

In this review, Chan and Narita focus on the various aspects of the senescence response and discuss the implication of senescence in tumor development and treatment, with particular emphasis on early stage tumors.
Keywords: senescence, cancer, epigenetics, inflammation
Abstract
Originally thought of as a stress response end point, the view of cellular senescence has since evolved into one encompassing a wide range of physiological and pathological functions, including both protumorignic and antitumorigenic features. It has also become evident that senescence is a highly dynamic and heterogenous process. Efforts to reconcile the beneficial and detrimental features of senescence suggest that physiological functions require the transient presence of senescent cells in the tissue microenvironment. Here, we propose the concept of a physiological “senescence life cycle,” which has pathological consequences if not executed in its entirety.
The concept of senescence has been evolving over recent decades, having originally been described as a stress response that leads to a persistent proliferative arrest even in the presence of growth factors. Since senescence can limit the propagation of damaged cells (which are at risk of malignant transformation), removing them from the proliferative pool (Campisi 2001; Lowe et al. 2004), it therefore has been considered to be an endogenous tumor suppressor mechanism or even a therapeutic goal, alternative to apoptosis. However, this is not an all-encompassing view of senescence. Known as the senescence-associated secretory phenotype (SASP), senescent cells typically secrete diverse factors that affect the tumor microenvironment. This is also important for stromal cell senescence, which can be either antitumorigenic or protumorigenic depending on the context (for review, see Pérez-Mancera et al. 2014). It is perhaps safe to say that senescence is an autonomous tumor suppressor but a nonautonomous tumor “modulator” (Hoare and Narita 2018).
Senescence has also been implicated in aging as one of the hallmarks of aging (López-Otín et al. 2013). It has been proposed that the age-dependent accumulation of senescent cells within tissue stem or progenitor cell compartments results in the decline of their regenerative capacity in multiple tissues (for review, see Sharpless and DePinho 2007). These studies have provided the first functional relevance for senescence in aging within body organs, but more recent evidence that enforced killing of senescent cells in mice extends their life span has firmly established the causal role of senescence in aging (Baker et al. 2011, 2016).
In parallel, recent evidence has described more physiological roles for senescent cells that extend beyond the cancer and aging contexts, with functions in shaping embryonic development (Chuprin et al. 2013; Muñoz-Espín et al. 2013; Storer et al. 2013), contributing to the maturation of certain cell types (Besancenot et al. 2010; Helman et al. 2016), and maintaining tissue integrity in adult organisms by facilitating wound healing (Berg and Robson 2003; Krizhanovsky et al. 2008; Jun and Lau 2010a; Demaria et al. 2014). Thus, despite once being considered a state of “functional death,” senescent cells do have diverse functionalities in vivo.
These functions of senescent cells in tumor suppression or tissue homeostasis are often reinforced by or dependent on their transient presence, respectively, within the microenvironment, where senescent cells are eventually removed by immune cells (Ito et al. 2017). At least in some contexts, senescence can be seen as a progressive process that is initiated by cellular activation, develops senescence effectors, modulates the tissue microenvironment, and, finally, recruits immune cells to mediate resolution (Krizhanovsky et al. 2008). Senescence-associated disorders can perhaps be viewed as a consequence of a failure in the execution of the “senescence life cycle” (Fig. 1). Misregulation of senescence within tissues can promote pathological conditions, including cancer and other age-related disorders (Burton and Krizhanovsky 2014; Muñoz-Espín and Serrano 2014; He and Sharpless 2017). In this review, we provide an overview of the relevance of senescence in tumor development, discussing both the autonomous and nonautonomous aspects of senescence. We recently reviewed the relationship between senescence and cancer hallmarks elsewhere (Hoare and Narita 2018). Here, we focus on the various aspects of the senescence response and discuss the implication of senescence in tumor development and treatment, with particular emphasis on early stage tumors.
Figure 1.
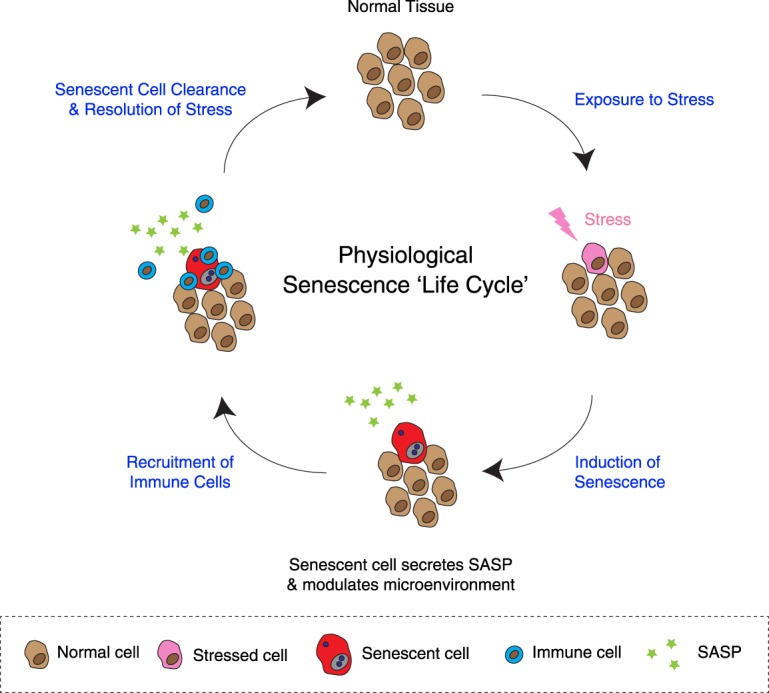
Senescence “life cycle.” Phases of the “senescence life cycle”: Cells undergo senescence in response to stress in normal (noncancerous) tissues. As part of the senescence program, the secretome is modified to include up-regulation of proinflammatory cytokines and chemokines, which modulate the tissue microenvironment. This recruits immune cells and facilitates clearance of senescent cells, mediating resolution and restoring tissue homeostasis. Deviation from this fail-safe mechanism can instead lead to age-related pathology or cancer. In some cases, cells become activated/proliferative before senescence establishment, and senescence signaling can be amplified locally through nonautonomous activities (for simplicity, these points are not reflected in the figure).
Senescence as cellular syndrome
The first observation of cellular senescence in vitro was described in replicative exhaustion, now known to be the result of telomere shortening (Hayflick 1965; Shay and Wright 2000). Since then, various stress-associated inducers of “premature” senescence have been identified, including DNA damage, oxidative stress, aberrant oncogene activation, and tumor suppressor gene loss (Kuilman et al. 2010; Campisi 2013). These cellular stressors evoke persistent DNA damage signaling and activation of the p53/p21 and p16/pRB tumor suppressor pathways, which mediate entry into and maintenance of senescence (d'Adda di Fagagna et al. 2003; Mallette et al. 2007; Rodier et al. 2009; Campisi and d'Adda di Fagagna 2007). Furthermore, a number of additional effector programs are involved in the senescence process, and the concept of senescence as a collective phenotype of multiple effectors instead of a single entity has been proposed. As such, due to the lack of a single biomarker that is both specific and unique to senescence, it is typically characterized using a combination of associated cellular changes, including chromatin reorganization, the gene expression profile, and secretome and metabolic pathways (Kuilman et al. 2010; Salama et al. 2014). In light of this, using an analogy from a clinical term, entry into senescence can perhaps be viewed as a “syndrome”: the incremental acquisition of associated phenotypes instead of a binary event where a defined set of changes manifests at the same time. This is consistent with the previously proposed concept of the differing depths of the senescence phenotype, which is modified by important cellular changes that follow an initial cell cycle arrest (Chen and Ozanne 2006; Passos et al. 2010; Baker and Sedivy 2013; Ivanov et al. 2013). This may further explain the well-described phenotypic heterogeneity of senescent cells, which manifests from the level of gene expression, where the phenotype depends on cell type, context, and the nature of the upstream stressor (Passos et al. 2007; van Deursen 2014; Marthandan et al. 2016; Hernandez-Segura et al. 2017). Senescent cell populations have also been described to possess a high degree of cell-to-cell variability that exceeds that of quiescent cells (Wiley et al. 2017), reinforcing the heterogeneity of senescence even at the single-cell level.
