

Abstract
The epithelial splicing regulatory proteins 1 and 2 (ESRP1 and ESRP2) control the epithelial‐to‐mesenchymal transition (EMT) splicing program in cancer. However, their role in breast cancer recurrence is unclear. In this study, we report that high levels of ESRP1, but not ESRP2, are associated with poor prognosis in estrogen receptor positive (ER+) breast tumors. Knockdown of ESRP1 in endocrine‐resistant breast cancer models decreases growth significantly and alters the EMT splicing signature, which we confirm using TCGA SpliceSeq data of ER+ BRCA tumors. However, these changes are not accompanied by the development of a mesenchymal phenotype or a change in key EMT‐transcription factors. In tamoxifen‐resistant cells, knockdown of ESRP1 affects lipid metabolism and oxidoreductase processes, resulting in the decreased expression of fatty acid synthase (FASN), stearoyl‐CoA desaturase 1 (SCD1), and phosphoglycerate dehydrogenase (PHGDH) at both the mRNA and protein levels. Furthermore, ESRP1 knockdown increases the basal respiration and spare respiration capacity. This study reports a novel role for ESRP1 that could form the basis for the prevention of tamoxifen resistance in ER+ breast cancer.
Keywords: alternative splicing, ER‐positive breast cancer, ESRP1, fatty acid metabolism, HTA
Subject Categories: Cancer, Metabolism, RNA Biology
Introduction
The estrogen receptor positive (ER+) subtype constitutes approximately 70% of all breast cancers. Despite the success of endocrine therapies in the early stage of ER+ breast cancer, at least 20% of patients will suffer a distant recurrence within 10 years 1, 2. More specifically, the intrinsic or acquired resistance to endocrine therapy limits its utility in these patients, leading to recurrence. Thus, it is crucial to identify the underlying molecular mechanisms of recurrence/resistance to improve the success of endocrine therapies and prevent breast cancer mortality.
Several studies have sought to understand the basis of recurrence by studying tamoxifen/endocrine therapy resistance models 3, 4, 5. Acquired resistance models, such as the stepwise increase in treatment with tamoxifen or fulvestrant in vitro, estrogen deprivation or the overexpression of a marker that confers endocrine resistance, have been used in vitro and in vivo to study this phenomenon. Crosstalk between ER and other signaling pathways and epigenetic mechanisms are well‐documented mechanisms of recurrence/resistance in endocrine resistance. Using Next‐Gen sequencing, the importance of ER mutations in endocrine resistance has been demonstrated 6, 7, 8. Other studies have evaluated the resistance mechanisms using ER+ tumors from patients treated before surgery and reported that FGFR1 amplification confers antiestrogen resistance to ER+ breast cancer and that the ERα pathway remains active in estrogen‐deprived ER+/FGFR1‐amplified breast cancers 9. Changes in biological processes such as proliferation and oxidative phosphorylation have also been reported to contribute to tamoxifen resistance 10.
Aberrant alternative splicing in cancer, including breast cancer, is an emerging field and may affect genes and proteins both at the expression and functional levels. These events are regulated by complex processes involving the core spliceosome machinery and multiple regulatory factors 11. Mutations in splicing factors or changes in expression levels of the proteins may contribute to aberrant alternative splicing. The splicing factor 3b subunit 1, SF3B1, was one of the mutated genes identified using next‐generation sequencing of breast tumors 12, 13, 14. We have previously demonstrated the upregulation of SF3B1 and SF3B3, another SF3B subunit, in acquired endocrine‐resistant models as well as in cases with Oncotype Dx high recurrence scores 15. However, only SF3B3 expression is correlated with a poor prognosis in the patients with ER+ breast cancer 15.
A number of splicing factors have been reported to affect the hallmarks of cancer 16. Epithelial splicing regulatory proteins (ESRPs—ESRP1 and ESRP2) are implicated in invasion, metastasis, and the regulation of the splicing program involved in the epithelial‐to‐mesenchymal transition (EMT) in cancer 17, 18, 19. ESRP1 promotes lung cancer metastasis by regulating CD44 splicing in ER‐negative 4T1 mouse mammary tumor cells 20. The relevance of ESRPs to breast cancer is not clear. In this study, we sought to determine the role of ESRPs in ER+ breast cancer and have identified a novel aspect of ESRP1 functionality in endocrine therapy‐resistant breast cancer.
Results
High ESRP1 expression correlates with worse prognosis in ER+ breast cancer
To determine the clinical relevance of ESRP1 and ESRP2 in breast cancer, we first correlated the gene expression levels with the overall survival (OS) using the BreastMark tool, which integrates the gene expression and survival data from 26 datasets based on 12 different microarray platforms 21. This analysis showed that a higher expression of ESRP1 correlated with a worse prognosis for ER+ breast cancer [Hazard ratio (HR) = 2.01 (1.53–2.64); Score (log rank) test = 26.4 on 1 df, P = 2.8 e‐07 (n = 934, number of events = 216)] (Fig 1A) using Cox proportional hazards regression analysis. However, ESRP1 expression was not prognostic in patients with estrogen receptor negative (ER−) breast cancers (Fig 1B; HR = 1.15 (0.8–1.68); Score (log rank) test = 0.58 on 1 df, P = 0.4481; n = 322, number of events = 130). Expression of ESRP2 was not associated with the overall survival of patients with ER+ tumors [Fig EV1A; HR = 1.117 (0.82–1.51); Score (log rank) test = 0.53 on 1 df, P = 0.47 (n = 708, number of events = 171)] as well as the overall survival of patients with ER‐negative tumors [Fig EV1B; HR = 1.37 (0.91–2.04); Score (log rank) test = 2.31 on 1 df, P = 0.13 (n = 253, number of events = 96)] in BreastMark datasets.
Figure 1. High ESRP1 expression correlates with poor prognosis in estrogen receptor positive (ER+) but not in estrogen receptor negative (ER−) breast cancer.
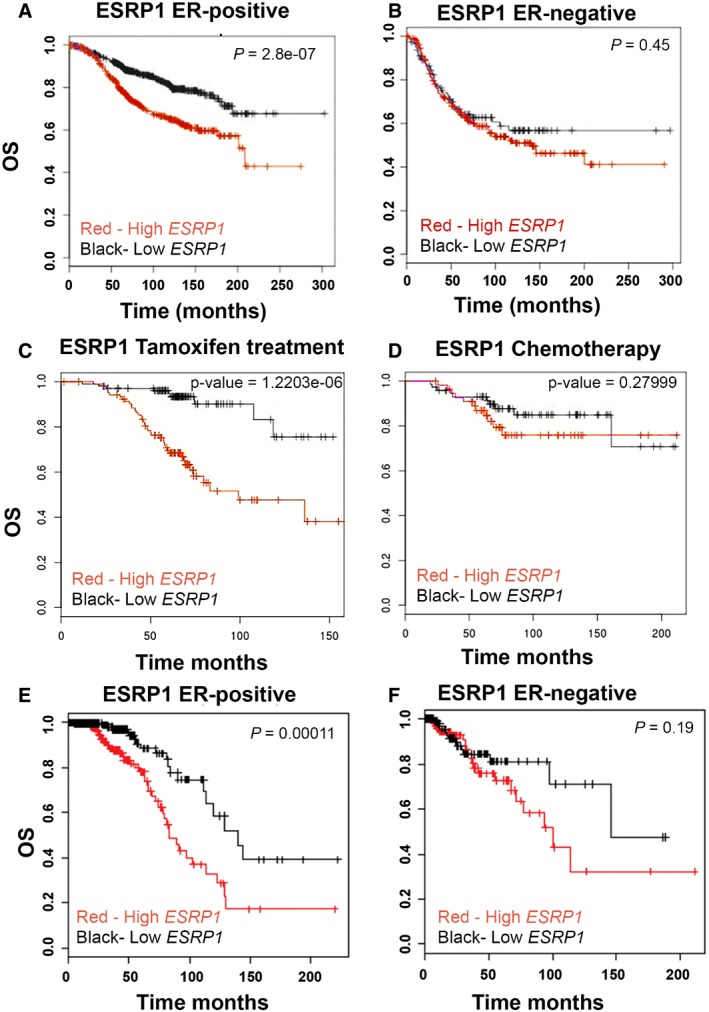
- BreastMark microarray platform; Kaplan–Meier curves of overall survival (OS), demonstrating that high expression of ESRP1 (red line) is associated with poor prognosis in ER+ breast cancer. A log rank test was used to calculate P = 2.8 e‐07 (n = 934, number of events = 216).
- BreastMark microarray platform; Kaplan–Meier curves of overall survival (OS) of ER− breast cancer. A log rank test was used to calculate P = 0.4481 (n = 322, number of events = 130).
- BreastMark microarray platform; correlation of ESRP1 expression with overall survival in tamoxifen‐treated patients. A log rank test was used to calculate P = 1.218, e‐06 (n = 210, number of events = 49).
- BreastMark microarray platform; correlation of ESRP1 expression with overall survival in chemotherapy‐treated patients. A log rank test was used to calculate the P = 0.28 (n = 129, number of events = 21)
- The Cancer Genome Atlas Breast Invasive Carcinoma (TCGA‐BRCA) RNA‐Seq dataset; Kaplan–Meier curves of overall survival (OS) in ER+ breast cancer, demonstrating that high expression of ESRP1 (red line) is associated with poor prognosis in ER+ breast cancer. A log rank test was used to calculate P = 0.00011 (n = 656, number of events = 62).
- The Cancer Genome Atlas Breast Invasive Carcinoma (TCGA‐BRCA) RNA‐Seq dataset; Kaplan–Meier curves of overall survival (OS) in ER− breast cancer. Red‐high ESRP1 expression; Black‐low ESRP1 expression. A log rank test was used to calculate P = 0.19 (n = 100, number of events = 17).
Figure EV1. High ESRP2 expression is not associated with prognosis in both ER+ and ER− breast tumors.
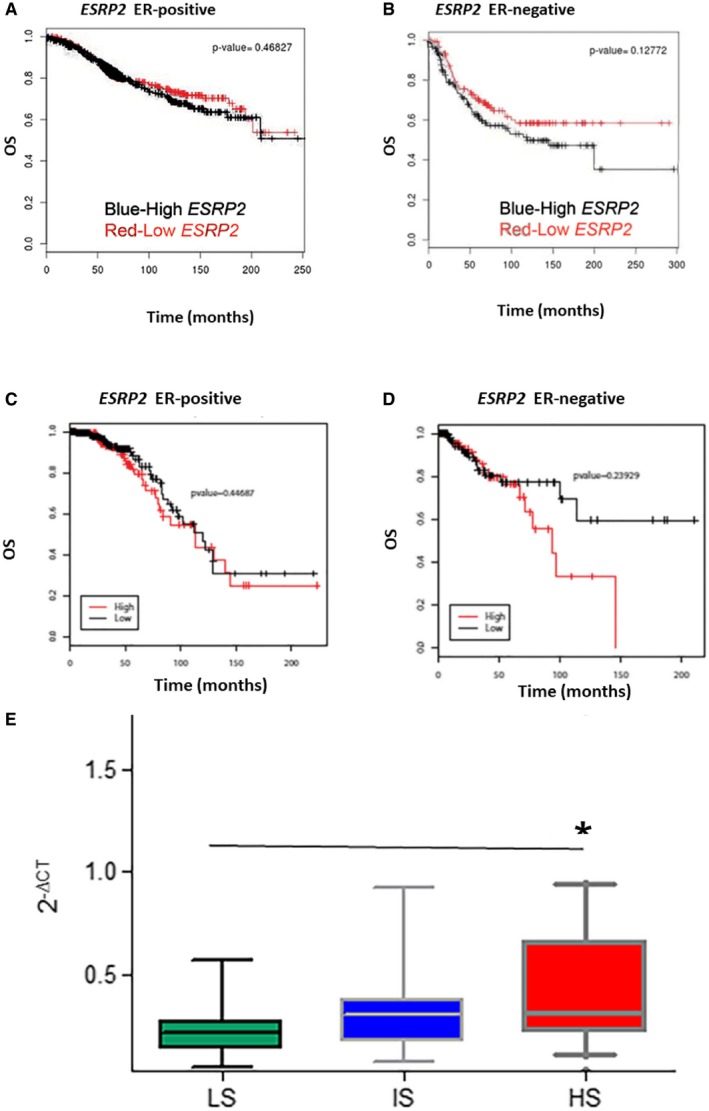
- BreastMark microarray platform; Kaplan–Meier curves of overall survival (OS) in ER‐positive (ER+) breast cancer. A log rank test was used to calculate the P = 0.47 (n = 708, number of events = 171) and P = 0.13 (n = 253, number of events = 96)].
- BreastMark microarray platform; Kaplan–Meier curves of overall survival (OS) in ER‐negative (ER−) breast cancer. A log rank test was used to calculate the P = 0.13 (n = 253, number of events = 96).
- TCGA Breast Invasive Carcinoma (BRCA) RNA‐Seq dataset; Kaplan–Meier curves of overall survival (OS) in ER+ breast cancer. A log rank test was used to calculate the P = 0.45 (n = 210, number of events = 49).
- TCGA Breast Invasive Carcinoma (BRCA) RNA‐Seq dataset; Kaplan–Meier curves of overall survival (OS) in ER− breast cancer. Red, high ESRP1 expression; black, low ESRP1 expression. A log rank test was used to calculate the P = 0.24 (n = 129, number of events = 21).
