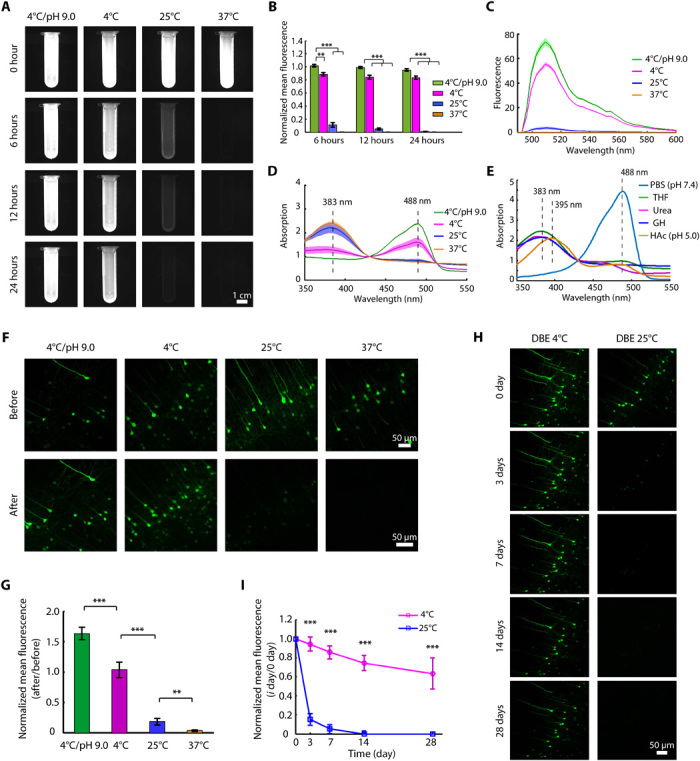
(A) Fluorescence images of recombinant EGFP dissolved in 30% THF during 24 hours of incubation under the indicated conditions. (B) Quantification of the normalized mean fluorescence intensity in (A) (n = 3). (C and D) Emission (C) and absorption (D) spectra of EGFP/THF solutions at 24 hours (n = 3). (E) Absorption spectra of recombinant EGFP treated by various solutions. GH, guanidine hydrochloride; HAc, acetic acid. (F and G) Confocal fluorescence images (F) and normalized mean fluorescence intensity quantification (G) of EGFP before and after clearing under the indicated conditions (n = 6). (H and I) The brain slices cleared under the “4°C/pH 9.0” condition were stored in DBE at 4° and 25°C. The images (H) and quantified normalized mean fluorescence (I) over time are shown (n = 6). All confocal images are maximum intensity projections (MIPs) of z stacks (40 to 60 μm thick) from the surface of brain slices. All values are means ± SD; statistical significance in (B), (G), and (I) (**P < 0.01 and ***P < 0.001) was assessed by an independent-sample t test and one-way analysis of variance (ANOVA), followed by Bonferroni or Dunnett’s T3 post hoc test.
Second, we corroborated the effect of the temperature and pH of THF on EGFP-labeled mouse brain sections (Thy1-GFP-M). We imaged neurons in the brain cortex before and after clearing and found that the lower temperature and alkaline clearing condition also led to greater EGFP fluorescence in brain tissues (Fig. 1, F and G). Furthermore, we stored the cleared samples (4°C/pH 9.0 group) in pure DBE at 4° and 25°C and found that in the 4°C condition, the EGFP fluorescence signals were maintained for weeks and that the storage time was notable longer than that at 25°C (Fig. 1, H and I). These results indicate that the lower temperature was beneficial for EGFP stability during long-term storage in DBE.
Hence, we selected 4°C/pH 9.0 and 4°C as suitable conditions for THF and DBE, respectively, during the clearing procedure, and these conditions resulted in a modified tissue-clearing protocol with improved fluorescence preservation termed FDISCO.
FDISCO preserves the fluorescence signals of multiple probes
FDISCO was investigated for the imaging of other FPs besides EGFP and for imaging multiple chemical fluorescent tracers. For enhanced yellow fluorescent protein (EYFP) and tdTomato, FDISCO demonstrated better fluorescence preservation than 3DISCO and uDISCO, similar to the results for EGFP (Fig. 2, A to C, and fig. S1, A and B). In particular, FDISCO was suitable for tdTomato fluorescence, which is incompatible with uDISCO clearing. Moreover, the endogenous fluorescence signals could still be detected after 28 days of storage (Fig. 2, D and E).
Fig. 2. Compatibility of FDISCO with multiple FPs and chemical fluorescent tracers.
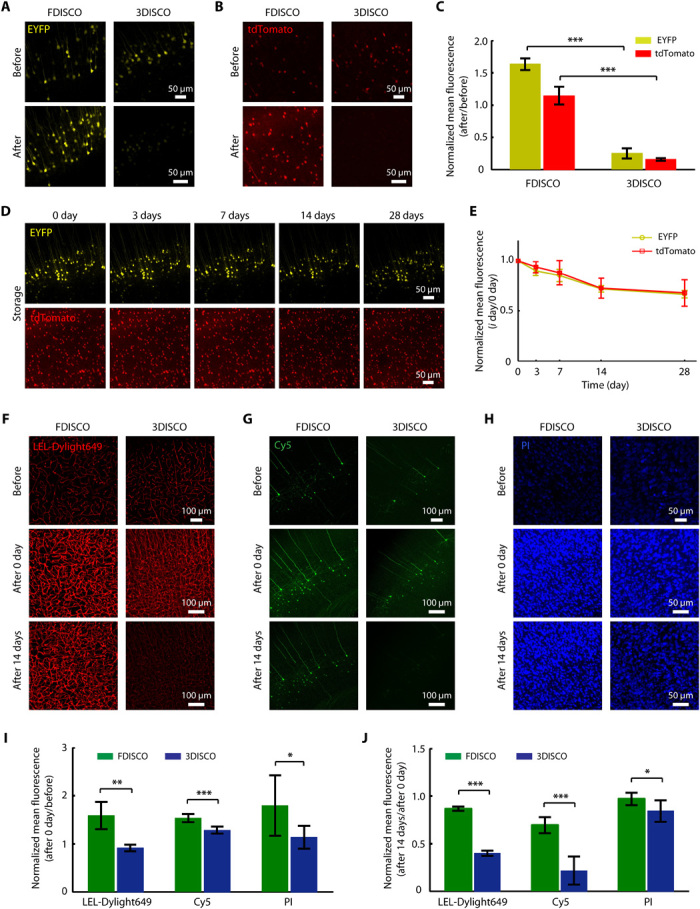
(A and B) Fluorescence images of EYFP (Thy1-YFP-H mouse) (A) and tdTomato (Sst-IRES-Cre::Ai14 mouse) (B) in 1-mm-thick brain slices before and after FDISCO clearing compared with 3DISCO clearing. (C) Fluorescence preservation quantification of EYFP and tdTomato after the clearing shown in (A) and (B). (D and E) EYFP and tdTomato images of FDISCO-cleared brain slices over time (D) and quantification data (E). (F to H) Fluorescence images of the following chemical fluorescent tracers after the clearing and storage procedures for FDISCO and 3DISCO: LEL-Dylight649 (F), antibody conjugated to Cy5 (G), and PI (H). (I and J) Quantification of the fluorescence preservation after clearing (I) and 14-day storage (G). All confocal images are MIPs of z stacks (40 to 60 μm thick) from the surface of slices. All values are means ± SD (n = 6); statistical significance in (C), (I), and (J) (*P < 0.05, **P < 0.01, and ***P < 0.001) was assessed by an independent-sample t test.
In addition to FPs, many valuable chemical fluorescent tracers have been used to label tissues (39). Here, we tested FDISCO with several types of tracers, including the DyLight 649 Lycopersicon esculentum lectin (LEL-Dylight649) and Alexa Fluor 647 anti-mouse CD31 antibody (CD31-A647) for blood vessel labeling, fluorescent dyes for nuclear staining [propidium iodide (PI)], and antibodies conjugated to different fluorophores for neuron immunostaining. For immunostaining, 1-mm-thick Thy1-GFP-M brain sections were stained with primary antibodies against GFP, followed by secondary antibodies conjugated to Cy3, Cy5, or Alexa Fluor 594 (A594).
By recording fluorescence images of cleared brain sections over time, we found that FDISCO allowed fine signal preservation of multiple chemical tracers during clearing and long-term storage (Fig. 2, F to J, and fig. S1, C and D). Compared with 3DISCO, FDISCO significantly improved the maintenance of all tested tracers, except CD31-A647 and A594, for which the differences were not significant. For LEL-Dylight649 and Cy5 in particular, which decay rapidly during the 3DISCO clearing or storage procedure, FDISCO improved the fluorescence preservation by more than 50%.