Senescence in tumors
As the induction of senescence is essentially the converse of the canonical unrestricted proliferation that defines cancer cells, much of the original research into the biology of senescence focused on its role in endogenous tumor suppression, which limits immortalization (telomere shortening-induced senescence) (see the next section) and excessive mitotic signaling (oncogene-induced senescence [OIS]).
Telomere shortening
The tumor-suppressive role of senescence was originally conceived in the context of telomere dysfunction (Shay and Wright 2005). Replicative senescence, for instance, can be considered to be a counterpart of immortalization in culture. Indeed, replicative senescence can be at least partially rescued by ectopic expression of telomerase in culture (Bodnar et al. 1998; Vaziri and Benchimol 1998), and most (>90%) human cancers aberrantly up-regulate telomerase (Kim et al. 1994; Shay and Bacchetti 1997; Jafri et al. 2016). The tumor-suppressive role of telomere shortening is recapitulated in telomerase deficiency in various tumor-prone mouse models (González-Suárez et al. 2000; Blasco 2005). However, telomerase deficiency alone in mice can lead to an increase in spontaneous cancer incidence mainly in rapidly proliferating cell types, exemplified by the unusually high incidence of teratocarcinomas, probably due to increased genome instability, a hallmark of cancer (Rudolph et al. 1999). Importantly, these exceptions, where short telomeres counterintuitively promote the incidence of cancer, tend to be associated with a p53 defect (Chin et al. 1999; Artandi et al. 2000).
This is consistent with the view that telomere dysfunction triggers a DNA damage response (DDR), which activates the p53 pathway (d'Adda di Fagagna et al. 2003; Deng et al. 2008). The downstream effects of this pathway can be apoptosis and/or senescence. However, which components are responsible for the tumor-suppressive activity of short telomeres in vivo? Using a mouse model of Burkitt's lymphoma with telomerase deficiency (Adams et al. 1985; Blasco et al. 1997), Feldser and Greider (2007) report that apoptosis is dispensable for short telomere-mediated tumor suppression. Provided that p53 is intact, mice remain resistant to tumor formation even following the blockade of apoptosis. Instead, microlymphomas, which stain positive for markers of senescence, are observed, demonstrating the importance of senescence as an effector of tumor suppression in this context (Feldser and Greider 2007).
These data suggest that the intrinsic limitation of proliferative capacity can contribute to a reduction in both tissue-regenerative capacity and cancer initiation (Chang 2005; Liu et al. 2011). This appears to support the idea of a trade-off between aging and cancer (Kirkwood and Austad 2000; Tyner et al. 2002), although this view considers only the autonomous aspect of senescence (see “Nonautonomous Effectors” below), and telomere shortening is just one way to trigger senescence.
Telomeres are notorious for DNA repair: It has been suggested that DNA damage in telomeres is effectively unrepairable, causing a persistent DDR and the formation of telomere-associated foci (TAFs) in aging- and stress-induced senescence (Takai et al. 2003; Fumagalli et al. 2012; Hewitt et al. 2012). This can occur independently of telomere length, as markers of DDR can accumulate at telomeres that are not yet critically shortened (Fumagalli et al. 2012). Although the chromosome body can also harbor persistent DNA damage after genotoxic stress (called DNA segments with chromatin alterations reinforcing senescence [DNA-SCARS]) (Rodier et al. 2011), live-cell imaging experiments have suggested previously that the majority of these persistent foci are associated with telomeres (Hewitt et al. 2012). These residual DNA damage foci appear to contribute to the maintenance of cell cycle arrest (Passos et al. 2010; Rodier et al. 2011), suggesting that telomere dysfunction-mediated senescence can also occur in contexts other than replicative stress.
Importantly, DNA damage can induce senescence even in cancer cells in some contexts (called therapy-induced senescence [TIS]) (Schmitt et al. 2002; Nardella et al. 2011; Acosta and Gil 2012), and TAF formation after irradiation is not affected by telomerase activity (Fumagalli et al. 2012; Hewitt et al. 2012), suggesting that cancer cells could possibly be subjected to this mechanism. However, the antitumor effect of TIS is less evident and highly context- and model-dependent (Dörr et al. 2013; Tato-Costa et al. 2016; Demaria et al. 2017; Yang et al. 2017b). This is perhaps in part due to the diversity of the SASP components and their pleiotropic activities as well as the heterogeneity and stochasticity of cancers and their response to therapies within the tissue microenvironment. This point is further discussed below (see “Nonautonomous Effectors”).
Senescence in preneoplasia
While replicative senescence limits cellular “immortalization,” an early step in tumorigenesis, an alternative autonomous tumor-suppressive function of senescence can be seen in OIS, where some “mitogenic” oncogenes (or loss of tumor suppressors, as exemplified by PTEN loss-induced cellular senescence [PICS]) can paradoxically induce persistent proliferative arrest (Braig and Schmitt 2006; Courtois-Cox et al. 2008; Pérez-Mancera et al. 2014). OIS was first described in human diploid fibroblasts in culture, where a phenotype similar to replicative senescence was observed following ectopic expression of constitutively active HRas (Serrano et al. 1997) or its downstream effectors: MEK (Lin et al. 1998) or RAF1 (Zhu et al. 1998). This senescent phenotype is accompanied by accumulation of the tumor suppressor proteins p53 and p16, and a concomitant ablation of these tumor suppressors leads instead to transformation, supporting the notion of OIS as an intrinsic tumor suppressor mechanism (Serrano et al. 1997; Lin et al. 1998). In mouse models, the tumor suppressor p19Arf has been shown to mediate the activation of p53 in response to oncogenic Ras (Palmero et al. 1998). Following the description of OIS in vitro, senescent cells harboring oncogenic mutations were observed to accumulate in premalignant lesions of various tissue types in both humans and mice (Braig et al. 2005; Chen et al. 2005; Collado et al. 2005; Narita and Lowe 2005; Lazzerini Denchi et al. 2005; Michaloglou et al. 2005; Collado and Serrano 2010).
In a cell-autonomous manner, the nature of the activating oncogenic signal influences the balance between entry into senescence and tumorigenesis, where oncogene dose and the presence of other cooperating mutations have been suggested to influence cellular outcome (Quintanilla et al. 1986; Tuveson et al. 2004; Sarkisian et al. 2007). The most widely used OIS model remains the classic culture model: overexpression of oncogenic HRas-G12V-induced senescence in human diploid fibroblasts (Serrano et al. 1997). This is highly robust and, particularly with the introduction of a 4OHT-inducible system (Young et al. 2009), useful for procedures that require a large number of cells, as are often required for genomic and proteomic approaches. However, one inherent caveat of this model is the level of Ras, which is supraphysiological. The natural Ras-driven tumorigenesis is typically initiated with a single-allelic oncogenic mutation (Hobbs et al. 2016).