- ESRP1 expression in breast cancer with poor prognosis. RT–qPCR assay showing that expression of ESRP1 is higher in patients with high recurrence scores in a Oncotype DX cohort n = 59 (19 LS, 20 IS, and 20 HS); LS, low score; IS, intermediate score; and HS, high score, * represents P < 0.05; data (mean ± SD) were statistically significant and calculated using two‐way ANOVA.
We further assessed the correlation of ESRP1 expression with the overall survival in tamoxifen‐treated patients using the same platform in the BreastMark database. A high expression of ESRP1 was associated with a shorter overall survival in the patients with ER+ tumors that were treated with tamoxifen [Fig 1C; HR = 5.021 (2.434–10.36); Score (log rank) test = 23.55 on 1 df, P = 1.218 e‐06 (n = 210, number of events = 49)]. On the other hand, the overall survival was independent of ESRP1 expression in the patients treated with chemotherapy alone (Fig 1D; HR = 1.599 (0.6773–3.773); Score (log rank) test = 1.17 on 1 df, P = 0.28 (n = 129, number of events = 21)). The effect of ESRP1 levels in the patients treated with a combination of tamoxifen and chemotherapy was not significant (data not shown), probably due to the small numbers in the cohort.
Analysis of the Cancer Genome Atlas Breast Invasive Carcinoma (TCGA‐BRCA) 12 cohort also revealed that high ESRP1 expression was associated with a significantly shorter overall survival in ER+ breast cancer patients [Fig 1E; P = 0.00011 (n = 656, number of events = 62)], but not in ER− cases [Fig 1F; P = 0.19 (n = 100, number of events = 17)]. The limitation of the TCGA dataset was that the treatment status was not available for all cases. The expression of ESRP2 was not associated with the overall survival in either the ER+ or ER− datasets using the TCGA cohorts (Fig EV1C and D). Collectively, these results show that ESRP1 expression (but not ESRP2) is prognostically important in the ER+ subtype of breast cancer.
The association of high ESRP1 expression with poor prognosis in ER+ breast cancer was further confirmed by quantitative RT‐qPCR in a cohort of patients with a high Oncotype Dx recurrence score (HS) (P = 0.01034) (Fig EV1E). Oncotype Dx is a commercial assay developed using the National Surgical Adjuvant Breast and Bowel Project (NSABP) B14 and B20 clinical trials that assess the recurrence of ER+ breast cancers 22. A high recurrence score usually predicts resistance to endocrine therapies.
To investigate the role of ESRP1 in ER+ preclinical models with poor prognosis, we next determined ESRP1 expression at the mRNA and protein levels in acquired tamoxifen‐resistant LCC2 cells and fulvestrant‐resistant (and cross‐resistant to tamoxifen) LCC9 cells compared to therapy‐sensitive parental MCF‐7‐AZ control cells (Fig EV2A). ESRP1 levels were significantly higher in the LCC2 cells (P = 0.0001) and LCC9 cells (P = 0.0001) than in the parental MCF‐7‐AZ cells in both the mRNA (Fig EV2A) and protein analyses (Fig EV2B). In addition, another poor prognostic ER+ cell line (T‐47D) displayed high ESRP1 levels at both levels (P = 0.0001). Together, these results support the hypothesis that ESRP1 plays an important role in recurrent and endocrine therapy‐resistant ER+ breast cancer.
Figure EV2. ESRP1 expression in ER+ breast cancer cell lines at the mRNA and protein levels.
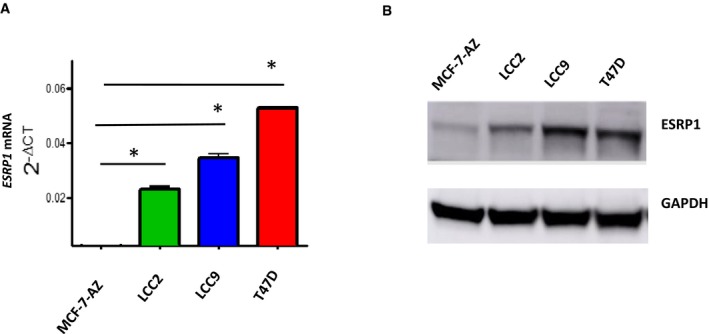
- RT–qPCR assay showing ESRP1 expression in endocrine‐resistant (MCF7/LCC2‐tamoxifen resistant, MCF7/LCC9‐fulvestrant and tamoxifen cross‐resistant) and T‐47D cells compared to parental MCF‐7‐AZ therapy‐sensitive cell lines. * represents P < 0.05; statistically significant. All RT–qPCR assays are performed in three independent biological replicates calculated using two‐way ANOVA (data (mean ± SD).
- Representative Western blot analysis of the indicated breast cancer cell lines (see details in Materials and Methods). All Western blot assays are performed in three independent biological replicates.
Source data are available online for this figure.
Knockdown of ESRP1 in ER+ breast cancer impacts cell and tumor growth in endocrine‐resistant breast cancer
To further understand the functional role of ESRP1 in recurrence/resistance to endocrine therapy, we first established stable ESRP1 knockdown in the LCC2 (tamoxifen‐resistant) and LCC9 (fulvestrant‐resistant) cell lines using a lentiviral shRNA approach. The knockdown resulted in a dramatic decrease in both the mRNA and protein levels in these cell lines (ESRP1 knockdown in LCC2 cells‐clone 2C1 and clone 2C3 cells, P < 0.05; ESRP1 knockdown in LCC9 cells‐clones 9C2, P = 0.0001 and clone 9C3 cells, P = 0.0021; Fig 2A and B) compared to their control counterparts (LCC2 empty vector‐2‐control and LCC9 empty vector‐9‐control). The clones with the highest knockdown (2C3 for LCC2; 9C2 for LCC9) were chosen to perform the functional and mechanistic studies. Knockdown was also performed in T‐47D cells, which have a higher level of ESRP1 compared to that of MCF‐7 cells. Overexpression of ESRP1 has been performed in MCF‐7 cells, which have lower levels of endogenous ESRP1. Appendix Fig S1A and B show the mRNA and protein expression levels for both models.
Figure 2. Knockdown of ESRP1 decreases malignant and tumor growth significantly in endocrine‐resistant ER+ breast cancer models.
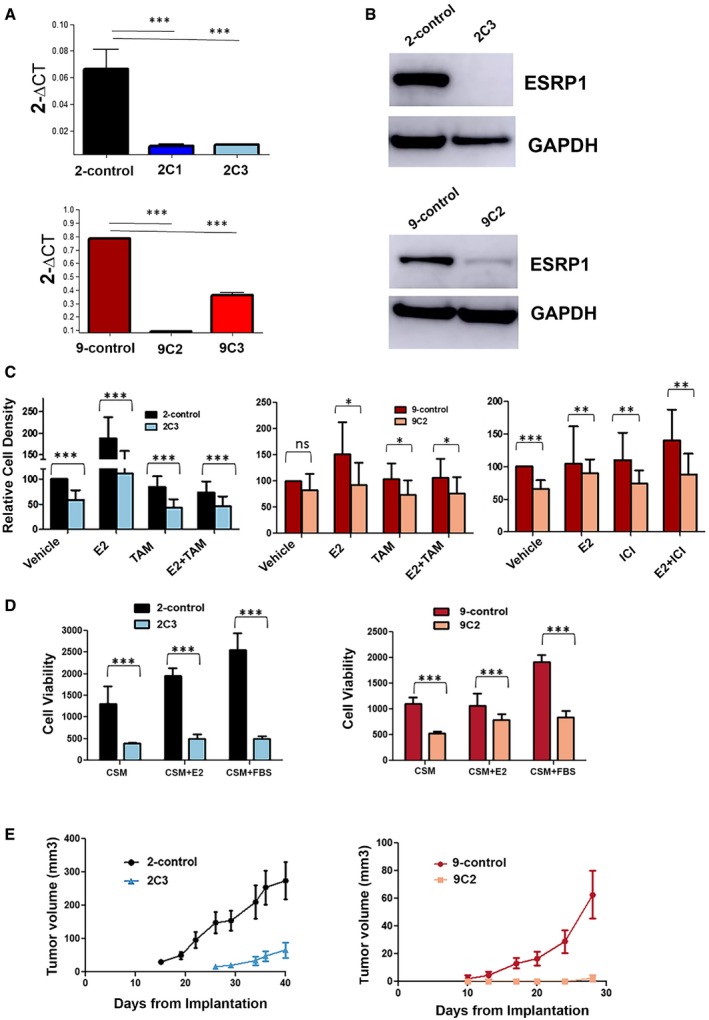
- RT–qPCR confirmed the stable knockdown of ESRP1 expression in endocrine‐resistant cells using a shRNA lentiviral system (Mission TRC human shRNA constructs‐Sigma); 2‐control (LCC2 cells transduced with pLKO.1 control vector) and 2C1 and 2C3 (LCC2 cells transduced with pLKO.1 shRNA ESRP1). Clones were selected for expression verification (clone 1 and clone 3); 9‐control (LCC9 cells transduced with pLKO.1 control vector), 9C2 and 9C3 (LCC9 cells transduced with pLKO.1 shRNA ESRP1). Clones were selected for expression verification (clone 2 and clone 3). * represents P < 0.05; statistically significant. Data (mean ± SD) were calculated using two‐way ANOVA based on three independent biological replicates.
- Western blot analysis using ESRP1 antibody GTX 131373 (GeneTex). GAPDH is used as the reference control.
- Relative cell density was determined by crystal violet assay. Cells were treated with TAM (4‐hydroxytamoxifen; 10−6 M) or ICI (ICI 182,780; 10−9 M) for 6 days in the presence and absence of E2 (β‐estradiol; 10−10 M). Data (mean ± SD) were calculated using two‐way ANOVA based on three independent biological replicates.
- Cell viability was determined using Cell Biolabs CytoSelect™ Cell Transformation Assay using 2‐control, 2C3, 9‐control, and 9C2 cells in CSM—charcoal‐stripped media, E2 (β‐estradiol; 10−10 M), and 10% fetal bovine serum. Data (mean ± SD) were calculated using two‐way ANOVA based on three independent biological replicates.
- Impact of ESRP1 knockdown on in vivo tumor growth. Five million cells (2‐control, 2C3, 9‐control, and 9C2) were implanted into mammary fat pads of athymic mice (five mice per group) in the presence of supplemental estrogen. Tumors were measured weekly using calipers for external measurements. Tumor volume was calculated as L × W2/2, where L is length and W is width (note the different scale for tumor volumes). Data (mean ± SD) were calculated using two‐way ANOVA (n = 5 mice per group).
Source data are available online for this figure.
To further analyze the impact of endocrine therapy on the ESRP1 knockdown cells, we determined the relative cell density in the presence and absence of β‐estradiol (E2), tamoxifen (TAM), and fulvestrant (ICI 182,780). These studies showed that ESRP1 knockdown in LCC2 alone resulted in a significant (P < 0.0001) reduction in the cell growth with a further decrease in the response to E2 and TAM or the combination of these two agents (Fig 2C). Similar results were observed in the LCC9 knockdown cells in response to fulvestrant (P < 0.0001) but to a lesser degree compared to those treated with tamoxifen (P < 0.01) (Fig 2C).
A significant reduction in the colony formation and cell growth was observed in soft agar at 8 days in both the knockdown models (Fig 2D). Despite the effect of ESRP1 knockdown on cell growth, no changes were observed in the cell cycle or apoptosis in vitro (Appendix Fig S2A and B). In vivo, knockdown of ESRP1 significantly suppressed the tumor growth in the mammary fat pad of orthotopic xenograft mice models of LCC2 and LCC9 (P < 0.05) (Fig 2E), confirming the role of ESRP1 in cell growth and proliferation in endocrine therapy‐resistant ER+ models.