Overall, these results indicate that FDISCO not only preserves the fluorescence of various FPs well but also is compatible with multiple chemical fluorescent tracers. This method should be applicable to samples labeled with multiple probes and should be particularly beneficial in studies requiring long-term storage after clearing.
Moreover, the strategy of low temperature adjustment proved to be effective for fluorescence preservation with other clearing protocols. The results showed that the EGFP and EYFP fluorescence intensities were higher for tissue clearing with ethanol and BABB at 4°C than for clearing at 25°C (fig. S2, A and B), and the lower temperature of the storage procedure also contributed to better fluorescence maintenance of cleared tissues for uDISCO clearing (fig. S2, C and D).
FDISCO performs good clearing capability
To test whether FDISCO retained the fine clearing capability of the original protocol, we used FDISCO to clear whole brains and compared the results with other existing whole-brain clearing methods, including 3DISCO, uDISCO, FluoClearBABB, Ethanol-ECi, CUBIC, and PACT (Fig. 3). We recorded bright-field images of the brains before and after clearing and measured the transmittance spectra of the brains cleared with the different protocols. We found that the lower temperature adjustment in FDISCO had no influence on the clearing time (Fig. 3B). FDISCO achieved high transparency similar to that of 3DISCO and uDISCO within days, while FluoClearBABB and Ethanol-ECi demonstrated lower transparency (Fig. 3, A and C). The CUBIC-cleared brains were blurry in some areas after a 2-week treatment, and PACT demonstrated high transparency but required more than 2 weeks (Fig. 3, A to C). In addition, we calculated the size reductions and found that there was no significant difference between FDISCO and 3DISCO (Fig. 3D). Tissue structure and fine neuronal morphology were largely preserved after FDISCO clearing (fig. S3E). We further compared the fluorescence preservation of these methods when applied to whole-brain imaging with LSFM. We quantified the level of fluorescence signals from the cortical neurons of the cleared brains (FDISCO, 24.41 ± 1.13; 3DISCO, 2.18 ± 0.50; uDISCO, 12.26 ± 0.82; FluoClearBABB, 11.88 ± 1.71; Ethanol-ECi, 2.29 ± 0.68; CUBIC, 6.14 ± 1.06; PACT, 8.99 ± 1.78), which showed that FDISCO could preserve the fluorescence signal significantly better than the other methods could (Fig. 3E). This fluorescence-preserving capability, together with the high tissue transparency and size reduction, allowed full-depth imaging through the entire central nervous system (CNS; whole brain and spinal cord) with LSFM (Fig. 3F and fig. S4, A and B).
Fig. 3. Comparison of the whole-brain clearing performance of FDISCO and other clearing methods.
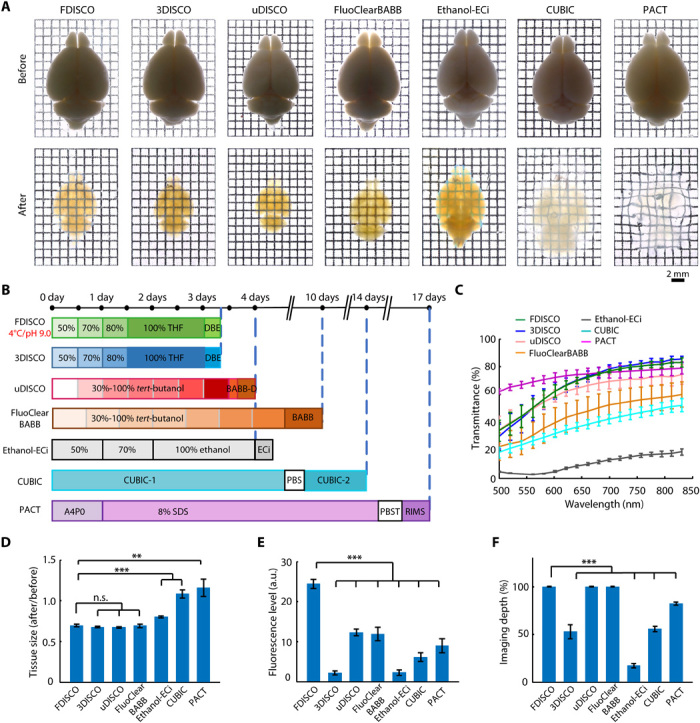
(A) Bright-field images of adult whole brains cleared with FDISCO, 3DISCO, uDISCO, FluoClearBABB, Ethanol-ECi, CUBIC, and PACT. (B) Whole-brain clearing protocol timeline. RIMS, refractive index matching solution. (C) Transmittance curves of the brain samples cleared with different clearing methods. (D) Linear expansion and shrinkage of whole brains during optical clearing. (E and F) Quantification of the fluorescence level in the cortex (E) and imaging depth of whole brains (F) cleared with different clearing methods, as assessed by LSFM imaging. All values are means ± SD (n = 5); the statistical significance in (D) to (F) (n.s., not significant, P > 0.05; **P < 0.01; and ***P < 0.001) was assessed by one-way ANOVA followed by Bonferroni or Dunnett’s T3 post hoc test. a.u., arbitrary units.
In addition to its application for the entire CNS, FDISCO achieved effective clearing of various specimens, including internal organs (stomach, liver, lung, and kidney), soft tissues (muscle), hard tissues (calcified bone), and mouse embryos and was also applicable to rat organs (fig. S3, A and B). For bone clearing by FDISCO, decalcification with EDTA-2Na (ethylenediaminetetraacetic acid disodium salt) was conducted prior to THF dehydration to improve the final transparency. FDISCO also enabled simple, effective whole-body clearing of an adult mouse without the organic solvent perfusion adopted by uDISCO. After whole-body clearing by FDISCO, we imaged the head of a Thy1-YFP-H mouse with LSFM and detected the neurons in the brain through the intact skull (fig. S3, C and D).
FDISCO allows 3D visualization of the nervous and vascular systems
As described in the literature, 3DISCO resulted in more quenching of fluorescence signals when longer incubation times in clearing solution were required for large samples, e.g., the entire CNS (26). This serious fluorescence decline impeded the visualization of neurons throughout the whole brain and spinal cord.
FDISCO allowed clearing of the entire CNS of an adult Thy1-GFP-M mouse and fine 3D visualization of the nervous system with LSFM imaging (fig. S4, A and B). We observed the neuronal structures in different regions of both the brain and the spinal cord, including the cortex, caudate putamen (CPu), hippocampus, cerebellum, and midbrain of the brain (Fig. 4, A and B, and movie S1), and the neurons in the gray matter and the axons in the white matter of the spinal cord (fig. S4, C to E). Compared with 3DISCO or uDISCO, FDISCO-treated samples revealed more neuronal details and higher levels of fluorescence imaging. We recorded the EGFP fluorescence of cleared brains at different time points after clearing and found that FDISCO could maintain the EGFP fluorescence at a fairly high level over 7 months, which allowed repeated imaging of the cleared samples (Fig. 4, C and D). The fluorescence level of FDISCO samples at each time point was significantly higher than those of 3DISCO and uDISCO samples (Fig. 4D). For mouse brains expressing EYFP (Thy1-YFP-H), FDISCO also demonstrated obviously better imaging quality (fig. S5). Moreover, we applied FDISCO to the Sst-IRES-Cre::Ai14 transgenic mouse brain, which selectively expressed tdTomato in dendritic inhibitory interneurons (40), and imaged the brain with LSFM. With the good fluorescence preservation of tdTomato, which is incompatible with uDISCO, FDISCO achieved fine reconstruction of neurons in different brain regions at single-cell resolution (fig. S6).
Fig. 4. LSFM imaging of neural structures in the mouse brain and gastrocnemius muscle after FDISCO clearing.