Evidence for the notion that oncogenic dose influences cellular outcome was provided by earlier studies showing that mouse embryonic fibroblasts (MEFs) expressing endogenous levels of the single-allelic Kras-G12V or Kras-G12D fail to undergo senescence, although they were not fully transformed (Guerra et al. 2003; Tuveson et al. 2004). A similar insensitivity to senescence induction by a moderate level of ectopic oncogenic Ras or its downstream effector, Raf, was also shown in human or immortalized mouse fibroblasts (Sewing et al. 1997; Deng et al. 2004). At the same time, mouse models developed to recapitulate the role of oncogenic Ras mutations in tumorigenesis have shown that while endogenous Kras-G12V expression leads to premalignant lesions in the pancreas (Hingorani et al. 2003) and preneoplastic hyperplasia in the lung and intestine, additional cellular alterations are typically required for progression to full malignancy (Tuveson et al. 2004). Furthermore, only a subset of cells is transformed by oncogenic KRas—and even then in a highly context-dependent manner (Jackson et al. 2001; Guerra et al. 2003; DuPage et al. 2009; Lee and Bae 2016).
These observations suggest that a low dose of oncogenic Ras is not sufficient to trigger senescence programs or malignant transformation. This was further supported by a mouse breast cancer model in which the level of a doxycycline-inducible ectopic Hras-G12V can be titrated (Sarkisian et al. 2007). Consistent with the earlier studies, this study also showed that high-level but not low-level Ras induces senescence in mammary glands in vivo. In addition, while low-level Ras (comparable with the level expressed from the endogenous Kras-G12D allele in the mouse pancreas model described above) is not immediately sufficient for cancer development, the mice eventually develop tumors. Interestingly, these tumors (derived from low-level Ras) are accompanied by the spontaneous up-regulation of oncogenic Ras to a level similar to that of high-level Ras, which induces senescence. Furthermore, they observed senescent mosaicism within those low-Ras-initiated tumors with spontaneous up-regulation of oncogenic Ras. Mechanisms for the spontaneous up-regulation of oncogenic Ras in this study were not clear, but a similar up-regulation of oncogenic Ras during cancer development has been reported in different tumor models (Quintanilla et al. 1986; Finney and Bishop 1993; Aguirre et al. 2003; Junttila et al. 2010).
It is possible that, even when it is initiated by a single copy mutation, Ras activity needs to be increased for full malignant transformation but that this is counteracted by senescence programs. Perhaps the OIS culture system models a tumor-suppressive event at this critical step of Ras-driven tumorigenesis (Fig. 2). Of note, the Ras pathway is regulated by diverse effectors; thus, its oncogenic activity can be up-regulated through multiple routes (Downward 2003; Calvisi et al. 2006; Courtois-Cox et al. 2006; Shaw et al. 2007; Vandal et al. 2014). It would be important to determine the correlation between the level of oncogenic activity and the senescence phenotype during the preneoplastic stage in those genetically engineered OIS models.
Figure 2.
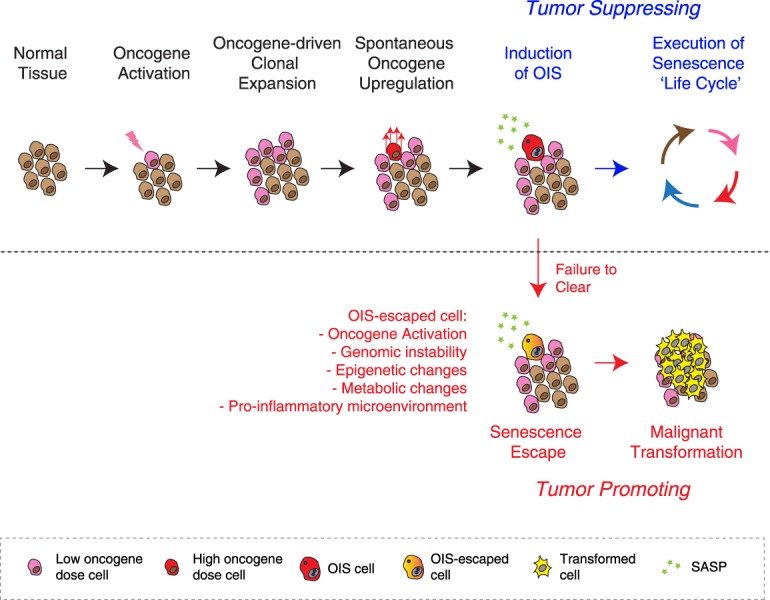
OIS as a model of spontaneous up-regulation of somatically mutated oncogenic signaling. Using oncogenic Ras as an example, an age-dependent increase of somatic mutation of oncogenes and their clonal expansion are common, but high-levels of oncogenic signaling are necessary for both OIS and full malignant transformation. Typically, spontaneous up-regulation of oncogenic signaling (to the levels sufficient for malignancy) triggers the OIS program, which is tumor-suppressive as long as the “senescence life cycle” is executed to completion. Conversely, failure to clear OIS cells can be tumor-promoting, as these cells are at risk of senescence escape, having acquired tumor-facilitating cellular changes as well as having shaped a protumorigenic microenvironment.
Autonomous senescence effectors
As a collective phenotype made up of numerous cellular effector programs, we discuss autonomous and nonautonomous effectors separately and focus here on chromatin and genomic alterations as representative of the autonomous effectors potentially contributing to the static nature of senescence arrest.
Epigenetics
It has been proposed that senescence, unlike quiescence (a state of physiological and readily reversible cell cycle arrest), uses distinct alterations in the chromatin landscape (Parry and Narita 2016). These epigenetic and chromatin alterations occur at various levels, including DNA methylation, histone marks and variants, chromatin accessibility, and noncoding RNAs (Pal and Tyler 2016; Parry and Narita 2016; Buschbeck and Hake 2017; Nacarelli et al. 2017). Among these, DNA methylation (5-methylcytosine at CpG), on the one hand, is known as a marker of constitutive heterochromatin, particularly at regions with repetitive sequences. On the other hand, although CpG islands, which are abundant regulatory elements in mammalian promoters, are usually hypomethylated, the CpG islands of some genes can be hypermethylated in abnormal conditions; e.g., hypermethylation of CpG islands at the promoters of tumor suppressors leads to their silencing and promotes tumorigenesis (Deaton and Bird 2011).
It has been long known that DNA methylation globally declines during senescence, forming the basis for the well-known “heterochromatin loss model”: the idea that a gradual breakdown of heterochromatin leads to the desilencing of otherwise repressed genes (or noncoding RNAs), contributing to senescence and aging (Villeponteau 1997). However, more recent studies using next-generation sequencing technology have revealed that the alterations in DNA methylation during senescence are not unidirectional. Using a model of replicative senescence in human fibroblasts, Cruickshanks et al. (2013) demonstrated that DNA methylation is globally reduced at heterochromatic regions but focally increased at a subset of CpG islands. Interestingly, the pattern of hypermethylated CpG islands in replicative senescence is reminiscent of that of cancers, leading to the hypothesis that senescence and cancer share features of epigenetic change. This suggests that cells that have escaped from senescence would be more susceptible to subsequent transformation (Cruickshanks et al. 2013).
This idea—that senescence-associated epigenetic alterations might contribute to a tumor-suppressive senescence phenotype but, at the same time, encompass oncogenic properties—appears to explain well the dual effect of senescence in cancer; i.e., senescence as a tumor suppressor and yet also as a risk factor for cancer. This might also provide a partial explanation for the recent study suggesting that senescence escapers exhibit a stem-like signature with high tumor-initiating potential (Milanovic et al. 2018). However, a recent study provides an alternative view. By directly comparing the DNA methylation profile between replicative senescent and transformed cells derived from the same cell line, Xie et al. (2018) identified a global reduction with focal increases in DNA methylation during senescence, consistent with the findings of the previous study. However, looking at individual genomic regions conversely suggested that methylation patterns evolved separately during tumorigenesis and progression to senescence: The former is stochastic and the latter is highly reproducible, thus suggesting that they are somewhat programmed. Based on these data, the investigators propose that transformation-associated changes in DNA methylation status do not stem from senescence escape but rather reflect alterations that can be associated independently with both aging and increased cancer risk (Xie et al. 2018). In contrast to replicative senescence, OIS-associated DNA methylation alterations appear to be minimal, underscoring the diversity of the senescence phenotype (Sakaki et al. 2017; Xie et al. 2018). Whichever is the case, the decline in DNA methylation of heterochromatic regions with repetitive sequences (e.g., transposons, satellite DNAs) in senescence or cancer potentially causes genome instability through a reactivation of transposons for instance. Indeed, expression of retrotransposable elements and satellite sequences has been shown to increase during replicative senescence (De Cecco et al. 2013b).