ESRP1 knockdown does not induce an EMT phenotype in ER+ breast cancer
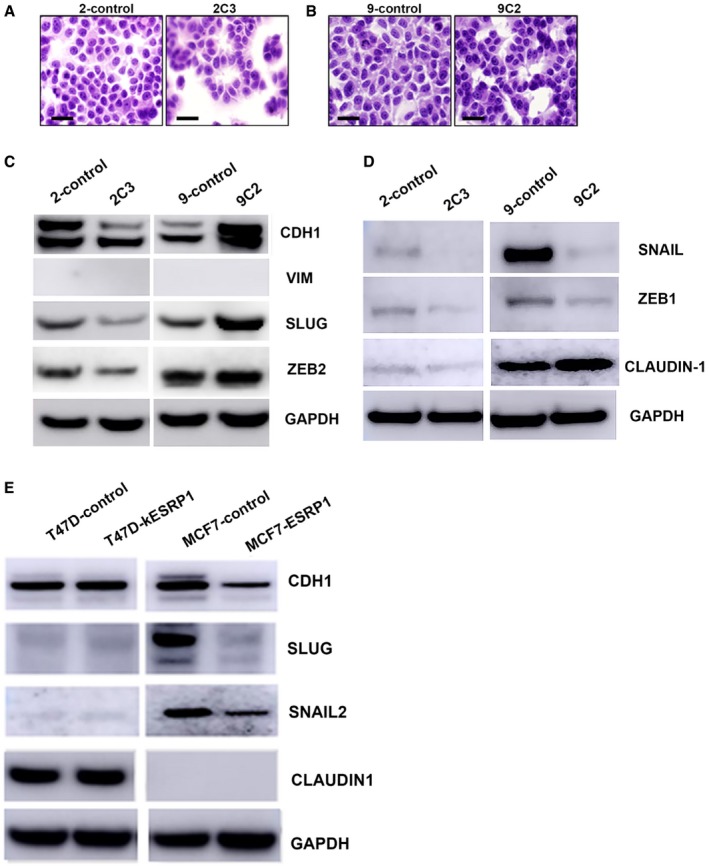
Lack of ESRP1 is known to induce EMT in epithelial cells. To better understand the impact of ESRP1 knockdown on EMT in ER+ breast cancer, we first assessed the morphology of the cells in both models. Knockdown of ESRP1 switched the cells to a more glandular (normal) pattern compared to that of the control cell lines. No mesenchymal patterns were observed in either model of ESRP1 knockdown (Fig 3A and B). We further analyzed the key EMT‐inducing transcription factors (EMT‐TFs) and proteins associated with the EMT process using Western blot analysis (Fig 3C). One of the hallmarks of EMT is the loss of E‐cadherin (encoded by CDH1) 23. E‐cadherin protein levels, representative of epithelialness, were not significantly altered in both the ESRP1 knockdown cells (Fig 3C and D). Vimentin was absent in these models. SLUG (SNAI2) and ZEB2 levels were down in the 2C3 cells but up in the 9C2 cells. SNAIL (SNAI1) and ZEB1, both inducers of EMT and repressors of CDH1, decreased in both the 2C3 and 9C2 knockdowns. Claudin‐1 levels, another regulator of EMT, remained unchanged in response to ESRP1 knockdown. ZO‐1 is not expressed in these models (data not shown). We also confirmed these results in another cell line knockdown model (T‐47D‐control and T‐47D‐kESRP1 knockdown) and in an ESRP1 overexpression model (MCF‐7‐control and MCF‐7‐ESRP1) (Fig 3E). These data show that the knockdown of ESRP1 does not induce key EMT players in ER+ breast cancer models. In addition, a change in in vitro invasive ability was not observed in either model in response to knockdown (Appendix Fig S3). This suggests that ESRP1's key role in recurrence/resistance in ER+ breast cancer may not be due to its impact on the EMT process.
Figure 3. Knockdown of ESRP1 does not induce EMT in models of resistance to endocrine therapy.
-
ACell morphology using high‐power magnification (40x) of the cultured cell lines under a microscope in LCC2 model‐2‐control and 2C3 cells. Scale bar: 50 μm.
-
BCell morphology using high‐power magnification (40×) of the cultured cell lines under a microscope in LCC9 model‐9‐control and 9C2 cells. Scale bar: 50 μm.
-
C, DProtein expression of key EMT‐TFs in ESRP1 knockdown cells (2C3 and 9C2) compared to endocrine‐resistant control cell lines (2‐control and 9‐control) using Western blotting analysis.
-
EProtein expression of key EMT‐TFs in ESRP1 knockdown cells (T‐47D‐kESRP1) and ESRP1‐overexpressing MCF‐7 cells (MCF‐7‐ESRP1) compared to their control counterparts (T‐47D‐control and MCF‐7‐control, respectively).
Source data are available online for this figure.
Preca et al 24 reported that ZEB1 overexpression in MCF10A downregulates ESRP1 and switches cells to CD44s, suggesting the importance of ZEB1 for the EMT phenotype. In addition, ER+ and luminal breast tumors mostly retain the CD44 variable exons 25. High expression of CD44s has also been shown to be essential for cells to undergo EMT 26. To further understand the impact of ESRP1 knockdown on the CD44 splice variants in our models, we assessed the CD44s versus CD44v switch in response to ESRP1 knockdown using RT–qPCR (Appendix Fig S4). We observed that the CD44s isoform is significantly dominant in the fulvestrant‐resistant knockdown (9C2) compared to CD44v2. However, this switch from CD44v2 to CD44s was not significant in the 2C3‐ESRP1 knockdown (tamoxifen‐resistant model). In 9C2‐ESRP1 knockdown cells, ZEB1 was not altered significantly compared to the levels in 9‐control cells. In the 2C3 and 2‐control cells, very low levels of ZEB1 were present and were not induced in response to ESRP1 knockdown. These data clearly show that ESRP1's role in our models is different from that outlined by Preca et al and is independent of EMT.
ESRP1 and epithelial splicing program
We next performed an Illumina paired‐end RNA sequencing (RNA‐Seq) of the 2C3 and 9C2‐ESRP1 shRNA knockdown cells and their control counterparts (2‐control and 9‐control). This revealed significant differences in the RNA profiles of these knockdown clones. The differentially regulated genes were determined for each set alone and the commonly overlapping sets based on a 2‐fold change and P < 0.05. Of the 1,178 significant genes, 484 were upregulated and 694 were downregulated in the 2C3 versus 2‐control cells. In the 9C2 versus 9‐control cells, 334 genes were significantly upregulated, while 255 were downregulated.
Prior studies in epithelial cells, including ER‐negative breast cancer cells, have reported that ESRP1 regulates EMT by promoting an epithelial splicing program 18, 19, 27. To explore the effect of knockdown on this program in ER+ breast cancer, we performed a multivariate analysis of transcript splicing (MATS) analysis 28, 29, 30 comparing the ESRP1 knockdown clones with corresponding control cells using Illumina paired‐end RNA‐Seq (Fig 4A). In the 2C3 knockdown versus 2‐control cells, we identified 535 differential ASEs in 432 genes (P < 0.05 and Δψ < 0.1; Fig 4A). The skipped exon (SE) events comprised the largest group (348 SE (65%) in 289 genes (66.9%)) and were followed by changes in 78 mutually exclusive exons (MXE—14.6%) in 36 genes (8.3%), 40 retained introns (RI—7.5%) in 40 genes (9.3%), 32 alternative 5′ splice sites (A5SS—6%) in 30 genes (6.9%), and 37 alternative 3′ splice sites (A3SS—6.9%) in 37 genes (8.6%). Similar data were obtained for the 9C2 knockdown versus 9‐control cells and resulted in the identification of 1,083 significant ASEs (Fig 4A). The 1,083 events (842 genes) were categorized as follows: 625 SE (57.7%) in 528 genes (62.7%), 216 MXE (19.9%) in 87 genes (10.3%), 91 RI (8.4%) in 86 genes (10.2%), 76 A5SS (7%) in 70 genes (8.3%), and 75 A3SS (6.9%) in 72 genes (8.4%).
Figure 4. A unique set of EMT genes are altered at the splicing level but not at the gene level.
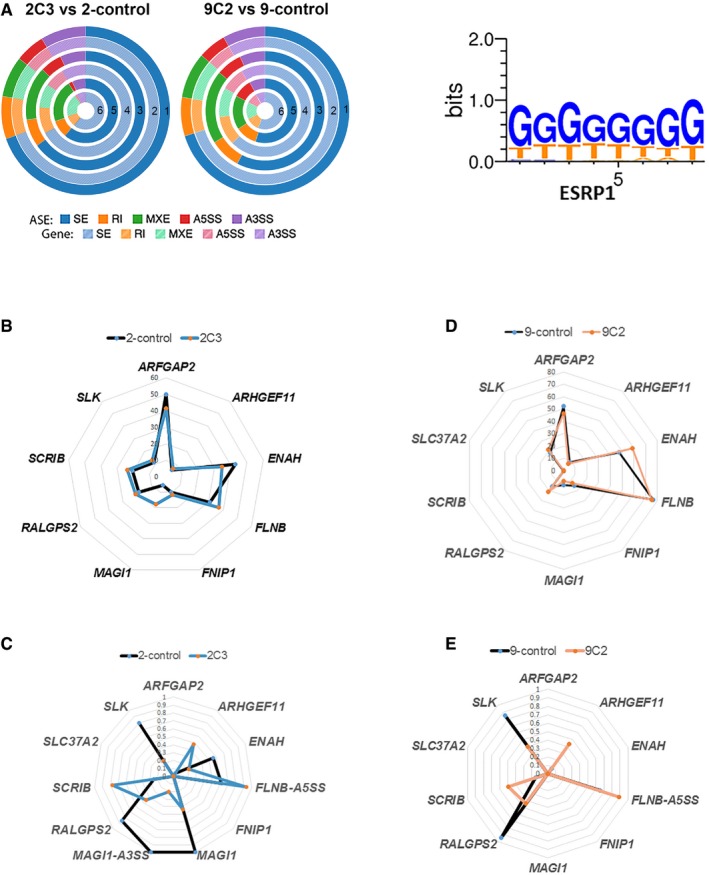
- Donut charts for RNA‐Seq analysis showing the differentially regulated alternative splicing events (ASEs) and their corresponding genes in ESRP1 knockdown cells compared to control cells; SE: skipped exon, RI: retained intron, MXE: mutually exclusive exons, A5SS: alternative 5′ splice site, and A3SS: alternative 3′ splice site. The “percent spliced in” (PSI or ψ) value was estimated. Differences in inclusion levels (∆ψ) between samples and their significance were calculated (P < 0.05 and IΔψI > 0.1). Circle 1: all ASEs; Circle 2: all genes corresponding to all ASEs; Circle 3: significant ASEs; Circle 4: significant genes corresponding to significantly altered ASEs; Circle 5: significant ASEs with ESRP1 motifs; Circle 6: significant genes corresponding to significantly altered ASEs with ESRP1 motifs. ESRP binding motifs were obtained from Dittmar et al 31 and analyzed based on the following assumption: The presence of the motif in the downstream region of an exon results in inclusion of the exon, while upstream or intraexonic location leads to exclusion of the exon.
- Radar plot showing that the EMT gene signature was not altered at the gene level in the ESRP1 knockdown model (LCC2 set).
- Radar plot showing that the EMT gene signature was altered at the ASEs level (SE) in the ESRP1 knockdown model (LCC2 set).
- Radar plot showing that the EMT gene signature was not altered at the gene level in the ESRP1 knockdown model (LCC9 set).
- Radar plot showing that the EMT gene signature was altered at the ASEs level (SE) in the ESRP1 knockdown model (LCC9 set).
Source data are available online for this figure.
We further analyzed the role of ESRP1 in alternative splicing by classifying the differential ASEs based on the ESRP motifs to determine direct control of the event by ESRP1. The ESRP binding motifs (Fig 4A) were obtained from Dittmar et al 31 and analyzed based on the following assumption: The presence of the motif in the downstream region of an exon results in the inclusion of the exon, while upstream or intraexonic locations lead to the exclusion of the exon. Among the significant events in the 2C3 knockdown versus 2‐control cells, 102 events (91 genes) harbored the predicted ESRP1 motifs consisting of 62 SE (61%) in 55 genes (60%), 22 MXE (22%) in 18 genes (20%), 9 RI (9%) in 9 genes (10%), 7 A3SS (7%) in 7 genes (8%), and 2 A5SS (2%) in 2 genes (2%). Of the 1,083 events in the 9C2 knockdown versus 9‐control cells, 241 (20.4%) in 221 genes (26.2%) had ESRP1 motifs consisting of 132 SE (54.8) in 124 genes (56.1%), 41 MXE (17%) in 32 genes (14.5%), 26 RI (10.8%) in 26 genes (11.8%), 23 A5SS (9.5%) in 21 genes (9.5%), and 19 A3SS (7.9%) in 18 genes (8.1%).
Warzecha et al 19 previously described a 10‐gene EMT splicing signature. We next analyzed the RNA‐Seq data to understand the impact of ESRP1 knockdown on this signature in these models. At the gene level, ESRP1 knockdown did not change the mRNA expression of these genes (Fig 4B and D). In contrast, the ASEs of EMT‐related exons in ARHGEF11 (SE), RALGPS2 (SE), SCRIB (SE), SLK (SE), and FLNB (A5SS) were present in both the LCC2 and LCC9 knockdown models, as indicated by the change in the splice index (Fig 4C and E). Furthermore, ENAH (SE) and MAGI1 (SE and A3SS) were identified only in the LCC2 model. The other three ASEs (FNIP1, ARFGAP2, and SLC37A2) were not significant in either of the cells.
To validate the RNA‐Seq analysis, we employed an alternative platform (Human Transcriptome Array 2.0 (HTA 2.0), Applied Biosystems/Thermo Fisher Scientific, Santa Clara, CA) that identifies ASEs using 10 probes per exon and 4 probes per exon–exon junction. This analysis using Transcriptome Analysis Console (TAC) 3.0 validated seven of the 10 EMT ASE patterns in both cell lines (Fig 5). In addition, the patterns for FLNB were confirmed in LCC2, but not in the LCC9 model.
Figure 5. Validation of the ASEs events of 10 EMT genes in response to ESRP1 knockdown using HTA 2.0 Splicing viewer (Transcriptome Analysis Console (TAC) Software‐Applied Biosystems/Thermo Fisher Scientific).