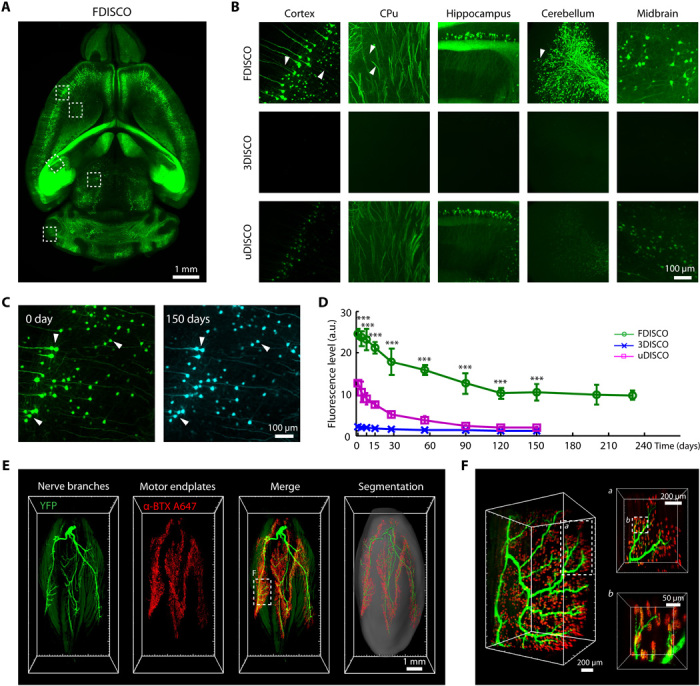
(A) Image of the whole brain (Thy1-GFP-M) cleared by FDISCO. (B) Comparison of the high-magnification images of the cleared brains assessed immediately after FDISCO, 3DISCO, and uDISCO clearing. The white arrowheads mark the tiny nerve fibers detected. For different clearing methods, the same imaging parameters and image processing methods were used for the same regions. (C) Images of cortical neurons in the FDISCO-cleared brain taken at 0 and 150 days after clearing, respectively. The neurons (e.g., white arrowheads) could still be viewed well after 150 days. (D) Fluorescence level quantification of cleared brains over time after FDISCO, 3DISCO, and uDISCO clearing (n = 4, 3, and 3, respectively). (E) 3D reconstruction and segmentation of nerve branches (green) and motor endplates (red) of the gastrocnemius muscle (Thy1-YFP-16) cleared by FDISCO. (F) High-magnification images of the dashed boxed region in (E). The images in (A) to (C) are the MIPs of 100-μm-thick z stacks. Values are means ± SD; the statistical significance in (D) (***P < 0.001) was assessed by one-way ANOVA, followed by the Bonferroni post hoc test.
In addition, we reconstructed the nerve branches and the motor endplates in muscle tissue by FDISCO (Fig. 4, E and F, and movie S2). The nerve branches and motor endplates in the gastrocnemius muscle were lighted by YFP (Thy1-YFP-16) and Alexa Fluor 647–conjugated α-BTX (bungarotoxin), respectively. The 3D visualization by FDISCO allowed the tracing of nerve branches and the locating of motor endplates readily, while 3DISCO could not. The morphologies of the individual motor endplates and the contact to the axon were identified (Fig. 4F).
3D visualization of the intact vascular system plays a valuable role in functional and pathological studies (41). We immunolabeled blood vessels with an anti-CD31 antibody by tail vein injection and obtained LSFM images of the FDISCO-cleared mouse brain and kidney (Fig. 5). The cerebral vasculature in the whole brain is shown in Fig. 5 (A to E), and blood vessels with various diameters in the cortex and hippocampus could be identified from reconstruction of the vascular system (movie S3). For kidney imaging, we obtained the 3D distribution of blood vessels and glomeruli in the kidney (Fig. 5, F to I, and movie S4). The glomeruli were mainly distributed in the renal cortex, and the number of glomeruli could be counted. Hence, FDISCO addresses the limitations of 3DISCO and uDISCO in imaging of various samples and allows successful visualization of detailed neuronal and vascular structures throughout intact organs.
Fig. 5. 3D visualization of the vasculature in the mouse brain and kidney after FDISCO clearing.
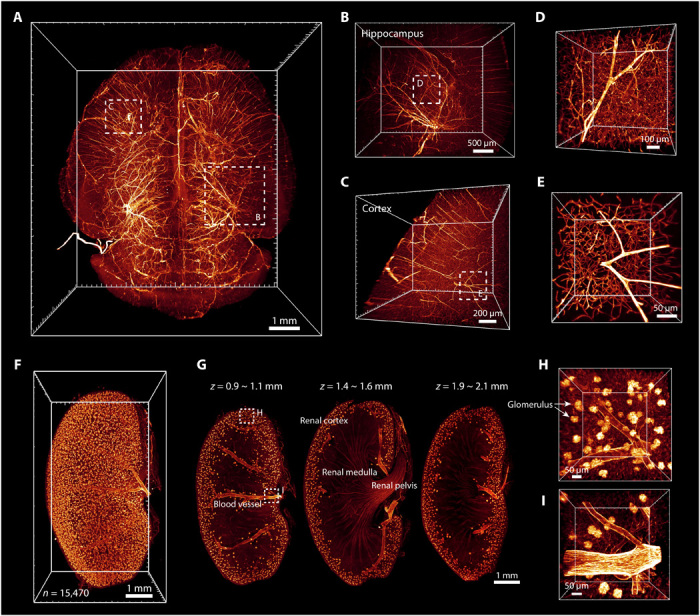
The vasculature was labeled by injection of CD31-A647 antibody. (A) 3D reconstruction of the vasculature in the whole brain after FDISCO clearing and LSFM imaging. (B to E) The details of blood vessels in the hippocampus (B) and cortex (C) are shown. High-magnification views of the dashed boxed regions in (B) and (C) are shown in (D) and (E), respectively. (F) 3D reconstruction of blood vessels and glomeruli in the kidney. The number of glomeruli was counted as 15,470 by Imaris software. (G) Images at gradient depth. The glomeruli were mainly distributed in the renal cortex. (H and I) High-magnification views of the dashed boxed regions in (G).
FDISCO allows the detection of weakly labeled neurons with virus
Given its high level of fluorescence preservation, FDISCO has the potential to detect labeled structures with weak fluorescence signals. Here, we applied FDISCO to visualize the neuronal connectivity of whole brains labeled with multiple viruses, including rabies virus (RV), pseudorabies virus (PRV), and adeno-associated virus (AAV).
First, we injected RV-DsRed into the ventral tegmental area (VTA) region of the right hemisphere for retrograde tracing of the projections onto the VTA, which receives connections from a wide range of brain regions (42). The distribution of DsRed-positive cells was found in various brain regions, including nearby regions such as the dorsal raphe nucleus (DR) and long-range neuronal connections such as those from the prefrontal cortex (PFC) (Fig. 6 and movie S5). Hence, FDISCO successfully visualized weakly labeled neuronal connectivity using RV-DsRed labeling throughout the whole brain, which is difficult to achieve by 3DISCO and uDISCO (fig. S7).
Fig. 6. 3D visualization of the RV-labeled neurons projecting to the VTA through the whole brain by FDISCO clearing.
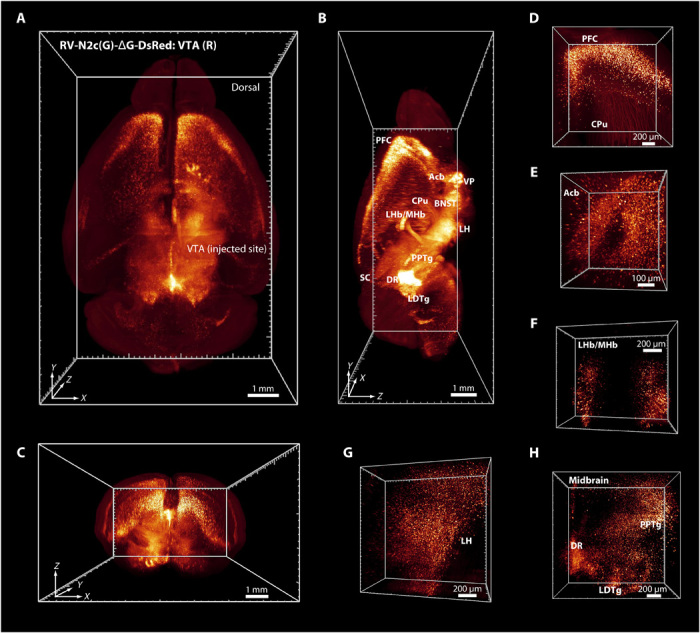
(A to C) Distribution of DsRed-positive cells in the whole brain [horizontal (A), sagittal (B), and coronal (C)]. The injection site in the VTA in the right hemisphere is marked in (A). Most regions projected to the VTA are marked in (B). (D to H) Several regions of RV-DsRed–positive cells are shown. Acb, accumbens nucleus; VP, ventral pallidum; SC, superior colliculus; BNST, bed nucleus of the stria terminalis; LDTg, laterodorsal tegmental nucleus; LH, lateral hypothalamic area; LHb/MHb, lateral/medial habenular nucleus; PPTg, pedunculopontine tegmental nucleus.