In an interesting contrast to DNA methylation, the profile of DNA accessibility (a marker for open chromatin, which can be an indicator of developmental maturity) (Stergachis et al. 2013) is also altered during replicative senescence: DNA accessibility exhibits a global increase with focal declines (De Cecco et al. 2013a). This appears to show a mirror image of the DNA methylation pattern, but whether or not DNA methylation and chromatin accessibility are physically or functionally related has not been tested. In OIS too, a general increase in chromatin accessibility has been reported, and different oncogenes induce different profiles of accessible regions upon senescence entry (Parry et al. 2018).
Chromatin dynamics
In addition to the alterations to the linear structure of the genome, widespread changes in higher-order chromatin structure have been observed during senescence. For example, senescence-associated heterochromatic foci (SAHFs) are readily visualized by microscopy as DAPI-dense foci in the nucleus (Narita et al. 2003; Zhang et al. 2005). A number of structural and functional factors that are required for SAHF formation have been identified; e.g., HMGA chromatin architectural proteins and histone cochaperones HIRA/ASF1a have been shown to be necessary for SAHF formation, whereas loss of linker histone H1 is correlated with SAHFs (Zhang et al. 2005; Funayama et al. 2006; Narita et al. 2006). It was initially proposed that SAHFs are involved in the stable silencing of a subset of genes, including cell cycle genes (Narita et al. 2003; Zhang et al. 2007; Sadaie et al. 2013), but it is also possible that inter-SAHF euchromatic regions actively take part in stable gene activation. It is important to note that SAHF formation is model-dependent, which may represent the heterogeneity of senescence. Interestingly, higher levels of p16 and HMGA, both of which are critical for SAHF formation, are correlated with the irreversibility of the senescence arrest (Beauséjour et al. 2003; Narita et al. 2006), reinforcing the existence of a spectrum of senescence, an idea associated with the “senescence syndrome” mentioned above.
In contrast to SAHFs, senescence-associated distension of satellites (SADS) represents a distension of α-satellite and satellite II sequences, which are heterochromatic and normally compacted at the (peri)centromere, during senescence. SADS appears to be more universal than SAHFs (Swanson et al. 2013). Interestingly, both of these newly formed heterochromatins (SAHFs) and unfolding of constitutive heterochromatins (SADS) appear not to require alterations in repressive marks H3K9me3 and H3K27me3 (Chandra et al. 2012; Swanson et al. 2013). Instead, these chromatin structural changes are mediated by a differential association with the nuclear membrane. In proliferating cells, heterochromatin typically localizes preferentially to the nuclear periphery and around the nucleolus, tethered by lamina-associated chromatin domains (LADs) and nucleolus-associated chromatin domains (NADs) (Padeken and Heun 2014). While NADs have been suggested to be conserved at least in replicative senescence (Dillinger et al. 2017), the level of Lamin B1 declines during senescence (Shimi et al. 2011; Freund et al. 2012; Sadaie et al. 2013; Shah et al. 2013; Criscione et al. 2016). Although the loss of Lamin B1 is not sufficient for senescence or SAHF induction, it liberates H3K9me3-rich heterochromatin from the nuclear periphery and facilitates SAHF formation (Sadaie et al. 2013). These observations have led to the model in which SAHFs are formed through the spatial repositioning of pre-existing repressive histone marks (Chandra et al. 2012, 2015). Although it is not clear how Lamin B1 loss promotes SADS formation, it has been shown that SADS often extends along or toward the nuclear periphery, and the researchers speculated that alterations in Lamin B1 and other structural proteins might contribute to this process (Swanson et al. 2013).
Another consequence of Lamin B1 down-regulation during senescence is compromised nuclear envelope integrity and the subsequent leakage of chromatin fragments from the nucleus into the cytoplasm as cytoplasmic chromatin fragments (CCFs). Although it was reported that Lamin B1 down-regulation during senescence is mediated by RB-dependent silencing (Shimi et al. 2011), Lamin B1 is also a substrate of autophagic degradation (see below; Dou et al. 2015; Lenain et al. 2015). Indeed, CCFs, together with Lamin B1, can be degraded by autophagy; thus, this process was proposed as a mechanism used by senescent cells to remove damaged genomic material (Ivanov et al. 2013; Dou et al. 2015, 2017). The loss of chromatin would impact on the chromatin landscape, gene expression, and long-term phenotype of senescent cells (O'Sullivan et al. 2010; Ivanov et al. 2013). However, emerging evidence indicates that leaked cytoplasmic DNA in turn can also trigger a discrete aspect of senescence, the nonautonomous activity, via activation of the cGAS/STING pathway, which is further discussed in the next section.
Nonautonomous effectors
In addition to their essential feature (the persistency of cell cycle arrest), senescent cells are actively engaged in communication with other cells and the extracellular matrix (ECM) within the tissue microenvironment. Several modes of senescence-associated cell–cell communication have been shown, including the SASP, cell fusion (Chuprin et al. 2013), cytoplasmic bridges (Biran et al. 2015), cell–cell contact (Nelson et al. 2012; Hoare et al. 2016; Ito et al. 2017; Parry et al. 2018), and secreted extracellular vesicles (Takasugi et al. 2017).
The SASP is the best characterized of these mechanisms (Kuilman and Peeper 2009; Malaquin et al. 2016), and the nonautonomous “functionality” of senescent cells has been mostly attributed to the SASP. The fact that senescent cells secrete soluble factors was long known, and expression of some factors, such as MMP3 and PAI1, had been used as a marker of senescence (Krtolica et al. 2001; Parrinello et al. 2005; Kortlever et al. 2006). Initially, the SASP was collectively assumed to have tumor-promoting effects (Campisi and d'Adda di Fagagna 2007) because senescent fibroblasts promote the tumorigenesis of coexisting premalignant epithelial cells (Krtolica et al. 2001). The SASP has since been demonstrated as a part of the senescence program, with diverse downstream effects. For example, PAI1 was shown to be a downstream effector of the p53 pathway during replicative senescence, which contributes to cell cycle arrest, while IL6 and IL8 (an inflammatory cytokine and chemokine, respectively) reinforce senescence arrest through paracrine activities (Kortlever et al. 2006; Acosta et al. 2008; Kuilman et al. 2008; Wajapeyee et al. 2008). Increasing evidence has revealed that the SASP is a highly dynamic process associated with diverse composites, regulatory mechanisms, and functionalities.
Inflammatory SASP and senescence surveillance
Among the diverse components, the best-characterized factors of the SASP are of the proinflammatory type, represented by IL1, IL6, and IL8, among others. Expression of these inflammatory SASP components is controlled by diverse factors, such as epigenetic and chromatin regulators, the translation machinery, and signaling molecules (Rodier et al. 2009; Freund et al. 2011; Takahashi et al. 2012; Herranz et al. 2015; Laberge et al. 2015; Aird et al. 2016; Capell et al. 2016; Hoare et al. 2016; Tasdemir et al. 2016; Dou et al. 2017). The central mode of SASP regulation is a hierarchical model, where the genes encoding the proximal inflammatory cytokines, such as IL1 and IL6, are induced by the inflammatory transcription factors NFkB and C/EBPβ cooperatively with cofactors such as BRD4 (Tasdemir et al. 2016). Importantly, these inflammatory cytokines in turn reinforce the activity of those transcription factors through a positive feedback loop; thus, the signaling is locally amplified (Kuilman et al. 2008; Orjalo et al. 2009; Acosta et al. 2013; Pérez-Mancera et al. 2014).