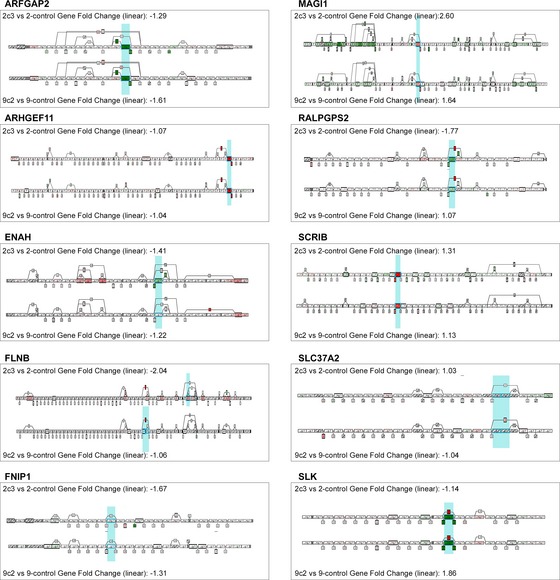
All Probe Selection Regions (PSRs) and Junctions are represented in the structural view with boxes that have the same size. An inclusion junction detects two neighboring PSRs. The PSRs detected by an inclusion junction are linked and graphically represented as dotted lines. An exclusion junction detects PSRs that are apart from each other. The PSRs detected by an exclusion junction are linked and graphically represented as dotted lines. Altered ASE for each corresponding gene has been highlighted as light blue representing the altered PSR. Red color indicates the inclusion of an exon, whereas green color represents the skipping of an exon. The blue column indicates the alternative splicing event at the listed location.Source data are available online for this figure.
EMT alternative splicing program in human ESRP1high ER+ breast cancers
Cell line models can be poor representations of cancer cell behavior in patients. To better understand the importance of splicing events, we analyzed them in the TCGA Breast Invasive Carcinoma (BRCA) dataset. TCGA SpliceSeq is a resource for the investigation of cross‐tumor and tumor‐normal alterations in the mRNA splicing patterns of TCGA RNA‐Seq data 32. The survival and splicing patterns in the cases (100 each) with the highest and lowest ESRP1 expression were analyzed. Accordingly, the exon skipping (SE) events comprised the largest group (290 SE corresponding 279 genes) shown in Fig 6A. This pattern was followed by alternative first exon (173 AP corresponding 173 genes), alternative last exon (89 AT corresponding 89 genes), retained intron (5 RI corresponding to 5 genes), alternative 5′ donor (33 AD corresponding to 33 genes), alternative 3′ acceptor (28 AA corresponding 28 genes), and mutually exclusive exon (5 ME corresponding to 5 genes) events. In the ESRP1_TCGA SpliceSeq BRCA analysis of the EMT signature, ASEs were also identified in five of the 10 genes (ARHGEF11, ENAH, FNIP1, SCRIB, and SLK; Fig 6B), indicating that the patterns of ASEs in the LCC2 and LCC9 models are identical to those described in the EMT gene signature (Table 1). These results validate that ESRP1 regulates the EMT‐splicing program in ER+ breast cancer. However, the EMT‐splicing program is not sufficient to induce an EMT phenotype in this breast cancer subtype.
Figure 6. TCGA SpliceSeq analysis using BRCA dataset in ESRP1 high and ESRP1 low tumors.
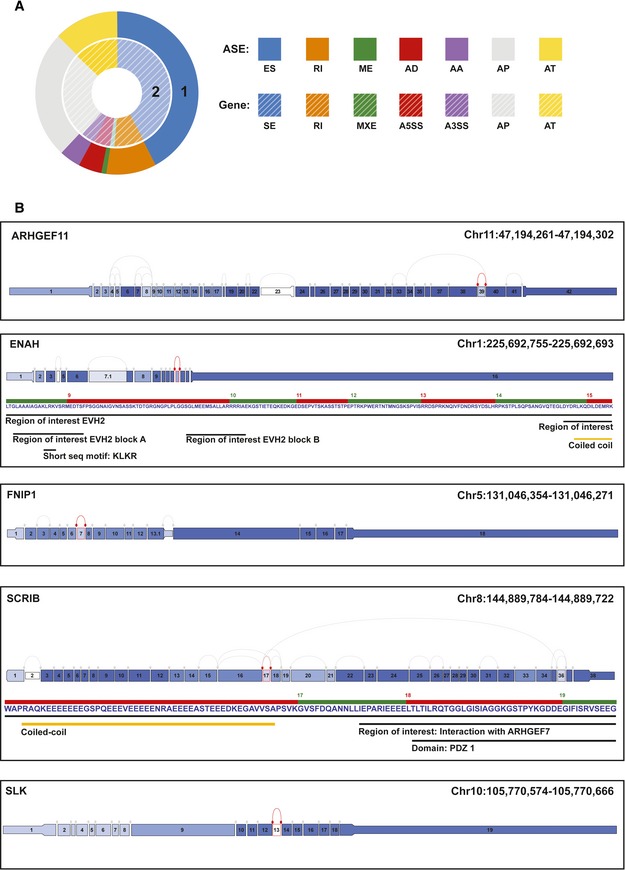
- The data were analyzed by categorizing the splice events into seven types: exon skip (ES), alternative 5′ donor (AD), alternative 3′ acceptor (AA), retained intron (RI), mutually exclusive exons (ME), alternative first exon (AP), and alternative last exon (AT). The analysis of splice events was performed using the following filter criteria: Min Gene RPKM ≥ 2, |dPSI| ≥ 0.1, P ≤ 0.02, Min Group Obs % > 0.85; RPKM‐reads per kilobase of transcript per million aligned reads; and |dPSI|‐absolute changes in percent splicing (dPSI, ∆Ψ). The events are presented as differential expression of splice events between ESRP1low and ESRP1high cases.
- TCGA splice graphs for the EMT signature genes in ESRP1high versus ESRP1low tumors. A splice graph of the gene's exons is shaded based on the expression level and shows the selected splice event outlined in red.
Source data are available online for this figure.
Table 1.
Alternative splicing events of the EMT 10‐gene splicing signature using three different platforms in ER+ ESRP1 low and high states
| Gene symbol | Exon location Warzecha et al 19 NCBI‐36‐hg18 | Exon location GRCh37 | Type of ASE in Warzecha et al in low ESRP1 | 2C3 versus 2‐control | 9C2 versus 9‐control | 2C3 versus 2‐control | 9C2 versus 9‐control | GA SpliceSeq (low versus high ESRP1) |
|---|---|---|---|---|---|---|---|---|
| RNA‐Seq | HTA | RNA‐Seq | ||||||
| ARFGAP2 | Chr11:47,150,836–47,150,877 | Chr11:47,194,261–47,194,302 | Skip | NS | NS | Skip | Skip | NS |
| ARHGEF11 | Chr1: 155,174,834–155,174,929 | Chr1;155,908,305–156,908,210 | Inc | Inc | Inc | Inc | Inc | Inc |
| ENAH | Chr1:223,759,316–223,759,378 | Chr1:225,692,755–225,692,693 | Skip | Skip | NS | Skip | NS | Skip |
| FLNB | Chr3:58,102,625–58,102,696 | Chr3:58,127,585–58,127,656 | Skip | NS | NS | NS | Skip | NS |
| FNIP1 | Chr5:131,074,170–131,074,253 | Chr5:131,046,354–131,046,271 | Skip | NS | NS | NS | NS | Skip |
| MAGI1 | Chr3:65,408,737–65,408,772 | Chr3:65,433,732–65,433,697 | Inc | Skip | NS | Inc | Inc | NS |
| RALGPS2 | Chr1:177,127,988–177,128,065 | Chr1:178,861,365–178,861,442 | Skip | Skip | Skip | Skip | Skip | NS |
| SCRIB | Chr8:144,961,710–144,961,772 | Chr8:144,889,784–144,889,722 | Inc | Inc | Inc | Inc | Inc | Inc |
| SLC37A2 | Chr11:124,461,310–124,461,366 | Chr11:124,956,100–124,956,156 | Skip | NS | NS | NS | NS | NS |
| SLK | Chr10:105,760,564–105,760,656 | Chr10:105,770,574–105,770,666 | Skip | Skip | Skip | Skip | Skip | Skip |
Novel functional role of ESRP1 knockdown on endocrine‐resistant breast cancer: impact of ESRP1 on metabolic pathways
To further identify and validate the functional role of ESRP1 knockdown on endocrine‐resistant breast cancer, we next analyzed the differentially expressed genes in the 2C3 and 9C2 knockdown models using the HTA 2.0 platform. In LCC2 versus 2C3 ESRP1 knockdown, the expression of 1,186 genes (1,263 transcripts) was significantly altered, while 413 genes (432 transcripts) were significantly regulated in the LCC9 versus 9C2 ESRP1 knockdown with an FDR < 0.1. Volcano plots representing the distribution of the fold changes (−2 > FC > 2, FDR < 0.1) of these genes are shown in Fig 7A. Of these significant genes, 34 downregulated and 68 upregulated (102 genes total) were shared by both the 2C3 and 9C2 ESRP1 knockdowns (Fig 7B). Using the DAVID Functional Annotation Clustering Tool, we identified the biological processes that were significantly altered in response to ESRP1 knockdown. The most significant annotation clusters downregulated by ESRP1 knockdown consisted of fatty acid metabolism/lipid metabolism (SCD, ACACA, FASN, ACAT2, PLCH1, and HPGD) and oxidoreductase processes (SCD, PHGDH, FASN, DHTKD1, and HPGD). Other clusters, such as the Ubl conjugation pathway (NEDD4, FBXO27, UBE2S, and UBE2T) and extracellular exosome cluster (genes including MUC1, TEX14, CD44 NEDD4, and ACACA), remained significant. In contrast, glycosylation processes (including B4GALT1, CRISP3, ATP1B1, PXDN, and MPZL) and some cytokine activity (TNFRSF11B, GDF15, BMP7, and TIMP1) were upregulated in ESRP1 knockdown cells versus control resistant cells. Surprisingly, no major EMT‐ or invasion‐related genes were significantly altered at the gene level. Taken together, we have demonstrated a novel functional impact of ESRP1 on the regulation of tumor growth at the functional and molecular levels independent of EMT.
Figure 7. Impact of ESRP1 knockdown on endocrine‐resistant breast cancer.
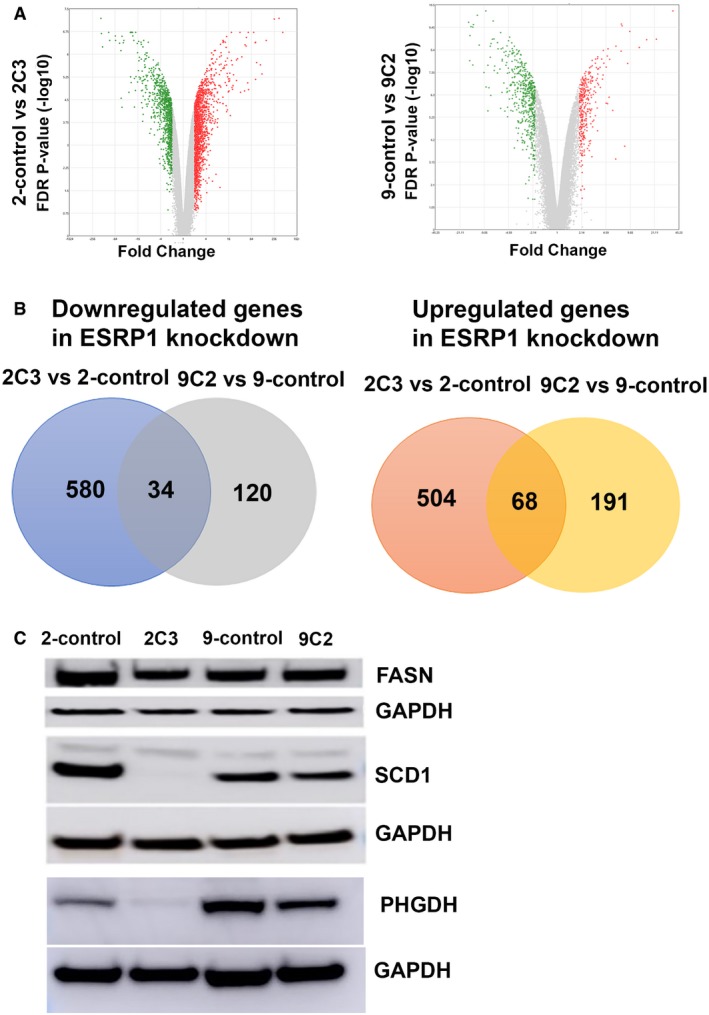
- Volcano plots showing the distribution of significant gene expression changes using the HTA 2.0 platform.
- Differential gene expression analysis of ESRP1 knockdown compared to resistant control cells using HTA 2.0 platform; downregulated genes in ESRP1 knockdown (left panel) and upregulated genes in ESRP1 knockdown (right panel) common in 2‐control versus 2C3, and 9‐control versus 9C2.
- Validation of the protein levels of FASN, SCD1, and PHGDH protein levels in ESRP1 knockdown versus control resistant cell lines using Western blot analysis. GAPDH was used as the loading control. The data are representative of three individual biological replicates.
Source data are available online for this figure.
We next compared the expression levels of key fatty acid metabolism/lipid metabolism and oxidoreductase processes at the gene, alternative splicing, and protein level. FASN, SCD, and PHGDH were altered at the alternative splicing and protein levels in the ESRP1 knockdown cells (Table 2 and Appendix Figs S5 and S6). The same exon inclusions based on the splicing index levels were significant in both the tamoxifen‐ and fulvestrant‐resistant models (see locations in Table 2). At the protein level, decreased expression of FASN and SCD1 in knockdown cells was observed, particularly in the tamoxifen‐resistant (2C3 versus 2‐control) model (Fig 7C). These results may be specific to acquired tamoxifen‐resistant cells, as FASN and SCD1 protein levels remained the same in response to ESRP1 knockdown in the T‐47D breast cancer cell line (Appendix Fig S1A). On the other hand, PHGDH levels decreased in both MCF‐7‐derived tamoxifen‐ and fulvestrant‐resistant cells as well as in T‐47D cells. The results from the overexpression of ESRP1 in the MCF‐7 model suggested that this by itself may not be sufficient to result in the altered expression of these metabolic genes.