In addition, FDISCO was used to image functionally connected neurons in the brain, which regulate the motor function of the skeletal muscle. PRV152-EGFP was injected into the flexor digitorum superficialis muscle and then underwent retrograde propagation through multisynaptic circuits in the CNS. The EGFP-positive cells were mainly found in the brainstem, hypothalamus, and primary motor cortex (fig. S8). The number and distribution of these cells in the indicated regions could be identified even with the weak fluorescence expression of PRV-delivered proteins.
We also applied FDISCO to clear a brain labeled with AAV. We injected the left and right primary motor cortices of mice with AAV-EYFP and AAV-mCherry, respectively. We observed that the motor neurons sent long axon bundles to the contralateral primary motor cortex and spinal cord throughout the brain (fig. S9 and movie S6). In the medulla oblongata, the descending motor axons from the bilateral cortex performed pyramidal decussation to cross from the ipsilateral side to the contralateral side and from the ventral side to the dorsal side and extended along the corticospinal tract in the spinal cord.
DISCUSSION
Here, we report FDISCO, an advanced solvent-based clearing method for imaging whole organs. FDISCO achieved excellent fluorescence preservation of various fluorescent probes by temperature and pH adjustments while providing a high level of tissue transparency and size reduction for large-volume imaging. When combined with LSFM, FDISCO performed well in the 3D imaging and visualization of neuronal and vascular structures in various whole organs, including the brain, spinal cord, kidney, and muscle. Because of its high level of fluorescence preservation, FDISCO allowed the detection of weakly labeled neuronal structures in the whole brain (e.g., RV/PRV labeling) and repeated revisiting of the cleared tissues over months.
Current tissue clearing protocols are divided into two groups based on the chemistry used for clearing: organic solvent–based clearing methods and aqueous-based clearing methods (8–10, 13, 14). Compared with aqueous-based clearing methods, such as CUBIC and PACT, FDISCO enables higher clearing efficiency and fluorescence levels and induces a substantial size reduction, which is advantageous for the imaging of very large samples. In addition, the low viscosity of the clearing agents and the hardness of the cleared tissue make FDISCO more convenient for LSFM imaging than most aqueous-based clearing methods (26). Compared with available organic solvent–based clearing methods, such as FluoClearBABB, uDISCO, and Ethanol-ECi, FDISCO can achieve a higher level of fluorescence preservation with less clearing time. These features make FDISCO applicable for samples with a low expression of endogenous fluorescence protein and for the repeated imaging of cleared samples during long-term storage. In particular, uDISCO is not applicable to tdTomato, which is a valuable FP for specific labeling, while FDISCO can preserve the tdTomato fluorescence well. Overall, given these advantages, FDISCO should be a better alternative for whole-organ imaging as a state-of-the-art method.
Usually, the optimal conditions of clearing methods include the maintenance of sample size to image the initial morphology of the tissue, but most clearing methods induce size changes (shrinkage or expansion) during or at the end of the clearing procedure (8, 9). It is difficult to define whether sample shrinkage or expansion after clearing is bad or good for imaging. For example, tissue expansion can be used to improve the imaging resolution by overcoming the limit of optic resolution (43, 44). Tissue shrinkage may be more suitable for large samples because the imaging volume suffers from the limit of the working distance of high–numerical aperture objectives (8, 26). Moreover, the tissue structures and cell morphology were largely preserved on tissue shrinkage in FDISCO clearing (fig. S3E). Thus, we reasoned that FDISCO-induced size reduction facilitates the imaging of large tissues.
As described in the literature (13), the organic solvents used in the clearing protocols suppress GFP fluorescence signals by two potential deactivation mechanisms: protonation of the chromophore and denaturation of the protein structure. Here, we explored this suppression. As shown in Fig. 1E, the absorption spectrum at acidic pH (pH 5.0) displayed a nonfluorescence absorption band at 395 nm, which was attributed to the protonated chromophore (32, 36), while the absorption spectrum of the EGFP/THF solution displayed a nonfluorescence peak at approximately 383 nm. The absorption spectrum change in response to THF was similar to the changes in response to urea and guanidine hydrochloride, which are well-known denaturants that unfold native proteins by interrupting the hydrogen bonding of protein molecules (Fig. 1E) (37, 38, 45). Hence, it is tempting to hypothesize that the EGFP fluorescence quenching by THF was due to irreversible protein denaturation. In addition, Becker et al. have indicated that the continuously generated peroxides in DBE could bind covalently to GFP and quench fluorescence permanently.
Furthermore, we demonstrated that the EGFP fluorescence in THF was more stable under alkaline conditions. The stability of GFP and the denaturation of GFP by various denaturants are highly dependent on the pH of the solution, and GFP becomes very sensitive to denaturation at lower pH (33, 34). An alkaline pH (such as pH 9.0) might reduce the sensitivity of GFP to denaturation by THF. As demonstrated above, a low temperature during the clearing procedure facilitated the preservation of EGFP fluorescence. Previous studies have indicated that the fluorescence intensity of EGFP is also thermally sensitive (46, 47). A higher temperature can increase the probability of GFP unfolding; moreover, the thermally induced enhancement of collisions between molecules would probably increase fluorescence quenching. The improved fluorescence preservation in this work may be attributed to the enhanced resistance of the protein to denaturation by the use of appropriate temperature and pH condition. The improved preservation of other FPs in our method may be due to a similar mechanism.
In addition, FDISCO could achieve high transparency of the whole brain similar to that of 3DISCO in the same time frame, indicating that the lower temperature in FDISCO had no influence on the clearing time needed, which might be due to the strong permeability of the clearing reagents. It is worth noting that the optimization strategy of temperature and pH adjustments for better fluorescence preservation should be applicable to other clearing methods. As mentioned before, the strategy of lowering temperature was also effective for other solvent-based clearing protocols, such as BABB and uDISCO, and previous studies have indicated that the lower temperatures could benefit fluorescence preservation for aqueous-based clearing protocols (13, 35). However, the application of a temperature of 4°C might be limited by the melting point of solvents (e.g., tert-butanol, 23° to 26°C), the solubility of reagents, and the penetration of high-viscosity clearing agents. Moreover, the strategy of pH adjustment for GFP fluorescence preservation has also been reported in various clearing protocols, such as a-uDISCO, FluoClearBABB, and CUBIC (18, 27, 48).
Although FDISCO could significantly reduce the fluorescence quenching of FPs and most tested tracers, it did not change the incompatibility with some specific tracers (such as DiI and SYTO 16). This incompatibility may be due to the interference of the interactions between the probes and desired binding targets by the organic solvent reagents used for clearing (39). The FDISCO-cleared samples could not be further studied by electron microscopy because the lipid structures were dissolved by the clearing agents (23). In addition, after FDISCO clearing, redness appeared in heme-rich tissues (such as rat liver) due to the residual blood in tissues, which decreased the tissue transparency and thereby blocked the penetration of light into deeper tissues. To address this problem, some decolorizing reagents, such as Quadrol solution (18, 29), can be used before clearing.
As an advanced solvent-based clearing method, FDISCO overcomes the limitation of fluorescence quenching, achieves a high level of fluorescence preservation, and has a potential for use in the imaging of FPs with low expression. We believe that this method provides a valuable alternative for the high-resolution imaging of large-volume tissues. FDISCO is expected to be used for the 3D visualization and quantitative analysis of various organs in multiple biological applications and to facilitate the study of clinical diagnoses and interventions for pathological diseases.
MATERIALS AND METHODS
Animals
We used the following animals in the study: wild-type mice (C57BL/6N, 10 to 14 weeks old, male), Thy1-GFP-M mice (8 to 12 weeks old, female), Thy1-YFP-H mice (7 to 12 weeks old, female), Thy1-YFP-16 mice (9 weeks old, male), Sst-IRES-Cre::Ai14 mice (6 months old, female), and Sprague-Dawley rats (8 weeks old, female). The animals used in this study were selected for each experiment based on their genetic background (wild-type or fluorescence transgenes). Animals were housed in a specific pathogen–free animal house under a 12/12-hour light/dark cycle and were provided food and water ad libitum. All animal care and experimental protocols were in accordance with the Experimental Animal Management Ordinance of Hubei Province, P. R. China, and the guidelines of Huazhong University of Science and Technology and were approved by the Institutional Animal Ethics Committee of Huazhong University of Science and Technology.