How is such a signaling cascade subsequently resolved? The major outcome of the inflammatory SASP in vivo is not just senescence reinforcement but also activation of an immune reaction (Fig. 1). The first in vivo evidence for this was derived from a p53-deficient liver cancer model, where cancer cells express a tetracycline-regulable p53 RNAi (Xue et al. 2007). Restoration of endogenous p53 after the cancer was established led to the induction of senescence and tumor regression, which was due not to apoptosis but to immune clearance of senescent liver tumor cells. Although this experiment was conducted in athymic nude mice, which are immunocompromised, their innate immune system is intact and sufficient for the clearance of senescent cells in this particular context. A follow-up study in mice lacking T and B cells reinforced the sufficiency of innate immune cells (in this case, natural killer cells) in mediating tumor-suppressive clearance of senescent cells (Iannello et al. 2013).
Subsequently, a similar elimination of senescent cells by innate immune cells has been shown in other models, such as chronic liver damage in mice (Krizhanovsky et al. 2008; Lujambio et al. 2013). Krizhanovsky et al. (2008) showed that the liver-damaging agent CCl4 induces senescence in the liver, but those senescent cells are mainly hepatic stellate cells (HSCs). HSCs are fibroblast-like cells that are normally in a quiescent state but become active and proliferative, followed by the development of senescence. Through the SASP, senescent HSCs can be eliminated by innate immune cells, including natural killer cells and tumor-inhibiting M1-state macrophages (Krizhanovsky et al. 2008; Lujambio et al. 2013). Interestingly, p53-deficient HSCs bypass senescence and secrete factors that stimulate macrophages into a tumor-promoting M2 state, suggesting a nonautonomous tumor suppressor activity for p53 (Lujambio et al. 2013). This study also highlights that the SASP derived from senescent stromal cells has a profound impact on the tumor microenvironment.
These “TIS” (i.e., restoration of endogenous p53) or damage-induced senescence studies established the role of innate immunity in senescence clearance, but a role for adaptive immune components in senescence surveillance was shown in a liver OIS model. As shown by Kang et al. (2011), ectopic expression of oncogenic NRas induces senescence in hepatocytes in mice, and those OIS hepatocytes are eliminated primarily by macrophages, but this process requires activation of mutant NRas-specific CD4+ T cells. Indeed, immune clearance of OIS hepatocytes was diminished when the experiment was conducted in immunocompromised mice, which eventually developed liver cancer (Kang et al. 2011). Interestingly, major histocompatibility complex class II (MHCII) is typically expressed by professional antigen-presenting cells (APCs), but hepatocytes appear to express MHCII to directly present antigens (Herkel et al. 2003). Indeed, a fraction of the OIS hepatocytes appears to express a higher level of MHCII, which, although not sufficient (i.e., professional APCs are necessary), is required for senescence surveillance (Kang et al. 2011). Similar up-regulation of MHCII was reported in melanocytic OIS models. In contrast to the liver model, however, MHCII on OIS melanocytes appears to be sufficient for T-cell activation (van Tuyn et al. 2017). These studies suggest that the senescence surveillance involves multileveled and distinct aspects of the immune system depending on the context, such as the senescence trigger and cell type.
Other SASP functionality
The functionality of the SASP is not limited to mediating immune clearance of senescent cells from the tissue. It also plays a role in the maintenance of tissue integrity. As mentioned above, upon liver damage, quiescent HSCs become activated and proliferate before developing senescence (Krizhanovsky et al. 2008). Activated HSCs produce ECM components, contributing to liver fibrosis. Importantly, the SASP components of senescent HSCs contain ECM degradation enzymes, thus countering the excessive fibrosis. Thus, tissue repair and integrity are controlled through a timely switch from a fibrogenic to a fibrolytic secretory program, and this switch is coupled with the eventual elimination of senescent cells by immune cells. A similar role for senescence in tissue repair and wound healing has been shown in other tissue damage models: the liver (Kong et al. 2012; Kim et al. 2013; Borkham-Kamphorst et al. 2014), kidney (Wolstein et al. 2010), skin (Jun and Lau 2010b; Pitiyage et al. 2011; Demaria et al. 2014), and heart (Zhu et al. 2013). Notably, Krizhanovsky et al. (2008) also showed in the liver model that persistence of senescent HSCs due to the failure of immune-mediated elimination of senescent HSCs rather promotes liver fibrosis, reinforcing the idea of a senescence life cycle. Association between fibrogenic senescence and chronic tissue damage/inflammation has also been reported in an idiopathic pulmonary fibrosis mouse model (Schafer et al. 2017).
More recently, a progressive switch of secretory factors has also been shown in the classical senescence model in culture, where NOTCH signaling is transiently activated during an early phase of senescence triggered by oncogenic HRas or DNA damage (Hoare et al. 2016). The NOTCH phase is correlated with a distinct secretome, representing TGFβ and other fibrogenic factors, and subsequent down-regulation of NOTCH signaling is required for switching to the typical inflammatory and fibrolytic SASP in the late phase of senescence. This is highly reminiscent of the case of HSC senescence (Krizhanovsky et al. 2008), but it remains to be elucidated whether Notch signaling is activated during the progressive development of HSC senescence. Mechanistically, it was shown that NOTCH signaling negatively regulates the expression of primary inflammatory cytokine production through suppressing the activity of the transcription factor C/EBPβ. Consistently, in the liver OIS model described above, the inhibition of NOTCH signaling in NRas-expressing hepatocytes facilitates their elimination (Ito et al. 2017).
A recent addition to the SASP functionality includes its role in cellular reprograming (for review, see Taguchi and Yamada 2017). Earlier studies had shown that the senescence machinery autonomously serves as a barrier to reprogramming into induced pluripotent stem cells (iPSCs) in response to expression of the four Yamanaka factors (Oct3/4, Sox2, Klf4, and c-Myc) (Banito et al. 2009; Hong et al. 2009; Kawamura et al. 2009; Krizhanovsky and Lowe 2009; Li et al. 2009; Marión et al. 2009; Utikal et al. 2009). However, using mice expressing those Yamanaka factors, recent studies have shown that senescent cells nonautonomously facilitate the reprogramming of surrounding cells through an NFkB-driven secretome, particularly IL6 (Mosteiro et al. 2016; Chiche et al. 2017). Consistently, Ritschka et al. (2017) showed that a transient or low-level, but not prolonged, exposure to the OIS SASP promotes cellular plasticity and regenerative capacity in mouse skin keratinocytes. It has been suggested that senescence arrest in tissue stem cell or progenitor cells reduces regenerative capacity in some tissues (Sharpless and DePinho 2007). In contrast to this autonomous effect of senescence, much like the case of cellular reprograming to iPSCs, senescent cells appear to nonautonomously contribute to maintaining the tissue stem cell niche.
The physiological roles of senescent cells in facilitating tissue repair in response to injury relies on their clearance after the cessation of the wound healing processes, as accumulation of senescent cells conversely favors the pathogenesis of aging and age-related disorders. Tumors have long been dubbed as “wounds that do not heal” (Dvorak 1986, 2015; Byun and Gardner 2013) due to similarities between the phases of wound healing and those of tumor stroma formation. Through the SASP (derived from either tumorous or stromal senescent cells), the chronic presence of senescent cells within a tumor microenvironment would maintain a chronically inflamed microenvironment that is inherently tumorigenic (Lecot et al. 2016; Baker et al. 2017). The SASP also modulates other key hallmarks of cancers through locally facilitating vascularization (Coppé et al. 2006), epithelial-mesenchymal transition (Coppé et al. 2008; Kuilman et al. 2008), tumor invasion (Kim et al. 2017; He et al. 2018), and cellular plasticity (Ritschka et al. 2017). Disruption of the senescence life cycle may create a tumorigenic microenvironment collectively through these SASP functionalities (Fig. 1).