Table 2.
Alternative splicing events of the genes using HTA validation in ER+ ESRP1 low and high states
| Gene symbol | 2C3 versus 2‐control | 9C2 versus 9‐control | Exon locationa GRCh37 | 2C3 versus 2‐control | 9C2 versus 9‐control |
|---|---|---|---|---|---|
| Gene level | ASEs | ||||
| FASN | Down | Down | Chr17:80,055,997–80,056,106 | Inc | Inc |
| SCD | Down | Down | Chr10;102,106,772–102,107,288 | Inc | Inc |
| PHGDH | Down | Down | Chr1:120,202,421–120,202,536 | Inc | Inc |
| CD44 | Down | Down | Multiple exon locations | Skip | Skip |
| CTNND1 (p120) | NS | NS | Multiple exon locations | Inc | Inc |
| FGFR1 | NS | NS | Multiple exon locations | Skip | No |
| FGFR2 | Up | NS | Chr10:123,298,106–123,239,535 | Inc | No event |
See Appendix Figs S5 and S6 for Alternative Splicing Events (ASEs) details specific to each gene.
To confirm the functional importance of ESRP1 in the regulation of cellular metabolism, we performed further experiments that analyzed the metabolic substrate flux in response to ESRP1 knockdown in resistant cells. The methods used to assess the glycolysis rate and the oxidation of major fuel substrates, glucose, glutamine, and fatty acids have been previously described 33. Using the XF Extracellular Flux analyzer, we measured the two major energy‐producing pathways of cell‐mitochondrial respiration through the oxygen consumption rate (OCR) and the extracellular acidification rate (ECAR), indicative of glycolysis, of the ESRP1 knockdown cells compared to their control resistant cells in real time. These analyses demonstrated that ESRP1 knockdown did not significantly alter the glycolysis rate (ECAR) in either the tamoxifen‐resistant or fulvestrant‐resistant models (Fig EV3A–D).
Figure EV3. Agilent Seahorse XFp Cell Energy Phenotype Assays in ESRP1 knockdown cells compared to their control resistant cell lines using Agilent Seahorse Cell XF Technology.
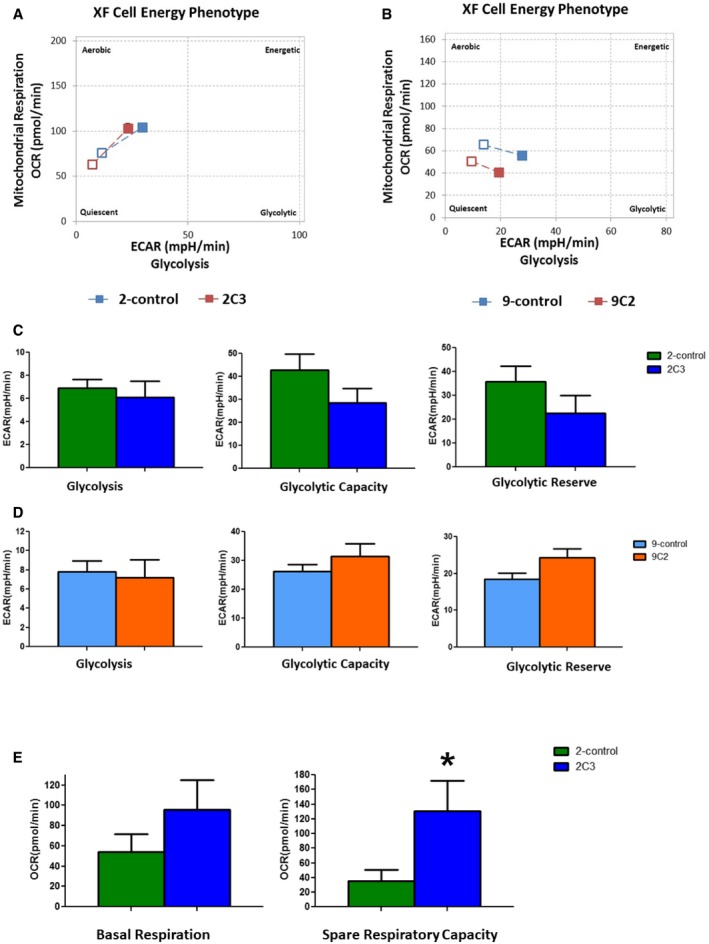
- Agilent Seahorse XF Cell Energy Phenotype Profile. The relative utilization of the two energy pathways of control and ESRP1 knockdown cells is determined under both baseline (Baseline Phenotype‐open squares) and stressed (Stressed Phenotype‐full squares) conditions—2‐control (blue squares) versus 2C3 (red squares).
- Agilent Seahorse XF Cell Energy Phenotype Profile. The relative utilization of the two energy pathways of control and ESRP1 knockdown cells is determined under both baseline (Baseline Phenotype‐open squares) and stressed (Stressed Phenotype‐full squares) conditions—9‐control (blue squares) versus 9C2 (red squares).
- Agilent Seahorse XF Glycolysis Stress representing extracellular acidification rate (ECAR) components‐glycolysis, glycolytic capacity, and glycolytic reserve—2‐control (green columns) and ‐2C3 ESRP1 knockdown (blue columns). Data (mean ± SD) are calculated using two‐way ANOVA based on the three independent biological replicates.
- Agilent Seahorse XF Glycolysis Stress representing extracellular acidification rate (ECAR) components—glycolysis, glycolytic capacity, and glycolytic reserve—9‐control (light blue columns) and 9C2 ESRP1 knockdown (orange columns). Data (mean ± SD) are calculated using two‐way ANOVA based on the three independent biological replicates.
- Mitochondrial respiration through oxygen consumption rate (OCR) components—basal respiration and spare respiration capacity. The data are representative of three independent sets. * represents P < 0.05; statistically significant. Data (mean ± SD) are calculated using two‐way ANOVA based on the three independent assays.
To compare the oxygen consumption rate (OCR) between the control and ESRP1 knockdown cells, we measured baseline respiration and spare respiration capacity, which is defined as the difference between the basal and maximum respiration for energy production through oxidative phosphorylation. ESRP1 knockdown significantly increased the basal respiration and spare respiration capacity in the tamoxifen‐resistant cells (P < 0.0001, Mann–Whitney U‐test) but not in the fulvestrant‐resistant cells (Fig EV3E). Furthermore, we showed a significant decrease in glucose uptake in the ESRP1 knockdown tamoxifen‐resistant cells and T‐47D ESRP1 knockdown cells (P < 0.05 for both cell lines; Student's t‐test). We did not observe any differences in the L‐lactate levels (Appendix Fig S7A and B).
Taken together, these results suggest that ESRP1‐mediated regulation of metabolic pathways might be more important for tamoxifen resistance than fulvestrant resistance. Further analysis will determine how ESRP1 controls the metabolic and OXPHOS pathways in tamoxifen resistance. Targeting these genes/pathways could provide novel therapeutic approaches for the control of recurrence/resistance to tamoxifen therapy in ER+ breast cancer.
Discussion
Accumulating evidence suggests the key role of splicing factors in cancer, including breast cancer 16, 34. Splicing factors, RNA‐binding proteins that interact with specific RNA sequences or motifs, may act as master regulators of gene expression and cellular behavior. Deregulation of their expression affects gene expression and alternative splicing of genes that contribute to breast cancer development and progression. In this study, we demonstrate for the first time that elevated levels of the splicing factor ESRP1 (epithelial splicing regulatory protein 1) are associated with poor outcomes in ER+ breast cancer using microarray‐based BreastMark and RNA‐Seq‐based TCGA datasets. These results are in contrast to a prior report from Lu et al, 27 who analyzed all (subtypes) breast cancers from the TCGA (n = 877). The larger number of cases in the provisional TCGA and subtype‐specific analysis could explain the observed differences in breast cancer. This led us to analyze an independent cohort of ER+ cancers with Oncotype Dx scores 22, an established surrogate of chemo‐ and endocrine resistance in breast cancer and a predictor for low and high recurrence cases. The correlation of high ESRP1 expression with poor prognosis (high recurrence score) was validated by RT–qPCR. Although ESRP2 (epithelial splicing regulatory protein 2), an associated protein with ESRP1, was also elevated in high recurrence cases, its expression did not correlate with outcomes in either the BreastMark or TCGA datasets. This led us to the conclusion that ESRP1, but not ESRP2, is an important player in the recurrence of ER+ breast cancer.
The biological relevance of ESRP1 in ER+ breast cancer was confirmed using ESRP1 knockdown in established resistant cell lines to endocrine therapy, representing models of poor prognosis. This analysis showed that the loss of ESRP1 in knockdown cells not only decreased the colony formation in vitro but also decreased the tumor growth in the xenograft models, highlighting the importance of ESRP1 in controlling cancer growth. Low ESRP1 expression has been associated with the development of EMT in ER‐negative breast cancer model (MDA‐MB‐231 cells) 17, 18, 19. In our study, we did not observe the development of a mesenchymal phenotype in response to knockdown of ESRP1. In contrast, glandular differentiation (more epithelial than parental cells) was maintained in the therapy‐resistant ER+ breast cancer cells. To confirm this rather surprising finding, we assessed the classical EMT proteins and the key EMT‐TFs in knockdown cells. Protein level data confirmed the absence of an EMT phenotype in the ESRP1 knockdown cells. Together, these data suggest that the induction of EMT by ESRP1 is contextual and may require the presence of appropriate background conditions.
ESRPs (ESRP1 and ESRP2) are major regulators of the EMT‐splicing program 17, 18, 19. Our analysis of the ESRP1 splicing program in these knockdown models confirmed that the alternative splicing patterns of most of the EMT‐splicing genes were consistent with prior publications. The TCGA Splice‐Seq in cases with low and high ESRP1 (n = 100 each) confirmed that some of the ASEs observed in the knockdown cells were identical to those observed in the human ER+ breast tumors with low ESRP1 levels, validating the active role of ESRP1 as a splicing factor in ER+ tumors. However, the lack of overt EMT at the cellular, functional, or pathway levels suggests the following: (i) In the absence of the appropriate settings, the ESRP1 splicing program may not be sufficient to cause morphological EMT; (ii) in contrast to the MDA‐MB‐231 model, ESRP1 knockdown in ER+ breast cancer promotes a differentiation effect; and (iii) high ESRP1 and epithelialness promotes cell growth and invasiveness. These findings are consistent with the dual role of splicing factors such as ESRP1 based on the tissue and cancer type 35. The results of our study, based on the analysis of human breast tumors, suggest that ESRP1‐driven aggressiveness in ER+ breast cancer is independent of EMT. This is consistent with data from Taube et al 36, who have documented that the core EMT gene signature is not associated with prognosis in breast cancer.
Furthermore, we identified a novel role for ESRP1 in ER+ breast cancer cells that impacts tumor progression through the regulation of genes involved in fatty acid/lipid metabolism and oxidation–reduction processes. In particular, knockdown of ESRP1 decreased the expression of key genes in these metabolic pathways. Dysregulation of cellular metabolism has been included as one of the emerging hallmarks of cancer 23. Altered metabolism can play a critical role in cancer progression. During oncogenesis, the metabolic programming of cancer is complex and requires multiple networks. The key element of this process is the switch from oxidative metabolism to glycolytic metabolism and the driving force of cancer proliferation and survival. In this study, we provide the first experimental evidence regarding the splicing factor ESRP1 contributing to the dysregulation of cellular metabolism.
Fatty acid synthase (FASN) and stearoyl‐CoA desaturase 1 (SCD1) are key proteins involved in the endogenous synthesis of fatty acids and function as important cofactors in various biological processes. Overexpression of FASN in breast cancer has been associated with significantly shorter disease‐free periods and overall survival 37, 38, 39, 40. Furthermore, FASN inhibition decreased cell proliferation and cell viability by promoting apoptosis in hormone‐dependent breast cancer cells 41, 42. In addition, SCD1 catalyzes the conversion of saturated fatty acids into monounsaturated fatty acids. The increased expression of SCD1 plays a crucial role in mediating glucose versus fatty acid metabolism in the development and progression of obesity 43. High SCD1 expression is also associated with shorter survival in ER+ and HER2+ breast cancer, but not in ER− breast cancers 44. We further confirmed the significant decrease in FASN and SCD1 protein expression in tamoxifen‐resistant ESRP1 knockdown cells rather than fulvestrant‐resistant ESRP1 knockdown cells.
We also verified a decrease in phosphoglycerate dehydrogenase (PHGDH) protein levels in both tamoxifen‐ and fulvestrant‐resistant models. PHGDH catalyzes the first step in the serine biosynthesis pathway. Suppression of PHGDH in cell lines with elevated PHGDH expression results in a strong decrease in cell proliferation and a reduction in serine synthesis 45. Increased expression of PHGDH was associated with breast cancer subtypes, and ectopic expression of PHGDH in mammary epithelial cells disrupted acinar morphogenesis and induced other phenotypic alterations that may predispose cells to transformation 46. Decreased expression impaired proliferation in amplified cell lines. Indeed, Samanta et al 47 reported that PHGDH knockdown sensitized both ER+ and ER− breast cancer lines to chemotherapy, resulting in increased mitochondrial reactive oxygen species (ROS) and apoptosis and loss of chemotherapy‐induced breast cancer stem cells (BCSC) enrichment. They suggested a role for PHGDH in the formation of secondary (recurrent or metastatic) tumors, with potential implications for therapeutic targeting of advanced cancers.