Perfusion and tissue preparation
Adult mice and rats were anesthetized with a mixture of 10% urethane and 2% chloral hydrate (0.8 ml/100 g) along with 0.3% xylazine (0.5 ml/100 g) via intraperitoneal injection. Thereafter, the animals were transcardially perfused with 0.01 M phosphate-buffered saline (PBS) (P3813, Sigma-Aldrich), followed by 4% paraformaldehyde (PFA; 158127, Sigma-Aldrich) in PBS. The tissue samples (such as the CNS, internal organs, muscles, and bones) were dissected. For mouse embryo collection, the day of the vaginal plug was defined as embryonic day 0.5 (E0.5), and embryos (E16.5) were removed from anesthetized mothers. All harvested samples were postfixed overnight at 4°C in 4% PFA and then rinsed several times with PBS before clearing. For tissue section collection, brains and kidneys were sliced into 1-mm-thick coronal blocks with a vibratome (Leica VT1000 S, Germany).
In vitro assay of FPs
To screen the temperature and pH conditions of THF (186562, Sigma-Aldrich) for dehydration, we first mixed recombinant EGFP (0.05 mg/ml, final concentration) with 30 volume % THF/dH2O solution and incubated the mixture for 24 hours under different conditions. The recombinant EGFP was a histidine-tagged fusion protein of EGFP that was expressed in E. coli and purified by Ni-affinity chromatography. The THF solution was adjusted to pH 9.0 with triethylamine (80134318, Sinopharm Chemical Reagent Co. Ltd., China). The pH was measured by a pH meter (Ohaus). The resulting fluorescence signals were measured with a custom-made wide-field optical imaging system over time. The absorption spectra were measured by a LAMBDA 950 UV/Vis/NIR spectrophotometer (PerkinElmer, USA). All absorption spectra were normalized to the absorbance measured at 430 nm in each sample for further analysis. The fluorescence spectrum excited by a 488-nm laser was measured on an inverted confocal fluorescence microscope (LSM 710, Zeiss, Germany) in lambda mode.
We also mixed the recombinant EGFP with PBS (pH 7.4, room temperature), acetic acid (dissolved in H2O with pH 5.0, room temperature), 8 M urea (60°C; 10023218, Sinopharm Chemical Reagent Co., Ltd., China), and 6 M guanidine hydrochloride (GH; room temperature; 30095516, Sinopharm Chemical Reagent Co., Ltd., China). The absorption spectra were measured after incubation for 2 hours, when the fluorescence of EGFP treated by HAc, urea, and GH almost disappeared.
FDISCO clearing procedure
FDISCO clearing consisted of two steps: dehydration and refractive index matching. Tissues were dehydrated with THF solutions (mixed with dH2O, pH adjusted to 9.0 with triethylamine) at a series of concentrations 50, 70, 80, and 100 volume % (twice or thrice). Pure DBE (108014, Sigma-Aldrich) was used as a refractive index matching solution to clear tissue after dehydration. All steps were performed at 4°C with slight shaking. During clearing, the tissues were placed in glass chambers covered with aluminum foil in the dark. The incubation time of each step depended on the tissue type and size (table S1).
For bone clearing, mouse tibias were decalcified with 0.1 M EDTA-2Na (dissolved in 0.01 M PBS) for 2 to 3 days at 37°C with slight shaking and then washed with PBS several times prior to THF treatment. For whole-body clearing, mouse hair was removed before perfusion, and the whole body was washed with PBS several times at 37°C to remove residual blood after PFA fixation.
After FDISCO clearing, the tissues were stored in DBE in airtight glass chambers at 4°C in the dark. As tissue transparency might decrease during long-term storage after clearing, prior to repeated imaging, the tissues should be transferred to 100 volume % THF for several hours and then incubated again in DBE until the tissues were transparent. The peroxides in THF and DBE were removed by column absorption chromatography with basic activated aluminum oxide (20001861, Sinopharm Chemical Reagent Co. Ltd., China) (24). The clearing agents were freshly prepared. All other clearing protocols, including BABB, 3DISCO, uDISCO, FluoClearBABB, Ethanol-ECi, CUBIC, and PACT, were performed following the original papers (16, 17, 22, 23, 26–28).
Vasculature labeling
We labeled the vasculature in C57BL/6N mouse brain and kidney with DyLight 649 L. esculentum (Tomato) lectin (LEL-Dylight649, DL-1178, Vector Laboratories) or Alexa Fluor 647 anti-mouse CD31 antibody (CD31-A647, 102416, BioLegend) by caudal vein injection. LEL-Dylight649 was diluted in sterile saline to a concentration of 0.5 mg/ml (0.1 ml per mouse) and used to label the blood capillaries in the brain. The Alexa Fluor 647 anti-mouse CD31 antibody (10 to 15 μg) was diluted in sterile saline (total volume of 200 μl) to trace blood vessels in various diameters. After injection, the animals were placed in a warm cage for 30 min prior to perfusion. The mouse brains and kidneys were collected after perfusion.
Motor endplate labeling
To label motor endplates in muscle, Alexa Fluor 647–conjugated α-BTX (Invitrogen, USA) was injected via the tail vein at a dose of 0.3 μg/g and a conjugation time of 1 hour prior to perfusion. The gastrocnemius muscle was cleared by FDISCO and then imaged with LSFM.
Immunostaining
The following antibodies were used in this study: primary antibody—GFP (1:500 dilution; AB3080, Millipore); secondary antibodies—Cy3 AffiniPure goat anti-rabbit immunoglobulin G (IgG) (H+L) (1:500 dilution; 111-165-003, Jackson ImmunoResearch), Cy5 AffiniPure goat anti-rabbit IgG (H+L) (1:500 dilution; 111-175-144, Jackson ImmunoResearch), and Alexa Fluor 594 goat anti-rabbit IgG (H+L) (1:500 dilution; A-11037, Life Technologies).
For immunostaining, 1-mm-thick Thy1-GFP-M brain slices were immunostained for GFP using the iDISCO protocol (25). For pretreatment with methanol, brain slices were washed twice with PBS for 1 hour and then placed in 50 and 80% methanol (in PBS) for 1 hour at each step and twice in 100% methanol for 1 hour. The sections were bleached with 5% H2O2 in 20% dimethyl sulfoxide (DMSO) (472301, Sigma-Aldrich)/methanol at 4°C overnight. After bleaching, the slices were washed with methanol for 1 hour twice, followed by 20% DMSO/methanol for 1 hour twice, 80 and 50% methanol for 1 hour at each step, PBS for 1 hour twice, and finally PBS/0.2% Triton X-100 (T8787, Sigma-Aldrich) (PBST) for 1 hour twice. For the immunostaining step, the slices were incubated in PBS/0.2% Triton X-100/20% DMSO/0.3 M glycine (50046, Sigma-Aldrich) at 37°C overnight, blocked in PBS/0.2% Triton X-100/10% DMSO/6% goat serum at 37°C for 1 day, washed in PBS/0.2% Tween 20 (P2287, Sigma-Aldrich) with heparin (10 mg/ml; PTwH) overnight, and finally incubated with primary antibody dilutions in PTwH/5% DMSO/3% goat serum at 37°C with slight shaking on an oscillator for 2 days. The slices were then washed for 1 day and incubated with secondary antibodies diluted in PTwH/3% goat serum at 37°C with slight shaking on an oscillator for 2 days. The slices were finally washed in PTwH for 2 days prior to imaging.
For nuclei staining, 1-mm-thick Thy1-GFP-M brain slices were incubated with PI (2 μg/ml; P1304MP, Life Technologies) in PBS/0.2% Triton X-100 for 1 day at room temperature with gentle oscillation and then washed with PBS.
Neuronal tracing by virus
We used RV-N2C (G)-ΔG-DsRed (RV-DsRed, BrainVTA, R03002), rAAV-hSyn-EYFP-WPRE-pA (AAV-EYFP, BrainVTA, PT-0102), rAAV-hSyn-mCherry-WPRE-pA (AAV-mCherry, BrainVTA, PT-0100), and PRV152-EGFP (a gift from the Department of Trauma and Orthopedics, Peking University People’s Hospital) for neuronal tracing in the present study. For virus injection, C57BL/6N mice (10 weeks old, male) were anesthetized and placed in a stereotaxic frame (RWD Life Science, China). To expose the brain area targeted for tracing neurons, a cranial window on the skull was created. Viruses were injected into the brain regions using a custom-established injector fixed with a pulled glass pipette. The injection site of RV-DsRed (300 nl) was targeted to the VTA with the following coordinates: bregma, −3.52 mm; lateral, 0.5 mm; ventral, 4.23 mm. AAV-EYFP and AAV-mCherry (200 nl) were injected into the primary motor cortex in the left and right brain hemispheres, respectively, with the following coordinates: bregma, 1.0 mm; lateral, 1.0 mm; ventral, 1.5 mm. The glass pipette was kept in situ for 10 min before being moved outside. After finishing the injection, the cranial window was covered, and the incision was closed. For PRV labeling, PRV152-EGFP (1.5 μl) was injected into the flexor digitorum superficialis muscle in the right forelimb. Then, the animals were placed in a warm cage to wake up and then transferred into a regular keeping room. The animals were kept for 10 days after RV, 28 days after AAV, and 4 days after PRV injection before performing perfusion.