Upstream regulators of SASP
The inflammatory SASP appears to be controlled through multiple effectors. For example, the SASP is typically promoted by a persistent DDR (Rodier et al. 2009; Ciccia and Elledge 2010). However, how the inflammatory cascade is initiated had not been clear until a recent series of publications showed that the cGAS–STING cytosolic DNA-sensing pathway, described in the previous section, is a critical upstream event to trigger the SASP (Dou et al. 2017; Glück et al. 2017; Li and Chen 2018; Takahashi et al. 2018). Evolved as a defense response to viral and microbial infections, this pathway promotes inherent antitumor immunity (Deng et al. 2014; Woo et al. 2014; Bose 2017). The cGAS–STING pathway, an intrinsic tumor suppressor, is deregulated in carcinomas (Xia et al. 2016), and low levels of cGAS or STING are correlated with poor prognosis in some cancer types (Song et al. 2017; Yang et al. 2017a). Interestingly, the same pathway appears to be provoked during senescence, sensing CCFs (Ivanov et al. 2013; Dou et al. 2015).
Upon binding to DNA, the DNA sensor cGAS produces the second messenger cGAMP, which activates STING. This typically leads to the engagement of two canonical downstream pathways: a type I interferon (IFN) response mediated by IRF3 and an inflammatory response through NFkB activation (Abe and Barber 2014; Barber 2015). However, type I IFNs are not necessarily overrepresented in the SASP, depending on the cell type (Dou et al. 2017). The investigators proposed that this is due in part to the activation of p38MAPK (another positive regulator of the SASP) (Freund et al. 2011), which can inhibit STING-mediated IFN induction (Dou et al. 2017). Thus, the cGAS–STING pathway is critical in the activation of the NFkB-driven SASP. Consistently, the loss of this pathway results in reduced senescence surveillance and increased tumorigenesis in the liver OIS model (Dou et al. 2017; Glück et al. 2017). As with the inflammatory SASP, it is conceivable that prolonged activation of cGAS–STING can also promote tumorigenesis and metastasis, possibly through chronic inflammation (Li and Chen 2018). Indeed, in an obesity-induced liver cancer model, the SASP derived from senescent HSCs promotes liver cancer (Yoshimoto et al. 2013), and this effect was diminished in Sting knockout mice (Dou et al. 2017).
Autophagy
It has been shown that the SASP is also controlled through macroautophagy (referred to here simply as autophagy), another effector of senescence (Hoare et al. 2011). Autophagy, an evolutionarily conserved catabolic machinery, involves the formation of autophagosomes and their fusion to lysosomes to form autolysosomes, where encircled macromolecules or even organelles are digested by lysosomal enzymes (Shen and Mizushima 2014). The bulk degradation of damaged macromolecules facilitates their turnover, and the degradation products can be an alternative energy source. Thus, basal autophagy is important for the quality control of cellular components and energy homeostasis. In addition to its basal activity, autophagy can also be acutely activated by not only metabolic (e.g., starvation) but also cytotoxic (e.g., DNA damage and oncogenic) stress. It is conceivable that stress-induced autophagy contributes to the degradation of damaged cellular components, but its long-term functional relevance is not entirely clear.
One of the best-known markers of senescence is an increase in senescence-associated β-galactosidase activity (SA-β-gal), which is derived from a lysosomal enzyme (Lee et al. 2006). Earlier studies have suggested that autophagy is activated during senescence, and SA-β-gal reflects increased lysosomal mass, at least in some contexts (Kurz et al. 2000; Gerland et al. 2003; Narita et al. 2011). Functionally, it has been suggested that autophagy contributes to the senescence phenotype in part through modulating the SASP (Patschan et al. 2008; Young et al. 2009). Interestingly, prolonged activation of autophagic protein degradation was shown to activate the anabolic counterpart of autophagy, mTOR, on the surface of (auto)lysosomes through degradation products (i.e., amino acids) (Yu et al. 2010; Efeyan et al. 2012). Increased lysosomal biogenesis and compartmentalization during senescence have been shown in several models (Young et al. 2009; Narita et al. 2011; Dörr et al. 2013). It was proposed that prolonged activation of autophagy leads to the simultaneous activation of mTOR (which is typically regulated in the direction opposite to autophagy), which facilitates protein synthesis (Narita et al. 2011). How mTOR signaling specifically activates the SASP was not clear, but recent studies have proposed multiple mechanisms for this through modulating translation and the stabilization of mRNA molecules encoding SASP components (Fig. 3; Herranz et al. 2015; Laberge et al. 2015; Tomimatsu and Narita 2015).
Figure 3.
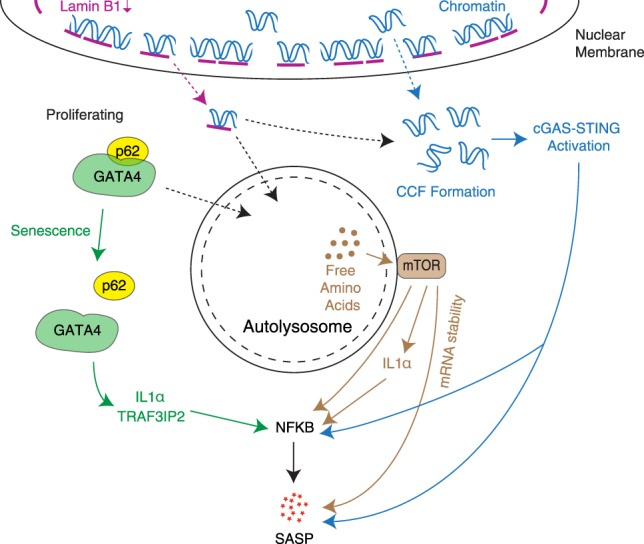
Multiple nonunidirectional levels through which autophagy-related processes in senescence affect regulation of SASP genes, often via NFkB activation. Activation of macroautophagy as an effector of senescence and spatial coupling of mTOR with autolysosomes lead to mTOR activation, which has been proposed to modulate SASP expression through multiple mechanisms. Autophagy-mediated degradation of Lamin B1 also promotes CCF formation, which up-regulates SASP genes through the cGAS–STING pathway. This has been suggested to occur by both nuclear membrane blebbing, which shuttles LADs to the cytoplasm, and loss of nuclear envelope integrity, which allows the escape of chromatin fragments. However, the activation of general autophagy during senescence is accompanied by an inhibition of p62-mediated selective autophagy, allowing stabilization of the GATA4 transcription factor, which regulates SASP genes via NFkB.
The positive regulation of senescence by autophagy was counterintuitive, considering the role of autophagy in cellular quality and fitness checks, although it is possible that increased autophagy activity serves as a prosurvival factor in senescent cells, which are known to be resistant to the cell death machinery (Dörr et al. 2013). The relevance of autophagy in senescence appears to be highly context-dependent (Kwon et al. 2017). It is probably important to consider the difference between basal and stress-induced autophagy: A lack of the former would lead to the accumulation of damaged macromolecules (such as p62-containing aggresomes) and damaged organelles (such as dysfunctional mitochondria). Thus, it is conceivable that loss of basal autophagy alone promotes senescence through cellular damage (Kwon et al. 2017). For instance, a role of basal autophagy in preventing senescence of muscle satellite cells, thus maintaining stemness, has been reported previously (García-Prat et al. 2016). However, in addition to this autonomous activity of autophagy, autophagy affects a nonautonomous aspect of senescence (i.e., the SASP) in the context of chronic stress-induced autophagy. In addition, autophagy also contributes to other senescence effectors, such as (epi)genetic modulation through Lamin B1 degradation (Dou et al. 2015; Lenain et al. 2015). As mentioned earlier in this review, autophagy-mediated degradation of Lamin B1 also leads to the formation of CCFs, which could activate the cGAS–STING–SASP axis (Fig. 3).