ESRP1 knockdown did not significantly alter the glycolysis rate (ECAR) in either the tamoxifen‐resistant or fulvestrant‐resistant models. However, ESRP1 knockdown significantly increased the basal respiration and spare respiration capacity, particularly in the tamoxifen‐resistant cells. These promising results warrant further investigation into the complex nature of ESRP1 knockdown in metabolic pathways and oxidative phosphorylation in tamoxifen resistance.
We next validated the changes in metabolic genes that were observed in the RNA‐Seq data at both the mRNA (probed‐based method HTA analysis) and protein levels (Western blot analysis) in response to ESRP1 knockdown, particularly in the tamoxifen‐resistant cells. Some of the variability between the RNA‐Seq and HTA analysis may be due to the differences between the two platforms. The depth of the RNA‐Seq (30 million) may not have been enough to obtain the same results. Indeed, Nazarov et al 48 reported that the stochastic variability was higher for the sequencing data than for microarray data due to the lack of reads for the short and low‐abundance genes. This usually reduces the number of differentially expressed genes and genes with predictive potential for RNA‐Seq compared to microarray data. HTA 2.0 is a probe‐based technology (10 probes per exon and 4 probes per exon‐exon splice junction) and is independent of the depth bias of RNA‐Seq.
In conclusion, this study documents novel roles of ESRP1 in modifying the behavior of ER+ cells. Although we could confirm its role in the alternative splicing of EMT‐related genes, this did not result in mesenchymal transformation. Knockdown was associated with epithelial differentiation and decreased growth by alternative mechanisms, such as changes in fatty acid and lipid metabolism. These data could form the basis of new avenues for the control of recurrence/resistance to therapies in ER+ breast cancer.
Materials and Methods
Analysis of publicly available databases
The prognostic value of ESRP1 and ESRP2 was evaluated using BreastMark, a tool for examining putative gene prognostic markers in breast cancer 21. Its algorithm integrates the gene expression and survival data from 26 datasets from 12 different microarray platforms corresponding to approximately 17,000 genes in up to 4,738 samples. The software allows different survival end points to be analyzed separately. Median expression was used to dichotomize the data, allowing stratification into high and low groups within each of the 26 individual datasets. The interface is available on a publicly accessible web server (BreastMark: Breast Cancer Survival Analysis Tool [http://glados.ucd.ie/BreastMark/index.html]). The software uses CGI to link the web server with the R/perl‐based algorithm. All calculations are carried out in real time.
Analysis of the Cancer Genome Atlas (TCGA)
Patients with breast cancer were categorized based on their ER status (n = 924; 656 ER−positive). The clinical information for each patient was also obtained. The normalized expression of ESRP1 and ESRP2 (Level 3 data) was analyzed in 924 breast cancer patients enrolled in The Cancer Genome Atlas (TCGA) database Breast Invasive Carcinoma (BRCA) (https://portal.gdc.cancer.gov/projects/TCGA-BRCA) study with subtype classification. To model survival, gene expression at or below median was considered low and above median was considered high. Overall survival was calculated from the date of initial diagnosis to disease‐specific deaths (patients whose vital status is termed dead) and months to last follow‐up (patients who are alive). The “survival” package in R (R Foundation for Statistical Computing) was used for statistical analyses (log rank test) and to generate Kaplan–Meier curves.
Oncotype DX samples
All protocols were reviewed and approved by the Institutional Review Board (IRB) of Indiana University. Samples and clinical records were anonymized prior to access by the authors and linked with a numerical identifier. The requirement for informed consent was waived by the IRB. Fifty‐nine archival formalin‐fixed, paraffin‐embedded (FFPE) tumor blocks were obtained from patients with ER‐positive node‐negative breast carcinomas at the Indiana University Simon Cancer Center based on their Oncotype DX RS (19 LS, 20 IS, and 20 HS). Demographic and clinical characteristics of the patients were acquired from medical charts as described previously 15.
Breast cancer cell lines
MCF‐7‐AZ control (endocrine therapy‐sensitive), MCF‐7‐LCC2 (LCC2; tamoxifen‐resistant), and MCF‐7‐LCC9 (LCC9; fulvestrant (ICI 182,780) and tamoxifen cross‐resistant) cell lines were kind gifts from Dr. R. Clarke (Georgetown University Medical School, Washington DC) 49, 50. MCF‐7 and T‐47D cell lines were purchased from American Type Culture Collection (ATCC, Manassas, VA). Cell lines were carefully maintained in a humidified tissue culture incubator at 37°C in a 5% CO2:95% air atmosphere, and stocks of the earliest passage cells were stored. The cell lines were grown in phenol red‐free DMEM containing 5% charcoal‐stripped fetal calf serum (CCS) and 100 mg/ml penicillin at least 4 days before the experiments were performed.
RNA isolation and sample quality assessment
Total RNA was extracted from 10‐μm‐thick sections of archival paraffin blocks using the RecoverAll™Total Nucleic Acid Isolation Kit (Thermo Fisher Scientific, Wilmington, DE) as described previously 15. For breast cancer cell lines, the RNeasy isolation kit was used according to the manufacturer's instructions (Qiagen, Germantown, MD). RNA was treated with Turbo DNase (Thermo Fisher Scientific, Wilmington, DE). The quality of RNA was assessed using the Nanodrop® ND‐1000 spectrophotometer (Thermo Fisher Scientific) and the Agilent 2100 Bioanalyzer (Agilent Technologies, Santa Clara, CA).
Quantitative reverse transcription–polymerase chain reaction (RT–qPCR) of breast cancer cell lines and FFPE samples
Total RNA was reverse‐transcribed using the High Capacity cDNA Reverse Transcription kit (Thermo Fisher Scientific) according the manufacturer's instructions. The mRNA levels of ESRP1 (Hs00936420_m1) were analyzed by real‐time RT–qPCR using TaqMan gene expression assays on an ABI Prism 7900 platform according to the manufacturer's instructions (Applied Biosystems/Thermo Fisher Scientific). Actin (ACTB; Hs00357333_g1) and GUSB (Hs99999908_m1) were used as endogenous controls for normalization purposes. All RT–qPCR mixtures from tumor blocks and breast cancer cell lines were performed in duplicate and triplicate, respectively. For breast cancer cell lines, all experiments were the average of three independent sets. The relative quantification of the gene expression changes was analyzed according to the DDCt method using the Applied Biosystems DataAssistTM Software v3.0. All graphs were generated using GraphPad Prism 5 software. The error bars were calculated and represented in terms of the mean ± SD.
ESRP1 knockdown in endocrine‐resistant cell lines
To stably knockdown ESRP1 expression in the LCC2, LCC9, and T‐47D cells, we performed lentiviral delivery of Mission TRC human shRNA constructs (pLKO.1 shRNA) according to the manufacturer's instructions (Sigma, St. Louis, MO). The following shRNA‐Mission plasmids were used: pLKO.1 control vector (SHC001) and shRNA ESRP1 (TRCN0000149820, TRCN0000146588). Briefly, 1 × 106 HEK 293 cells were transfected with 2.6 μg each of shRNA‐ ESRP1 and control shRNA plasmids and 10 μl packaging mix (Sigma, St. Luis, MO) using 16 μl FuGENE HD transfection reagent (Roche, Indianapolis, IN). After 48‐h incubation, the medium containing the newly formed viral particles was collected, filtered with millex‐HV 0.45‐μm filters (Millipore, Burlington, MA), and added to 1.6 ×104 LCC2, LCC9 or T‐47D cells together with 8 μg/ml polybrene solution (Sigma). After 24 h of infection, the cells were washed with PBS and MEM containing 2 μg/ml puromycin (Invitrogen). Infected cells were selected in Puromycin media and further passaged in culture to obtain stable clones that represent ESRP1 knockdown cells.
Overexpression of ESRP1 in MCF‐7 breast cancer cell line was performed using GenScript's GenEZ™ORF ESRP1 construct (ESRP1; NM_017697). GenScript's GenEZ™ORF ESRP1 was cloned into the mammalian expression cloning vector pcDNA3.1+/C‐(K)‐DYK using CloneEZ™cloning technology according to the manufacturer's instructions (GenScript Piscataway, NJ). Stable cells with the control vector (pcDNA3.1+/C‐(K)‐DYK vector only) or vector with ESRP1 construct were generated in MCF‐7 cells. Knockdown or overexpression of ESRP1 expression was verified using RT–qPCR and Western blot assays as described in the Materials and Methods.
Anchorage‐independent growth assay
Anchorage‐independent growth was determined using the soft agar colony formation (CytoSelect™ Cell Transformation Assay; Cell Biolabs Inc., San Diego, CA) according to the manufacturer's instructions. Briefly, cells were incubated 8 days in semisolid agar media before being solubilized, lysed, and detected by the CyQuantR® GR Dye in a fluorescence plate reader under charcoal‐stripped media (CSM) or in response to E2 (10−10 M, β‐estradiol) or 10% fetal bovine serum (FBS).
Crystal violet assay
Human breast cancer cells (1 × 103 cells/ml) in DMEM containing 5% charcoal‐stripped media were plated in 24‐well tissue culture plates. On day 1 after plating and every 3 days thereafter, cells were treated with E2 alone (10−10 M, Sigma, St. Louis, MO), TAM alone (4‐hydroxytamoxifen; 10−6 M, Sigma, St. Louis, MO), or in combination with E2 and TAM. The LCC9 set was also tested for ICI alone (ICI 182,780‐fulvestrant, 10−9 M, and in combination with E2 and ICI). On day 6, the media were aspirated, and the cells were stained with crystal violet (0.2% crystal violet staining solution in buffered formalin pH = 7.0). Cells were permeabilized using citrate buffer, and absorbance was read at 560 nm on a plate reader.
Morphology assessment
Cells were grown in 8‐well slide chambers (MatTek®) in CSS media for 4 days. The slides were then fixed in 60% ethanol and stained with H&E stains. The slides were examined under an Olympus BX41 microscope, and images were obtained using a DP‐72 camera and CellSens™software at 40× magnification.
Orthotopic xenograft models
Six‐ to eight‐week‐old female athymic nude mice (Harlan Sprague Dawley, Indianapolis, IN) were acclimatized for 3–7 days. All animals were housed in an SPF (specific pathogen‐free) facility at Indiana University. A controlled‐release E2 pellet (0.72 mg E2, 60‐day formulation; Innovative Research of America, Sarasota, FL) was injected subcutaneously (s.c.) via a sterile 14‐gauge trocar 24 h before tumor implantation. Five million cells for both control cells (2‐control and 9‐control) and ESRP1 knockdown cells (2C3 and 9C2) were implanted into mammary fat pads. Tumors were measured weekly using calipers for external measurements. Tumor volume was calculated as L × W2/2, where L is length and W is width. All animal experiments were performed under a protocol approved by the Indiana University Institutional Animal Care and Use Committee (IU IACUC). The investigators were blinded to group allocation during data collection and analysis.
RNA sequencing with rRNA‐depletion TruSeq RNA sample prep kit, library construction, cluster generation, and HiSeq 2500
Paired‐end RNA sequencing (RNA‐Seq) was performed using the Illumina HiSeq 2500 platform by SeqWright Genomic Services (Houston, TX) according to the manufacturer's instructions (Illumina, San Diego, CA). Briefly, the first step involved library preparation using the TruSeq Stranded Total RNA kit. Ribosomal RNAs (rRNAs) were depleted from total RNA using Ribo‐Zero Kits. After purification and RNA fragmentation, cleaved RNA fragments were copied into first strand cDNA using reverse transcriptase and random primers, followed by second‐strand cDNA synthesis using DNA Polymerase I and RNase H. A single “A” base was added to cDNA fragments with subsequent ligation of the adapter. The products were purified and enriched to create the final cDNA library. Using the TruSeq Paired‐End (PE) Cluster kit, library samples were amplified to create clonal clusters, and RNA deep sequencing (TruSeq SBS kit‐200 cycles) was performed on the HiSeq 2500 with the RNA isolated from each sample as described above.
Quality control of protocols and analysis tools using TruSeq RNA sequencing (Illumina‐Hi Seq Platform)
The quality control analysis of the sample library and quantification of the DNA library templates were validated according to the manufacturer's instructions. Briefly, the quality control of the library sample was checked for the size, purity, and concentration using the Agilent 2100 Bioanalyzer (Agilent Technologies, Santa Clara, CA). In addition, quantitative standards such as RNA spike‐ins were used to calibrate quantification, sensitivity, coverage, and linearity. Sequencing was performed using total RNAs to a depth of 30 million reads.
Computational methods for RNA‐Seq data analysis
Data from total RNA‐Seq were processed as described below. Raw FASTQ sequences were generated and demultiplexed using the Illumina CASAVA pipeline. The FastQC (http://www.bioinformatics.babraham.ac.uk/projects/fastqc) and FASTX toolkits (http://hannonlab.cshl.edu/fastx_toolkit) were used for a quality check to ensure that the FASTQ reads were in the entirely normal range and to preprocess the reads prior to mapping. Briefly, the FASTX clipper tool was employed to remove the Illumina 3 prime adaptor sequences. The postclipped reads were mapped to the human genome (hg19), and the RNA‐Seq‐based gene expression levels (FPKM; fragments per kilobase of exon per million fragments mapped) were subsequently calculated using the TopHat/Cufflinks 51, 52 framework to increase the mapping coverage and to generate the SAM/BAM files.