Measurement of light transmittance
The light transmittance of the cleared whole brain (Fig. 3C) was measured with a visible near-infrared optical fiber spectrometer (USB4000, Ocean Optics, USA). A circular spot of light (diameter, 5 mm) was irradiated on the central part of cleared brain samples from the dorsal side and was measured from the ventral side. The blank value was measured as the transmittance of the clearing reagents without a sample. The light transmittance of the sample normalized to the blank value, which was 100%, was defined as the relative transmittance. Each value was determined as an average of three measurements.
Fluorescence microscopy
Confocal microscopy
The brain and kidney slices were mounted on glass coverslips and incubated in final clearing solution (such as DBE). Then, the cortex regions of the brain slices were imaged with an inverted confocal fluorescence microscope (LSM 710, Zeiss, Germany) equipped with a Fluar 10×/0.5 objective (dry; working distance, 2.0 mm) and Plan-Apochromat 20×/0.8 objective (dry; working distance, 0.55 mm). The z-step interval was 5 μm.
Light sheet fluorescence microscopy
Large samples (such as brain, spinal cord, CNS, kidney, and muscle) were imaged using a light sheet fluorescence microscope (Ultramicroscope, LaVision BioTec, Germany) equipped with an sCMOS camera (Andor Neo), a 2×/0.5 objective lens equipped with a dipping cap, and an Olympus MVX10 zoom microscope body (magnification range of ×0.63 to ×6.3). The cleared tissues were mounted on the sample holder and incubated with the final clearing solution (such as DBE) in the sample reservoir. For entire scanning of whole organs, the z-step interval was 5 or 10 μm, and for image acquisition in the regions of interest, an interval in the range of 2 to 5 μm was used.
Imaging data processing
The obtained images were analyzed with ImageJ and Imaris software. All raw image data were collected in a lossless TIFF (Tag Image File Format) format (8-bit images for confocal microscopy and 16-bit images for LSFM data). The MIP of the z stack was performed with sequential images in ImageJ software. The MIP of the z stack was performed from 0 μm to a certain depth (40 to 60 μm) for each brain slice. The z stack ranges used for MIP were kept consistent for each slice before and after clearing. The LSFM images were transformed to 8-bit images with ImageJ and then imported into Imaris. 3D-rendered images and movies were visualized and captured with Imaris. The locations and counts of cells in the brain (fig. S8, G to I), the motor endplates in the muscle (Fig. 4E), and the glomeruli in the kidney (Fig. 5F) in the images were determined by the “Spots” function of Imaris. In addition, the tracing of nerve fibers for the muscle was performed by the “FilamentTracer” function of Imaris (Fig. 4E).
Quantifications
Measurement of tissue size change
The measurements of sample linear expansion or shrinkage were based on the bright-field images. The brain was outlined to measure the area using the “polygon-selections” function of ImageJ software. The linear size change value was quantified by the square root of area size change (Fig. 3D).
Imaging depth quantification
The z depth of LSFM images supposed to be “d” was measured from the surface to the inner part of the brain until the last identifiable cell over the background. The length of the cleared brain (dorsal to ventral) was supposed to be “D”. The imaging depth was quantified as “d/D” (Fig. 3F).
Normalized mean fluorescence quantification
To assess the fluorescence preservation of the clearing methods, we quantitatively calculated the normalized mean fluorescence intensity of fluorescence images at the indicated time points during clearing. For each brain sample at each time point, the MIP of the image stack (40 to 60 μm thick) was acquired from the surface of the brain slice in the cortex. Next, the signal and background area were separated from each z projection image by a threshold function. As the intensity of the signal area includes the signal intensity and the background intensity (26), the mean fluorescence intensity M of an image was calculated as follows
| (1) |
I and N are the sum of the pixel intensity and pixel number, respectively, of the signal or background. The mean fluorescence intensity of an image is supposed to be “Mb” before clearing, “M0” at 0 day after clearing, and “Mi” at i day during long-term storage. The fluorescence intensity preservation during clearing (0 day) was normalized to the intensity before clearing as “M0/Mb” [Figs. 1G and 2 (C and I) and figs. S1 (B and D), S2B, and S7B], and the fluorescence preservation at each time point (i day) during long-term storage was normalized to the intensity at 0 day after clearing as “Mi/M0” [Figs. 1I and 2 (E and J) and figs. S1D and S2D]. The fluorescence preservation of recombinant EGFP in the brain samples was quantitatively calculated during long-term storage (Fig. 1B).
Fluorescence level quantification
The fluorescence level (Figs. 3E and 4D) was expressed as the signal-to-background ratio and quantified as described in the uDISCO paper (26). From each brain scan, a 100-μm MIP of image stacks was acquired at the same cortex region. To calculate the mean intensity of the signal for each z projection, we used the threshold function of ImageJ software to separate the signal from the background and then measured the mean intensity of the signal by the “analyze particles” function. The mean intensity of the background was obtained by averaging the values of five regions without a signal in the z projection. The fluorescence level value was calculated by dividing the mean signal intensity above the background.
Normalized signal profile
In the ImageJ software, a straight line (length, 200 μm) was drawn across the specific signal, and the fluorescence signal was measured by the “plot profile” function. The intensity of the background was measured as the value of the baseline. Next, the fluorescence signal profile was normalized by the background intensity (fig. S5C).
Statistical analysis
Data analyses and graph construction were performed using the SPSS software. The sample sizes for technical replicates are presented in the figure legends, and samples were randomly assigned to groups for experiments. Data are presented as means ± SD. For analysis of the statistical significance, the data distribution in each experiment was checked for normality using the Shapiro-Wilk test. The homogeneity of variance for each group was evaluated by the Levene test. P values were calculated using an independent-sample t test (two-sided) to compare data between two groups [Figs. 1I and 2 (C, I, and J) and figs. S1 (B and D) and S2 (B and D)]. One-way ANOVA was used to compare more than two groups of data. If the variance was homogeneous, we used the Bonferroni post hoc test for multiple comparisons (Figs. 1B, 3E, and 4D and figs. S4E and S7B); otherwise, Dunnett’s T3 post hoc test was used (Figs. 1G and 3, D and F). In this study, P < 0.05 was considered significant (*P < 0.05, **P < 0.01, and ***P < 0.001).
Supplementary Material
Acknowledgments
We thank M. Zhu for discussions on the absorption and emission spectra of EGFP solutions; B. Jiang (Peking University People’s Hospital), T. Xu, and X. Li for providing the transgenic mice; S. Han (Peking University People’s Hospital), P. Zhao, and Z. Chen for virus injection; and Y. Dai and B. Dai for the help with recombinant EGFP experiments. We also thank the Optical Bioimaging Core Facility of WNLO-HUST for support with data acquisition. Funding: This study was supported by the National Key Research and Development Program of China (grant no. 2017YFA0700501), the National Nature Science Foundation of China (grant nos. 61860206009, 81870934, 31571002, 81701354, and 91749209), the Science Fund for Creative Research Group of China (grant no. 61721092), the Project funded by the China Postdoctoral Science Foundation (grant nos. 2017M612463 and 2018T110772), the Fundamental Research Funds for the Central Universities, HUST (grant no. 2018KFYXKJC026), and the Director Fund of WNLO. Author contributions: D.Z., Y.Q., and T.Y. designed the study. Y.Q. and T.Y. designed and performed most of the experiments. J.X., P.W., Y.M., J.Z., and Y.L. contributed to animal preparation and clearing of the samples. Y.Q. performed the imaging. Y.Q. and T.Y. prepared the figures and wrote the manuscript. D.Z. supervised the project and revised the manuscript. H.G. and Q.L. participated in the discussions of the results and commented on the manuscript. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/5/1/eaau8355/DC1
Fig. S1. Comparison of fluorescence preservation of FPs and chemical fluorescent tracers between FDISCO and other clearing methods.