Another layer of regulation of the SASP by autophagy was identified recently. Autophagy is generally considered to be a nonselective bulk degradation machinery but involves some level of selectivity through autophagy receptor proteins, such as p62/SQSTM1 (Komatsu and Ichimura 2010). Kang et al. (2015) have shown that the transcription factor GATA4 positively controls the NFkB-driven SASP. They found that GATA4 is a substrate of p62-mediated selective autophagy, which, in contrast to general autophagy, is inhibited during senescence, indicating that selective autophagy can negatively regulate the inflammatory SASP. Thus, overall, autophagy appears to affect the SASP at multiple levels in a nonunidirectional manner (Fig. 3).
Conclusion
Senescence blocks the proliferation of unnecessary or unwanted cells in both physiological and pathological contexts. Due to this autonomous aspect, senescence has been proposed to be a therapeutic goal of cancer therapy. While the senescence arrest can be an intrinsic tumor suppressor mechanism, particularly at the early stages of tumorigenesis, this view turns out to be too simplistic. Senescent cells are metabolically active and have a profound impact (often deleterious in the long term) on their neighbors through their nonautonomous activities. Interestingly, however, these autonomous and nonautonomous programs appear to be mechanistically linked. While the causal relevance of epigenetic changes during senescence remains elusive, the distinct epigenetic landscape in senescence has been correlated with the SASP (Aird et al. 2016; Tasdemir et al. 2016; Parry et al. 2018). Loss of Lamin B1 appears to orchestrate high-order chromatin structural alterations, genomic instability, and the formation of CCFs, which in turn triggers the SASP (Sadaie et al. 2013; Shah et al. 2013; Dou et al. 2015, 2017). Autophagy, an autonomous effector by nature, also affects the SASP (Fig. 3). Further understanding of how the “irreversibility” of cell cycle exit is mechanistically and functionally coupled with nonautonomous effectors of senescence will be of great interest. Georgilis et al. (2018) recently identified a number of druggable targets, the inhibition of which can induce senescence without the inflammatory SASP. Such decoupling between cell cycle arrest and the SASP might provide a promising therapeutic modality for prosenescence cancer therapy, with reduced complications from the SASP.
Alternatively, senescence itself has been proposed recently to be a therapeutic target rather than a goal. After the seminal work by van Deursen and colleagues (Baker et al. 2011) showing that the genetic ablation of senescent cells provides beneficial effects on healthy life span in mice, a number of small molecules that selectively kill senescent cells (collectively called senolytics) have been developed (Zhu et al. 2015, 2016; Yosef et al. 2016; Baar et al. 2017; Kirkland and Tchkonia 2017). Mostly, these reagents have been used (Demaria et al. 2017) in the context of aging and tissue damage (Xu et al. 2018). In the cancer context, it was shown that killing stromal senescent cells induced by chemotherapy reduces the associated side effects, such as cardiac and liver dysfunction and general fatigue (Baar et al. 2017; Demaria et al. 2017).
However, considering the potential adverse effects of the SASP derived from TIS, administering “senolytics adjuvants” in conjunction with prosenescence therapy in cancer might be a promising approach. In fact, it was shown that targeting the metabolic pathways increased in senescent cells (e.g., autophagy or glucose utilization) in TIS lymphoma cells leads to the selective killing of TIS cells and an improved treatment outcome (Dörr et al. 2013). A more recent study also suggests that selective targeting of TIS cells using sugar-coated (thus sensitive to β-galactosidase) nanoparticles encapsulating cytotoxic drugs is beneficial in xenograft models (Muñoz-Espín et al. 2018).
Senescence—or even the SASP—is mostly tumor-suppressive in the early stages, particularly when the senescent cells are eliminated by the immune system. This intrinsic phase might also be exploited for tumor therapy (Fig. 1). For example, it was shown that genetic inhibition of Notch signaling in the hepatocyte OIS model facilitates senescence surveillance (Hoare et al. 2016). We propose that OIS might be most relevant at the stage of oncogenic “amplification” (genetically or functionally) after somatic mutations, which are often found in normal tissues (Fig. 2; Risques and Kennedy 2018). Selective targeting of cells at this stage might also be beneficial for tumor therapy or prevention.
Acknowledgments
We thank all members of the Narita laboratory for helpful discussion, particularly Andrew Young for editing. The University of Cambridge, Cancer Research UK, and Hutchison Whampoa supported this work. The M.N. laboratory is funded by a Cancer Research UK Cambridge Institute core grant (C14303/A17197). M.N. is also supported by a Cancer Research UK Early Detection Pump Priming award (C20/A20976), the Medical Research Council (MR/R010013/1), and the Tokyo Tech World Research Hub Initiative.
Footnotes
Article is online at http://www.genesdev.org/cgi/doi/10.1101/gad.320937.118.
References
- Abe T, Barber GN. 2014. Cytosolic-DNA-mediated, STING-dependent proinflammatory gene induction necessitates canonical NF-κB activation through TBK1. J Virol 88: 5328–5341. 10.1128/JVI.00037-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Acosta JC, Gil J. 2012. Senescence: a new weapon for cancer therapy. Trends Cell Biol 22: 211–219. 10.1016/j.tcb.2011.11.006 [DOI] [PubMed] [Google Scholar]
- Acosta JC, O'Loghlen A, Banito A, Guijarro MV, Augert A, Raguz S, Fumagalli M, Da Costa M, Brown C, Popov N, et al. 2008. Chemokine signaling via the CXCR2 receptor reinforces senescence. Cell 133: 1006–1018. 10.1016/j.cell.2008.03.038 [DOI] [PubMed] [Google Scholar]
- Acosta JC, Banito A, Wuestefeld T, Georgilis A, Janich P, Morton JP, Athineos D, Kang T-W, Lasitschka F, Andrulis M, et al. 2013. A complex secretory program orchestrated by the inflammasome controls paracrine senescence. Nat Cell Biol 15: 978–990. 10.1038/ncb2784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adams JM, Harris AW, Pinkert CA, Corcoran LM, Alexander WS, Cory S, Palmiter RD, Brinster RL. 1985. The c-myc oncogene driven by immunoglobulin enhancers induces lymphoid malignancy in transgenic mice. Nature 318: 533–538. 10.1038/318533a0 [DOI] [PubMed] [Google Scholar]
- Aguirre AJ, Bardeesy N, Sinha M, Lopez L, Tuveson DA, Horner J, Redston MS, DePinho RA. 2003. Activated Kras and Ink4a/Arf deficiency cooperate to produce metastatic pancreatic ductal adenocarcinoma. Genes Dev 17: 3112–3126. 10.1101/gad.1158703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aird KM, Iwasaki O, Kossenkov AV, Tanizawa H, Fatkhutdinov N, Bitler BG, Le L, Alicea G, Yang T-L, Johnson FB, et al. 2016. HMGB2 orchestrates the chromatin landscape of senescence-associated secretory phenotype gene loci. J Cell Biol 215: 325–334. 10.1083/jcb.201608026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Artandi SE, Chang S, Lee SL, Alson S, Gottlieb GJ, Chin L, DePinho RA. 2000. Telomere dysfunction promotes non-reciprocal translocations and epithelial cancers in mice. Nature 406: 641–645. 10.1038/35020592 [DOI] [PubMed] [Google Scholar]