Identifying differential AS events
Differential alternative splicing events between the knockdown and control cell lines (2C3 versus 2‐control and 9C2 versus 9‐control) were identified using MATS.3.0.8 28, 29, 30, 53. Differential splicing events (defined as P < 0.05 and ∆ψ < 5%) were identified between the control and knockdown sets.
Identifying ESRP motifs
The splicing map of ESRPs is largely dependent on their binding to the proximal regions of the target exons, viz upstream, downstream, or exonic. The position weight matrices (PWM) of the ESRP binding motifs enriched in the 250 nt upstream, 250 nt downstream, and exonic regions of ESRP‐regulated exons were obtained from Warzecha et al and Dittmar et al 17, 18, 19, 31. These motifs were then screened in the upstream, downstream, and exonic regions of differentially present exons in the control versus knockdown cell lines using FIMO, a motif prediction tool available in the MEME suite 53. The motifs identified at P < 0.001 were considered to be significant.
Human Transcriptome Array 2.0 assay and validation
The RNAs from the control and knockdown cells were sent to the Applied Biosystems/Thermo Fisher Scientific Service laboratory (Santa Clara, CA). The Ambion® Whole‐Transcript (WT) Expression Kit and the GeneChip WT Terminal Labeling and Controls Reagent Kits were used to prepare the samples. GeneChip Human Transcriptome Array 2.0 was performed according to Applied Biosystems/Thermo Fisher Scientific’s instructions. Probe cell intensity (CEL) data generated from the Human Transcriptome Arrays were analyzed in Transcriptome Analysis Console (TAC) Software to obtain a list of the differentially expressed genes and alternative splicing events. TAC software also provided the visualization of genes, exons, junctions, and transcript isoforms. TAC Software was downloaded free from the Applied Biosystems/Thermo Fisher Scientific website. To ensure uniform coverage of the transcriptome, the GeneChip Human Transcriptome Array 2.0 was designed with approximately ten probes per exon and four probes per exon‐exon splice junction. The probes are all arranged into probe sets that translate and summarize the data into gene‐level, exon‐level, and splice‐junction probe sets.
TCGA SpliceSeq validation using BRCA dataset
We validated our predictions using the whole exome sequencing data of breast cancer patients deposited in The Cancer Genome Atlas Breast Invasive Carcinoma (TCGA‐BRCA). We identified 100 Luminal A samples with extremely low levels of ESRP1 (ESRP1 low) and 100 Luminal B samples with high levels of ESRP1 (ESRP1 high). The splicing pattern in these cases was analyzed in TCGA SpliceSeq using BRCA dataset (http://projects.insilico.us.com/TCGASpliceSeq/). This resource is designed for the investigation of cross‐tumor and tumor‐normal alterations in mRNA splicing patterns of TCGA‐BRCA RNA‐Seq data 32.
DAVID functional analysis
The functional enrichment analyses were performed using the DAVID functional annotation tool (http://david.abcc.ncifcrf.gov/). We identified differentially expressed genes in HTA between the LCC2 (2‐control versus 2C3) and LCC9 (9‐control versus 9C2) cells (FDR < 0.1) and looked for enrichment in the following annotation categories: cellular components (GOTERM_CC_FAT), molecular functions (GOTERM_MF_FAT) or biological processes (GOTERM_BP_FAT). The clusters are ordered by group enrichment score. Significant annotation groups were determined by group enrichment scores ≤ 0.05 (equivalent to 1.3 in minus log scale).
Agilent Seahorse XFp cell energy phenotype assays
The Seahorse XFp Cell Energy Phenotype Test kit is an assay that simultaneously measures the two major energy‐producing pathways in live cells, mitochondrial respiration, and glycolysis, allowing the rapid determination of energy phenotypes of cells and investigation of metabolic switching. Based on the results of the Seahorse XFp Cell Energy Phenotype Assay, either the Agilent Seahorse XF Cell Mito Stress test and/or Agilent Seahorse XF Glycolysis Stress are sequentially performed. The XF Cell Mito Stress test determines mitochondrial function by directly measuring the oxygen consumption rate (OCR) of cells. The Agilent Seahorse XF Glycolysis Stress test directly measures the acidification rate and reports this as ECAR. All OCR and ECAR assays were performed according to the instructions of Agilent Technologies (Santa Clara, CA). The results presented are the combination of three independent assays, and two‐way ANOVA analyses were performed using GraphPad software (P < 0.05, statistically significant).
Western blot analysis
The protein lysates were prepared, and equal amounts of protein were subjected to SDS–PAGE and Western blot analysis as described previously 54. The Bio‐Rad DC‐Protein assay kit (Bio‐Rad, Hercules, CA) was used to determine protein concentrations. Blots were incubated with antibodies against CDH1, VIM, SLUG, SNAIL, SNAIL2, CLAUDIN 1, ZEB1, and ZEB2 (Cell Signaling Technology, Danvers, MA), FASN (BD Biosciences, San Jose, CA), SCD1 (Alpha Diagnostic International Inc., San Antonio, TX), and PHGDH (Epigentek, Farmingdale, NY). Antibodies against GAPDH were used as the loading control (GeneTex, Inc., Irvine, CA). Protein bands were visualized by SuperSignal™West Pico PLUS Chemiluminescent Substrate kit (Amersham, Piscataway, NJ) and Amersham Imager 600 GE Healthcare Life Sciences (GE Healthcare Bio‐Sciences, Pittsburgh, PA). The data are representative of three individual assay sets.
Statistical analysis
In vitro experiments
Two‐way ANOVA tests were used for statistical analysis by GraphPad Prism 5.0 and Microsoft Excel. All results are representative of three independent biological replicates and expressed as mean values SD. In all cases, differences were considered to be statistically significant at P < 0.05.
Kaplan–Meier curves
BreastMark: Breast Cancer Survival Analysis Tool uses the software CGI (the web server with the R/perl‐based algorithm) to calculate the P‐values for the endpoint “overall survival” using log rank test. TCGA‐BRCA Kaplan–Meier curves: A log rank test was used to calculate P‐values for the endpoint “overall survival” using the “survival” package in R (R Foundation for Statistical Computing).
Cell cycle analysis
Cell cycle distribution was measured in LCC9 and LCC2 cells transduced with ESRP1 shRNA or nontargeting control shRNA. Cells were harvested at 24 and 48 h after transfection, suspended in PBS, and fixed in 70% ethanol. Then, the DNA content was evaluated after propidium iodide staining. Fluorescence‐activated cell‐sorting analysis was carried out using a FACScan flow cytometer (Beckton Dickinson) and CellQuest software.
Apoptosis assay
The PE Annexin V Apoptosis Detection Kit I (BD Biosciences) was used with flow cytometry according to the manufacturer's instructions. Briefly, ESRP1 shRNA and control shRNA cells (5 × 105 cells/ml) were harvested, washed in PBS, and pelleted by centrifugation. The cells were suspended in 1× binding buffer, and then, 5 μl Annexin V fluorescein isothiocyanate was added. After 15 min, 10 μl propidium iodide was added, and the suspension was incubated in the dark at room temperature for 15 min. Binding buffer (1×; 400 μl) was added to the suspension and gently vortexed. The cells were analyzed using a FACScan flow cytometer. The numbers show the percentages of cells in each quadrant (LL—lower left: intact cells; LR—lower right: early apoptotic cells; UL—upper left: necrotic cells; and UR—upper right: late apoptotic or necrotic cells).
Invasion assay
The Boyden in vitro invasion assay was performed as described previously 55. Briefly, cells were plated in phenol red free MEM with 5% charcoal‐stripped fetal calf serum for 48 h and switched to 1% serum media for 24 h. The cells were transferred to the upper chamber of the phenol red free Matrigel‐coated transwell plates and allowed to migrate toward 1% FBS, 10% FBS or E2 (17 beta‐estradiol; 10−10 M) in the lower chamber for 72 h. The cell invasion was evaluated after DiffQuick staining by counting the cells in four randomly chosen fields of filters. The results are representative of the three individual experiments performed in triplicate.
Quantitative reverse transcription–polymerase chain reaction (RT–qPCR) of CD44 splice variants
Total RNAs were reverse‐transcribed using the High Capacity cDNA Reverse Transcription kit (Thermo Fisher Scientific, Wilmington, DE) according to the manufacturer's instructions. The mRNA levels of CD44‐all transcript (HS99999195_m1), CD44s (Hs01081473_m1), and CD44v2‐v10 (Hs01075866_m1) were chosen based on the study by Olsson et al 25 and analyzed by real‐time RT–qPCR using TaqMan gene expression assays on an ABI Prism 7900 platform according to the manufacturer's instructions (Applied Biosystems/Thermo Fisher Scientific). Actin (ACTB; Hs00357333_g1) was used as an endogenous control for normalization purposes. All RT–qPCR mixtures from the indicated breast cancer cell lines were performed in triplicate (*see Materials and Methods for statistical analysis). For the breast cancer cell lines, all the experiments were the average of three independent sets. The relative quantification of the gene expression changes was analyzed according to the ΔΔC t method using the Applied Biosystems DataAssistTM Software v3.0. All graphs were generated using GraphPad Prism 5 software (two‐way ANOVA). The error bars were calculated and represented in terms of mean ± SD.
Glucose uptake and L‐lactate levels
The glucose uptake assay was performed using the Glucose Uptake Assay Kit (Abcam, #ab136955), and the lactate production assay was performed using the L‐Lactate Assay Kit (Abcam, #ab65331) according to the manufacturer's protocol. The results were normalized by the protein amounts in each assay.
Author contributions
YG‐P and SSB: Concept, design, interpretation, and execution of the entire project. YN, CPG, and SCJ: Bioinformatics analyses and interpretation. XG, GN, YG, and EV: Performed the cellular and molecular experiments. MR: Performed and interpreted the TCGA SpliceSeq analysis. YG‐P, YN, SCJ, MR, and SSB: manuscript writing. All authors approved the manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
Source Data for Expanded View and Appendix
Review Process File
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5
Source Data for Figure 6
Source Data for Figure 7
Acknowledgements
The authors would like to acknowledge the assistance of Chris Kubu, Ryan Estep, Michelle Staples, and Kristin Kelley from Applied Biosystems/Thermo Fisher Scientific for their support in the performance of HTA analysis. The animal studies and data analysis were performed by In Vivo Therapeutics Core (IVT) at Indiana University Simon Cancer Center (IUSCC). Sunil Badve is a Komen Scholar. This work was funded partly by the Susan G. Komen for the Cure® and 100 Voices of Hope to Sunil Badve.
EMBO Reports (2019) 20: e46078
Contributor Information
Yesim Gökmen‐Polar, Email: ypolar@iu.edu.
Sunil S Badve, Email: sbadve@iupui.edu.
Data availability
The RNA‐Seq data (.bam files) for LCC2 (2‐control and ESRP1 knockdown‐2C3 cell lines) and LCC9 (9‐control and ESRP1 knockdown‐9C2 cell lines) sets were deposited at GEO under accession GSE125355 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE125355).