Fig. S2. Applicability of low temperature for increasing the endogenous fluorescence preservation of the BABB and uDISCO methods.
Fig. S3. Clearing performance of FDISCO for whole organs and whole body.
Fig. S4. Fluorescence imaging of the spinal cord after FDISCO, 3DISCO, and uDISCO clearing in Thy1-GFP-M mice.
Fig. S5. Comparison of whole-brain imaging for Thy1-YFP-H mouse brains after FDISCO, 3DISCO, and uDISCO clearing.
Fig. S6. LSFM imaging of the tdTomato-labeled whole brain after FDISCO clearing.
Fig. S7. Fluorescence of RV-DsRed–labeled mouse brains cleared by FDISCO, 3DISCO, and uDISCO.
Fig. S8. 3D visualization of the PRV-labeled neuronal projections in the brain by FDISCO clearing and LSFM imaging.
Fig. S9. 3D visualization of AAV-labeled descending motor axons from the primary motor cortex by FDISCO clearing.
Table S1. FDISCO protocols for various tissues.
Movie S1. Imaging of FDISCO-cleared mouse brain (Thy1-GFP-M).
Movie S2. 3D reconstruction and rendering of nerve branches and motor endplates in the gastrocnemius muscle.
Movie S3. 3D visualization of the vasculature in the whole brain.
Movie S4. Reconstruction of the glomeruli in the intact kidney.
Movie S5. 3D visualization of the neurons projecting to VTA by RV injection.
Movie S6. Tracing of descending motor axons from both the left and right primary motor cortices by AAV labeling.
REFERENCES AND NOTES
- 1.Miyamichi K., Amat F., Moussavi F., Wang C., Wickersham I., Wall N. R., Taniguchi H., Tasic B., Huang Z. J., He Z., Callaway E. M., Horowitz M. A., Luo L., Cortical representations of olfactory input by trans-synaptic tracing. Nature 472, 191–196 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zingg B., Hintiryan H., Gou L., Song M. Y., Bay M., Bienkowski M. S., Foster N. N., Yamashita S., Bowman I., Toga A. W., Dong H.-W., Neural networks of the mouse neocortex. Cell 156, 1096–1111 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Oh S. W., Harris J. A., Ng L., Winslow B., Cain N., Mihalas S., Wang Q., Lau C., Kuan L., Henry A. M., Mortrud M. T., Ouellette B., Nguyen T. N., Sorensen S. A., Slaughterbeck C. R., Wakeman W., Li Y., Feng D., Ho A., Nicholas E., Hirokawa K. E., Bohn P., Joines K. M., Peng H., Hawrylycz M. J., Phillips J. W., Hohmann J. G., Wohnoutka P., Gerfen C. R., Koch C., Bernard A., Dang C., Jones A. R., Zeng H., A mesoscale connectome of the mouse brain. Nature 508, 207–214 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ragan T., Kadiri L. R., Venkataraju K. U., Bahlmann K., Sutin J., Taranda J., Arganda-Carreras I., Kim Y., Seung H. S., Osten P., Serial two-photon tomography for automated ex vivo mouse brain imaging. Nat. Methods 9, 255–258 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kasthuri N., Hayworth K. J., Berger D. R., Schalek R. L., Conchello J. A., Knowles-Barley S., Lee D., Vázquez-Reina A., Kaynig V., Jones T. R., Roberts M., Morgan J. L., Tapia J. C., Seung H. S., Roncal W. G., Vogelstein J. T., Burns R., Sussman D. L., Priebe C. E., Pfister H., Lichtman J. W., Saturated reconstruction of a volume of neocortex. Cell 162, 648–661 (2015). [DOI] [PubMed] [Google Scholar]
- 6.Gong H., Xu D., Yuan J., Li X., Guo C., Peng J., Li Y., Schwarz L. A., Li A., Hu B., Xiong B., Sun Q., Zhang Y., Liu J., Zhong Q., Xu T., Zeng S., Luo Q., High-throughput dual-colour precision imaging for brain-wide connectome with cytoarchitectonic landmarks at the cellular level. Nat. Commun. 7, 12142 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Conchello J.-A., Lichtman J. W., Optical sectioning microscopy. Nat. Methods 2, 920–931 (2005). [DOI] [PubMed] [Google Scholar]
- 8.Richardson D. S., Lichtman J. W., Clarifying tissue clearing. Cell 162, 246–257 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Susaki E. A., Ueda H. R., Whole-body and whole-organ clearing and imaging techniques with single-cell resolution: Toward organism-level systems biology in mammals. Cell Chem. Biol. 23, 137–157 (2016). [DOI] [PubMed] [Google Scholar]
- 10.Silvestri L., Costantini I., Sacconi L., Pavone F. S., Clearing of fixed tissue: A review from a microscopist’s perspective. J. Biomed. Opt. 21, 081205 (2016). [DOI] [PubMed] [Google Scholar]
- 11.V. V. Tuchin, Optical Clearing of Tissues and Blood (SPIE Press, 2005). [Google Scholar]
- 12.Zhu D., Larin K. V., Luo Q., Tuchin V. V., Recent progress in tissue optical clearing. Laser Photon. Rev. 7, 732–757 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tainaka K., Kuno A., Kubota S. I., Murakami T., Ueda H. R., Chemical principles in tissue clearing and staining protocols for whole-body cell profiling. Annu. Rev. Cell Dev. Biol. 32, 713–741 (2016). [DOI] [PubMed] [Google Scholar]
- 14.Yu T., Qi Y., Gong H., Luo Q., Zhu D., Optical clearing for multiscale biological tissues. J. Biophotonics 11, e201700187 (2018). [DOI] [PubMed] [Google Scholar]
- 15.Chung K., Wallace J., Kim S.-Y., Kalyanasundaram S., Andalman A. S., Davidson T. J., Mirzabekov J. J., Zalocusky K. A., Mattis J., Denisin A. K., Pak S., Bernstein H., Ramakrishnan C., Grosenick L., Gradinaru V., Deisseroth K., Structural and molecular interrogation of intact biological systems. Nature 497, 332–337 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yang B., Treweek J. B., Kulkarni R. P., Deverman B. E., Chen C.-K., Lubeck E., Shah S., Cai L., Gradinaru V., Single-cell phenotyping within transparent intact tissue through whole-body clearing. Cell 158, 945–958 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Susaki E. A., Tainaka K., Perrin D., Kishino F., Tawara T., Watanabe T. M., Yokoyama C., Onoe H., Eguchi M., Yamaguchi S., Abe T., Kiyonari H., Shimizu Y., Miyawaki A., Yokota H., Ueda H. R., Whole-brain imaging with single-cell resolution using chemical cocktails and computational analysis. Cell 157, 726–739 (2014). [DOI] [PubMed] [Google Scholar]
- 18.Tainaka K., Kubota S. I., Suyama T. Q., Susaki E. A., Perrin D., Ukai-Tadenuma M., Ukai H., Ueda H. R., Whole-body imaging with single-cell resolution by tissue decolorization. Cell 159, 911–924 (2014). [DOI] [PubMed] [Google Scholar]
- 19.Hama H., Hioki H., Namiki K., Hoshida T., Kurokawa H., Ishidate F., Kaneko T., Akagi T., Saito T., Saido T., Miyawaki A., ScaleS: An optical clearing palette for biological imaging. Nat. Neurosci. 18, 1518–1529 (2015). [DOI] [PubMed] [Google Scholar]
- 20.Lai H. M., Liu A. K. L., Ng H. H. M., Goldfinger M. H., Chau T. W., DeFelice J., Tilley B. S., Wong W. M., Wu W., Gentleman S. M., Next generation histology methods for three-dimensional imaging of fresh and archival human brain tissues. Nat. Commun. 9, 1066 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li W., Germain R. N., Gerner M. Y., Multiplex, quantitative cellular analysis in large tissue volumes with clearing-enhanced 3D microscopy (Ce3D). Proc. Natl. Acad. Sci. U.S.A. 114, E7321–E7330 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dodt H.-U., Leischner U., Schierloh A., Jährling N., Mauch C. P., Deininger K., Deussing J. M., Eder M., Zieglgänsberger W., Becker K., Ultramicroscopy: Three-dimensional visualization of neuronal networks in the whole mouse brain. Nat. Methods 4, 331–336 (2007). [DOI] [PubMed] [Google Scholar]
- 23.Ertürk A., Becker K., Jährling N., Mauch C. P., Hojer C. D., Egen J. G., Hellal F., Bradke F., Sheng M., Dodt H.-U., Three-dimensional imaging of solvent-cleared organs using 3DISCO. Nat. Protoc. 7, 1983–1995 (2012). [DOI] [PubMed] [Google Scholar]
- 24.Becker K., Jährling N., Saghafi S., Weiler R., Dodt H.-U., Chemical clearing and dehydration of GFP expressing mouse brains. PLOS ONE 7, e33916 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Renier N., Wu Z., Simon D. J., Yang J., Ariel P., Tessier-Lavigne M., iDISCO: A simple, rapid method to immunolabel large tissue samples for volume imaging. Cell 159, 896–910 (2014). [DOI] [PubMed] [Google Scholar]
- 26.Pan C., Cai R., Quacquarelli F. P., Ghasemigharagoz A., Lourbopoulos A., Matryba P., Plesnila N., Dichgans M., Hellal F., Ertürk A., Shrinkage-mediated imaging of entire organs and organisms using uDISCO. Nat. Methods 13, 859–867 (2016). [DOI] [PubMed] [Google Scholar]
- 27.Schwarz M. K., Scherbarth A., Sprengel R., Engelhardt J., Theer P., Giese G., Fluorescent-protein stabilization and high-resolution imaging of cleared, intact mouse brains. PLOS ONE 10, e0124650 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Klingberg A., Hasenberg A., Ludwig-Portugall I., Medyukhina A., Männ L., Brenzel A., Engel D. R., Figge M. T., Kurts C., Gunzer M., Fully automated evaluation of total glomerular number and capillary tuft size in nephritic kidneys using lightsheet microscopy. J. Am. Soc. Nephrol. 28, 452–459 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jing D., Zhang S., Luo W., Gao X., Men Y., Ma C., Liu X., Yi Y., Bugde A., Zhou B. O., Zhao Z., Yuan Q., Feng J. Q., Gao L., Ge W.-P., Zhao H., Tissue clearing of both hard and soft tissue organs with the PEGASOS method. Cell Res. 28, 803–818 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ariel P., A beginner’s guide to tissue clearing. Int. J. Biochem. Cell Biol. 84, 35–39 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Becker K., Hahn C. M., Saghafi S., Jährling N., Wanis M., Dodt H.-U., Reduction of photo bleaching and long term archiving of chemically cleared GFP-expressing mouse brains. PLOS ONE 9, e114149 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tsien R. Y., The green fluorescent protein. Annu. Rev. Biochem. 67, 509–544 (1998). [DOI] [PubMed] [Google Scholar]
- 33.Alkaabi K. M., Yafea A., Ashraf S. S., Effect of pH on thermal- and chemical-induced denaturation of GFP. Appl. Biochem. Biotechnol. 126, 149–156 (2005). [DOI] [PubMed] [Google Scholar]
- 34.Saeed I. A., Ashraf S. S., Denaturation studies reveal significant differences between GFP and blue fluorescent protein. Int. J. Biol. Macromol. 45, 236–241 (2009). [DOI] [PubMed] [Google Scholar]
- 35.Yu T., Qi Y., Zhu J., Xu J., Gong H., Luo Q., Zhu D., Elevated-temperature-induced acceleration of PACT clearing process of mouse brain tissue. Sci. Rep. 7, 38848 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Xiong H., Zhou Z., Zhu M., Lv X., Li A., Li S., Li L., Yang T., Wang S., Yang Z., Xu T., Luo Q., Gong H., Zeng S., Chemical reactivation of quenched fluorescent protein molecules enables resin-embedded fluorescence microimaging. Nat. Commun. 5, 3992 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ward W. W., Cody C. W., Hart R. C., Cormier M. J., Spectrophotometric identity of the energy transfer chromophores in renilla and Aequorea green fluorescent proteins. Photochem. Photobiol. 31, 611–615 (1980). [Google Scholar]
- 38.Niwa H., Inouye S., Hirano T., Matsuno T., Kojima S., Kubota M., Ohashi M., Tsuji F. I., Chemical nature of the light emitter of the Aequorea green fluorescent protein. Proc. Natl. Acad. Sci. U.S.A. 93, 13617–13622 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lai H. M., Ng W.-L., Gentleman S. M., Wu W., Chemical probes for visualizing intact animal and human brain tissue. Cell Chem. Biol. 24, 659–672 (2017). [DOI] [PubMed] [Google Scholar]
- 40.Kim Y., Yang G. R., Pradhan K., Venkataraju K. U., Bota M., García Del Molino L. C., Fitzgerald G., Ram K., He M., Levine J. M., Mitra P., Huang Z. J., Wang X.-J., Osten P., Brain-wide maps reveal stereotyped cell-type-based cortical architecture and subcortical sexual dimorphism. Cell 171, 456–469.e22 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Xiong B., Li A., Lou Y., Chen S., Long B., Peng J., Yang Z., Xu T., Yang X., Li X., Jiang T., Luo Q., Gong H., Precise cerebral vascular atlas in stereotaxic coordinates of whole mouse brain. Front. Neuroanat. 11, 128 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Watabe-Uchida M., Eshel N., Uchida N., Neural circuitry of reward prediction error. Annu. Rev. Neurosci. 40, 373–394 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Murakami T. C., Mano T., Saikawa S., Horiguchi S. A., Shigeta D., Baba K., Sekiya H., Shimizu Y., Tanaka K. F., Kiyonari H., Iino M., Mochizuki H., Tainaka K., Ueda H. R., A three-dimensional single-cell-resolution whole-brain atlas using CUBIC-X expansion microscopy and tissue clearing. Nat. Neurosci. 21, 625–637 (2018). [DOI] [PubMed] [Google Scholar]
- 44.Ku T., Swaney J., Park J.-Y., Albanese A., Murray E., Cho J. H., Park Y.-G., Mangena V., Chen J., Chung K., Multiplexed and scalable super-resolution imaging of three-dimensional protein localization in size-adjustable tissues. Nat. Biotechnol. 34, 973–981 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jung K.-C., Park J.-B., Maeng P.-J., Kim H.-J., Fluorescence quenching of green fluorescent protein during denaturation by guanidine. Bull. Korean Chem. Soc. 26, 413–417 (2005). [Google Scholar]
- 46.dos Santos A. M., Thermal effect on Aequorea green fluorescent protein anionic and neutral chromophore forms fluorescence. J. Fluoresc. 22, 151–154 (2012). [DOI] [PubMed] [Google Scholar]
- 47.Zhang C., Liu M.-S., Xing X.-H., Temperature influence on fluorescence intensity and enzyme activity of the fusion protein of GFP and hyperthermophilic xylanase. Appl. Microbiol. Biotechnol. 84, 511–517 (2009). [DOI] [PubMed] [Google Scholar]
- 48.Li Y., Xu J., Wan P., Yu T., Zhu D., Optimization of GFP fluorescence preservation by a modified uDISCO clearing protocol. Front. Neuroanat. 12, 67 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/5/1/eaau8355/DC1
Fig. S1. Comparison of fluorescence preservation of FPs and chemical fluorescent tracers between FDISCO and other clearing methods.
Fig. S2. Applicability of low temperature for increasing the endogenous fluorescence preservation of the BABB and uDISCO methods.
Fig. S3. Clearing performance of FDISCO for whole organs and whole body.
Fig. S4. Fluorescence imaging of the spinal cord after FDISCO, 3DISCO, and uDISCO clearing in Thy1-GFP-M mice.
Fig. S5. Comparison of whole-brain imaging for Thy1-YFP-H mouse brains after FDISCO, 3DISCO, and uDISCO clearing.
Fig. S6. LSFM imaging of the tdTomato-labeled whole brain after FDISCO clearing.
Fig. S7. Fluorescence of RV-DsRed–labeled mouse brains cleared by FDISCO, 3DISCO, and uDISCO.
Fig. S8. 3D visualization of the PRV-labeled neuronal projections in the brain by FDISCO clearing and LSFM imaging.
Fig. S9. 3D visualization of AAV-labeled descending motor axons from the primary motor cortex by FDISCO clearing.
Table S1. FDISCO protocols for various tissues.
Movie S1. Imaging of FDISCO-cleared mouse brain (Thy1-GFP-M).
Movie S2. 3D reconstruction and rendering of nerve branches and motor endplates in the gastrocnemius muscle.
Movie S3. 3D visualization of the vasculature in the whole brain.
Movie S4. Reconstruction of the glomeruli in the intact kidney.
Movie S5. 3D visualization of the neurons projecting to VTA by RV injection.
Movie S6. Tracing of descending motor axons from both the left and right primary motor cortices by AAV labeling.