References
- 1. Ali S, Coombes RC (2002) Endocrine‐responsive breast cancer and strategies for combating resistance. Nat Rev Cancer 2: 101–112 [DOI] [PubMed] [Google Scholar]
- 2. Deroo BJ, Korach KS (2006) Estrogen receptors and human disease. J Clin Invest 116: 561–570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Osborne CK, Schiff R (2011) Mechanisms of endocrine resistance in breast cancer. Annu Rev Med 62: 233–247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Sweeney EE, McDaniel RE, Maximov PY, Fan P, Jordan VC (2012) Models and mechanisms of acquired antihormone resistance in breast cancer: significant clinical progress despite limitations. Horm Mol Biol Clin Invest 9: 143–163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Nardone A, De Angelis C, Trivedi MV, Osborne CK, Schiff R (2015) The changing role of ER in endocrine resistance. Breast 24(Suppl 2): S60–S66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Jeselsohn R, De Angelis C, Brown M, Schiff R (2017) The evolving role of the estrogen receptor mutations in endocrine therapy‐resistant breast cancer. Curr Oncol Rep 19: 35 [DOI] [PubMed] [Google Scholar]
- 7. Robinson DR, Wu YM, Vats P, Su F, Lonigro RJ, Cao X, Kalyana‐Sundaram S, Wang R, Ning Y, Hodges L et al (2013) Activating ESR1 mutations in hormone‐resistant metastatic breast cancer. Nat Genet 45: 1446–1451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Giltnane JM, Hutchinson KE, Stricker TP, Formisano L, Young CD, Estrada MV, Nixon MJ, Du L, Sanchez V, Ericsson PG et al (2017) Genomic profiling of ER(+) breast cancers after short‐term estrogen suppression reveals alterations associated with endocrine resistance. Sci Transl Med 9: eaai7993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Formisano L, Stauffer KM, Young CD, Bhola NE, Guerrero‐Zotano AL, Jansen VM, Estrada MM, Hutchinson KE, Giltnane JM, Schwarz LJ et al (2017) Association of FGFR1 with ERalpha maintains ligand‐independent ER transcription and mediates resistance to estrogen deprivation in ER(+) breast cancer. Clin Cancer Res 23: 6138–6150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Huber‐Keener KJ, Liu X, Wang Z, Wang Y, Freeman W, Wu S, Planas‐Silva MD, Ren X, Cheng Y, Zhang Y et al (2012) Differential gene expression in tamoxifen‐resistant breast cancer cells revealed by a new analytical model of RNA‐Seq data. PLoS ONE 7: e41333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Irimia M, Blencowe BJ (2012) Alternative splicing: decoding an expansive regulatory layer. Curr Opin Cell Biol 24: 323–332 [DOI] [PubMed] [Google Scholar]
- 12. Cancer Genome Atlas N (2012) Comprehensive molecular portraits of human breast tumours. Nature 490: 61–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Ellis MJ, Ding L, Shen D, Luo J, Suman VJ, Wallis JW, Van Tine BA, Hoog J, Goiffon RJ, Goldstein TC et al (2012) Whole‐genome analysis informs breast cancer response to aromatase inhibition. Nature 486: 353–360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Maguire SL, Leonidou A, Wai P, Marchio C, Ng CK, Sapino A, Salomon AV, Reis‐Filho JS, Weigelt B, Natrajan RC (2015) SF3B1 mutations constitute a novel therapeutic target in breast cancer. J Pathol 235: 571–580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Gokmen‐Polar Y, Neelamraju Y, Goswami CP, Gu X, Nallamothu G, Janga SC, Badve S (2015) Expression levels of SF3B3 correlate with prognosis and endocrine resistance in estrogen receptor‐positive breast cancer. Mod Pathol 28: 677–685 [DOI] [PubMed] [Google Scholar]
- 16. Oltean S, Bates DO (2014) Hallmarks of alternative splicing in cancer. Oncogene 33: 5311–5318 [DOI] [PubMed] [Google Scholar]
- 17. Warzecha CC, Sato TK, Nabet B, Hogenesch JB, Carstens RP (2009) ESRP1 and ESRP2 are epithelial cell‐type‐specific regulators of FGFR2 splicing. Mol Cell 33: 591–601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Warzecha CC, Shen S, Xing Y, Carstens RP (2009) The epithelial splicing factors ESRP1 and ESRP2 positively and negatively regulate diverse types of alternative splicing events. RNA Biol 6: 546–562 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Warzecha CC, Jiang P, Amirikian K, Dittmar KA, Lu H, Shen S, Guo W, Xing Y, Carstens RP (2010) An ESRP‐regulated splicing programme is abrogated during the epithelial‐mesenchymal transition. EMBO J 29: 3286–3300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Yae T, Tsuchihashi K, Ishimoto T, Motohara T, Yoshikawa M, Yoshida GJ, Wada T, Masuko T, Mogushi K, Tanaka H et al (2012) Alternative splicing of CD44 mRNA by ESRP1 enhances lung colonization of metastatic cancer cell. Nat Commun 3: 883 [DOI] [PubMed] [Google Scholar]
- 21. Madden SF, Clarke C, Gaule P, Aherne ST, O'Donovan N, Clynes M, Crown J, Gallagher WM (2013) BreastMark: an integrated approach to mining publicly available transcriptomic datasets relating to breast cancer outcome. Breast Cancer Res 15: R52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Paik S, Shak S, Tang G, Kim C, Baker J, Cronin M, Baehner FL, Walker MG, Watson D, Park T et al (2004) A multigene assay to predict recurrence of tamoxifen‐treated, node‐negative breast cancer. N Engl J Med 351: 2817–2826 [DOI] [PubMed] [Google Scholar]
- 23. Hanahan D, Weinberg RA (2011) Hallmarks of cancer: the next generation. Cell 144: 646–674 [DOI] [PubMed] [Google Scholar]
- 24. Preca BT, Bajdak K, Mock K, Sundararajan V, Pfannstiel J, Maurer J, Wellner U, Hopt UT, Brummer T, Brabletz S et al (2015) A self‐enforcing CD44s/ZEB1 feedback loop maintains EMT and stemness properties in cancer cells. Int J Cancer 137: 2566–2577 [DOI] [PubMed] [Google Scholar]
- 25. Olsson E, Honeth G, Bendahl PO, Saal LH, Gruvberger‐Saal S, Ringner M, Vallon‐Christersson J, Jonsson G, Holm K, Lovgren K et al (2011) CD44 isoforms are heterogeneously expressed in breast cancer and correlate with tumor subtypes and cancer stem cell markers. BMC Cancer 11: 418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Brown RL, Reinke LM, Damerow MS, Perez D, Chodosh LA, Yang J, Cheng C (2011) CD44 splice isoform switching in human and mouse epithelium is essential for epithelial‐mesenchymal transition and breast cancer progression. J Clin Invest 121: 1064–1074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Lu ZX, Huang Q, Park JW, Shen S, Lin L, Tokheim CJ, Henry MD, Xing Y (2015) Transcriptome‐wide landscape of pre‐mRNA alternative splicing associated with metastatic colonization. Mol Cancer Res 13: 305–318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Park JW, Tokheim C, Shen S, Xing Y (2013) Identifying differential alternative splicing events from RNA sequencing data using RNASeq‐MATS. Methods Mol Biol 1038: 171–179 [DOI] [PubMed] [Google Scholar]
- 29. Shen S, Park JW, Huang J, Dittmar KA, Lu ZX, Zhou Q, Carstens RP, Xing Y (2012) MATS: a Bayesian framework for flexible detection of differential alternative splicing from RNA‐Seq data. Nucleic Acids Res 40: e61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Shen S, Park JW, Lu ZX, Lin L, Henry MD, Wu YN, Zhou Q, Xing Y (2014) rMATS: robust and flexible detection of differential alternative splicing from replicate RNA‐Seq data. Proc Natl Acad Sci USA 111: E5593–E5601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Dittmar KA, Jiang P, Park JW, Amirikian K, Wan J, Shen S, Xing Y, Carstens RP (2012) Genome‐wide determination of a broad ESRP‐regulated posttranscriptional network by high‐throughput sequencing. Mol Cell Biol 32: 1468–1482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Ryan M, Wong WC, Brown R, Akbani R, Su X, Broom B, Melott J, Weinstein J (2016) TCGASpliceSeq a compendium of alternative mRNA splicing in cancer. Nucleic Acids Res 44: D1018–D1022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Pike Winer LS, Wu M (2014) Rapid analysis of glycolytic and oxidative substrate flux of cancer cells in a microplate. PLoS ONE 9: e109916 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Silipo M, Gautrey H, Tyson‐Capper A (2015) Deregulation of splicing factors and breast cancer development. J Mol Cell Biol 7: 388–401 [DOI] [PubMed] [Google Scholar]
- 35. Hayakawa A, Saitoh M, Miyazawa K (2016) Dual roles for epithelial splicing regulatory proteins 1 (ESRP1) and 2 (ESRP2) in cancer progression. Adv Exp Med Biol 925: 33–40 [DOI] [PubMed] [Google Scholar]
- 36. Taube JH, Herschkowitz JI, Komurov K, Zhou AY, Gupta S, Yang J, Hartwell K, Onder TT, Gupta PB, Evans KW et al (2010) Core epithelial‐to‐mesenchymal transition interactome gene‐expression signature is associated with claudin‐low and metaplastic breast cancer subtypes. Proc Natl Acad Sci USA 107: 15449–15454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Alo PL, Visca P, Marci A, Mangoni A, Botti C, Di Tondo U (1996) Expression of fatty acid synthase (FAS) as a predictor of recurrence in stage I breast carcinoma patients. Cancer 77: 474–482 [DOI] [PubMed] [Google Scholar]
- 38. Alo PL, Visca P, Trombetta G, Mangoni A, Lenti L, Monaco S, Botti C, Serpieri DE, Di Tondo U (1999) Fatty acid synthase (FAS) predictive strength in poorly differentiated early breast carcinomas. Tumori 85: 35–40 [DOI] [PubMed] [Google Scholar]
- 39. Kuhajda FP, Jenner K, Wood FD, Hennigar RA, Jacobs LB, Dick JD, Pasternack GR (1994) Fatty acid synthesis: a potential selective target for antineoplastic therapy. Proc Natl Acad Sci USA 91: 6379–6383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Doria ML, Ribeiro AS, Wang J, Cotrim CZ, Domingues P, Williams C, Domingues MR, Helguero LA (2014) Fatty acid and phospholipid biosynthetic pathways are regulated throughout mammary epithelial cell differentiation and correlate to breast cancer survival. FASEB J 28: 4247–4264 [DOI] [PubMed] [Google Scholar]
- 41. Lupu R, Menendez JA (2006) Targeting fatty acid synthase in breast and endometrial cancer: an alternative to selective estrogen receptor modulators? Endocrinology 147: 4056–4066 [DOI] [PubMed] [Google Scholar]
- 42. Menendez JA, Lupu R (2017) Fatty acid synthase (FASN) as a therapeutic target in breast cancer. Expert Opin Ther Targets 21: 1001–1016 [DOI] [PubMed] [Google Scholar]
- 43. Poudyal H, Brown L (2011) Stearoyl‐CoA desaturase: a vital checkpoint in the development and progression of obesity. Endocr Metab Immune Disord Drug Targets 11: 217–231 [DOI] [PubMed] [Google Scholar]
- 44. Holder AM, Gonzalez‐Angulo AM, Chen H, Akcakanat A, Do KA, Fraser Symmans W, Pusztai L, Hortobagyi GN, Mills GB, Meric‐Bernstam F (2013) High stearoyl‐CoA desaturase 1 expression is associated with shorter survival in breast cancer patients. Breast Cancer Res Treat 137: 319–327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Possemato R, Marks KM, Shaul YD, Pacold ME, Kim D, Birsoy K, Sethumadhavan S, Woo HK, Jang HG, Jha AK et al (2011) Functional genomics reveal that the serine synthesis pathway is essential in breast cancer. Nature 476: 346–350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Locasale JW, Grassian AR, Melman T, Lyssiotis CA, Mattaini KR, Bass AJ, Heffron G, Metallo CM, Muranen T, Sharfi H et al (2011) Phosphoglycerate dehydrogenase diverts glycolytic flux and contributes to oncogenesis. Nat Genet 43: 869–874 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Samanta D, Park Y, Andrabi SA, Shelton LM, Gilkes DM, Semenza GL (2016) PHGDH expression is required for mitochondrial redox homeostasis, breast cancer stem cell maintenance, and lung metastasis. Cancer Res 76: 4430–4442 [DOI] [PubMed] [Google Scholar]
- 48. Nazarov PV, Muller A, Kaoma T, Nicot N, Maximo C, Birembaut P, Tran NL, Dittmar G, Vallar L (2017) RNA sequencing and transcriptome arrays analyses show opposing results for alternative splicing in patient derived samples. BMC Genomics 18: 443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Brunner N, Boysen B, Jirus S, Skaar TC, Holst‐Hansen C, Lippman J, Frandsen T, Spang‐Thomsen M, Fuqua SA, Clarke R (1997) MCF7/LCC9: an antiestrogen‐resistant MCF‐7 variant in which acquired resistance to the steroidal antiestrogen ICI 182,780 confers an early cross‐resistance to the nonsteroidal antiestrogen tamoxifen. Cancer Res 57: 3486–3493 [PubMed] [Google Scholar]
- 50. Brunner N, Frandsen TL, Holst‐Hansen C, Bei M, Thompson EW, Wakeling AE, Lippman ME, Clarke R (1993) MCF7/LCC2: a 4‐hydroxytamoxifen resistant human breast cancer variant that retains sensitivity to the steroidal antiestrogen ICI 182,780. Cancer Res 53: 3229–3232 [PubMed] [Google Scholar]
- 51. Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R, Salzberg SL (2013) TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol 14: R36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Trapnell C, Roberts A, Goff L, Pertea G, Kim D, Kelley DR, Pimentel H, Salzberg SL, Rinn JL, Pachter L (2012) Differential gene and transcript expression analysis of RNA‐Seq experiments with TopHat and Cufflinks. Nat Protoc 7: 562–578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Grant CE, Bailey TL, Noble WS (2011) FIMO: scanning for occurrences of a given motif. Bioinformatics 27: 1017–1018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Gokmen‐Polar Y, Mehta R, Tuzmen S, Mousses S, Thorat MA, Sanders KL, Turbin D, Leung S, Huntsman DG, Sledge GW Jr et al (2010) Differential subcellular expression of protein kinase C betaII in breast cancer: correlation with breast cancer subtypes. Breast Cancer Res Treat 124: 327–335 [DOI] [PubMed] [Google Scholar]
- 55. Gökmen‐Polar Y, Toroni RA, Hocevar BA, Badve S, Zhao Q, Shen C, Bruckheimer E, Kinch MS, Miller KD (2011) Dual targeting of EphA2 and ER restores tamoxifen sensitivity in ER/EphA2‐positive breast cancer. Breast Cancer Res Treat 127: 375–384 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Expanded View Figures PDF
Source Data for Expanded View and Appendix
Review Process File
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5
Source Data for Figure 6
Source Data for Figure 7
Data Availability Statement
The RNA‐Seq data (.bam files) for LCC2 (2‐control and ESRP1 knockdown‐2C3 cell lines) and LCC9 (9‐control and ESRP1 knockdown‐9C2 cell lines) sets were deposited at GEO under accession GSE125355 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE125355).