- Baar MP, Brandt RMC, Putavet DA, Klein JDD, Derks KWJ, Bourgeois BRM, Stryeck S, Rijksen Y, van Willigenburg H, Feijtel DA, et al. 2017. Targeted apoptosis of senescent cells restores tissue homeostasis in response to chemotoxicity and aging. Cell 169: 132–147.e16. 10.1016/j.cell.2017.02.031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker DJ, Sedivy JM. 2013. Probing the depths of cellular senescence. J Cell Biol 202: 11–13. 10.1083/jcb.201305155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker DJ, Wijshake T, Tchkonia T, LeBrasseur NK, Childs BG, van de Sluis B, Kirkland JL, van Deursen JM. 2011. Clearance of p16Ink4a-positive senescent cells delays ageing-associated disorders. Nature 479: 232–236. 10.1038/nature10600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker DJ, Childs BG, Durik M, Wijers ME, Sieben CJ, Zhong J, Saltness RA, Jeganathan KB, Verzosa GC, Pezeshki A, et al. 2016. Naturally occurring p16Ink4a-positive cells shorten healthy lifespan. Nature 530: 184–189. 10.1038/nature16932 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker DJ, Alimirah F, van Deursen JM, Campisi J, Hildesheim J. 2017. Oncogenic senescence: a multi-functional perspective. Oncotarget 8: 27661–27672. 10.18632/oncotarget.15742 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banito A, Rashid ST, Acosta JC, Li S, Pereira CF, Geti I, Pinho S, Silva JC, Azuara V, Walsh M, et al. 2009. Senescence impairs successful reprogramming to pluripotent stem cells. Genes Dev 23: 2134–2139. 10.1101/gad.1811609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barber GN. 2015. STING: infection, inflammation and cancer. Nat Rev Immunol 15: 760–770. 10.1038/nri3921 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beauséjour CM, Krtolica A, Galimi F, Narita M, Lowe SW, Yaswen P, Campisi J. 2003. Reversal of human cellular senescence: roles of the p53 and p16 pathways. EMBO J 22: 4212–4222. 10.1093/emboj/cdg417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berg J, Robson MC. 2003. Arresting cell cycles and the effect on wound healing. Surg Clin North Am 83: 509–520. 10.1016/S0039-6109(02)00195-0 [DOI] [PubMed] [Google Scholar]
- Besancenot R, Chaligné R, Tonetti C, Pasquier F, Marty C, Lécluse Y, Vainchenker W, Constantinescu SN, Giraudier S. 2010. A senescence-like cell-cycle arrest occurs during megakaryocytic maturation: implications for physiological and pathological megakaryocytic proliferation. PLoS Biol 8: e1000476 10.1371/journal.pbio.1000476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biran A, Perelmutter M, Gal H, Burton DGA, Ovadya Y, Vadai E, Geiger T, Krizhanovsky V. 2015. Senescent cells communicate via intercellular protein transfer. Genes Dev 29: 791–802. 10.1101/gad.259341.115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blasco MA. 2005. Telomeres and human disease: ageing, cancer and beyond. Nat Rev Genet 6: 611–622. 10.1038/nrg1656 [DOI] [PubMed] [Google Scholar]
- Blasco MA, Lee HW, Hande MP, Samper E, Lansdorp PM, DePinho RA, Greider CW. 1997. Telomere shortening and tumor formation by mouse cells lacking telomerase RNA. Cell 91: 25–34. 10.1016/S0092-8674(01)80006-4 [DOI] [PubMed] [Google Scholar]
- Bodnar AG, Ouellette M, Frolkis M, Holt SE, Chiu C-P, Morin GB, Harley CB, Shay JW, Lichtsteiner S, Wright WE. 1998. Extension of life-span by introduction of telomerase into normal human cells. Science 279: 349–352. 10.1126/science.279.5349.349 [DOI] [PubMed] [Google Scholar]
- Borkham-Kamphorst E, Schaffrath C, Van de Leur E, Haas U, Tihaa L, Meurer SK, Nevzorova YA, Liedtke C, Weiskirchen R. 2014. The anti-fibrotic effects of CCN1/CYR61 in primary portal myofibroblasts are mediated through induction of reactive oxygen species resulting in cellular senescence, apoptosis and attenuated TGF-β signaling. Biochim Biophys Acta 1843: 902–914. 10.1016/j.bbamcr.2014.01.023 [DOI] [PubMed] [Google Scholar]
- Bose D. 2017. cGAS/STING pathway in cancer: Jekyll and Hyde story of cancer immune response. Int J Mol Sci 18: 2456 10.3390/ijms18112456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braig M, Schmitt CA. 2006. Oncogene-induced senescence: putting the brakes on tumor development. Cancer Res 66: 2881–2884. 10.1158/0008-5472.CAN-05-4006 [DOI] [PubMed] [Google Scholar]
- Braig M, Lee S, Loddenkemper C, Rudolph C, Peters AHFM, Schlegelberger B, Stein H, Dörken B, Jenuwein T, Schmitt CA. 2005. Oncogene-induced senescence as an initial barrier in lymphoma development. Nature 436: 660–665. 10.1038/nature03841 [DOI] [PubMed] [Google Scholar]
- Burton DGA, Krizhanovsky V. 2014. Physiological and pathological consequences of cellular senescence. Cell Mol Life Sci 71: 4373–4386. 10.1007/s00018-014-1691-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buschbeck M, Hake SB. 2017. Variants of core histones and their roles in cell fate decisions, development and cancer. Nat Rev Mol Cell Biol 18: 299–314. 10.1038/nrm.2016.166 [DOI] [PubMed] [Google Scholar]
- Byun JS, Gardner K. 2013. Wounds that will not heal: pervasive cellular reprogramming in cancer. Am J Pathol 182: 1055–1064. 10.1016/j.ajpath.2013.01.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calvisi DF, Ladu S, Gorden A, Farina M, Conner EA, Lee J-S, Factor VM, Thorgeirsson SS. 2006. Ubiquitous activation of Ras and Jak/Stat pathways in human HCC. Gastroenterology 130: 1117–1128. 10.1053/j.gastro.2006.01.006 [DOI] [PubMed] [Google Scholar]
- Campisi J. 2001. Cellular senescence as a tumor-suppressor mechanism. Trends Cell Biol 11: S27–S31. 10.1016/S0962-8924(01)82148-6 [DOI] [PubMed] [Google Scholar]
- Campisi J. 2013. Aging, cellular senescence, and cancer. Annu Rev Physiol 75: 685–705. 10.1146/annurev-physiol-030212-183653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campisi J, d'Adda di Fagagna F. 2007. Cellular senescence: when bad things happen to good cells. Nat Rev Mol Cell Biol 8: 729–740. 10.1038/nrm2233 [DOI] [PubMed] [Google Scholar]
- Capell BC, Drake AM, Zhu J, Shah PP, Dou Z, Dorsey J, Simola DF, Donahue G, Sammons M, Rai TS, et al. 2016. MLL1 is essential for the senescence-associated secretory phenotype. Genes Dev 30: 321–336. 10.1101/gad.271882.115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandra T, Kirschner K, Thuret J-Y, Pope BD, Ryba T, Newman S, Ahmed K, Samarajiwa SA, Salama R, Carroll T, et al. 2012. Independence of repressive histone marks and chromatin compaction during senescent heterochromatic layer formation. Mol Cell 47: 203–214. 10.1016/j.molcel.2012.06.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandra T, Ewels PA, Schoenfelder S, Furlan-Magaril M, Wingett SW, Kirschner K, Thuret J-Y, Andrews S, Fraser P, Reik W. 2015. Global reorganization of the nuclear landscape in senescent cells. Cell Rep 10: 471–483. 10.1016/j.celrep.2014.12.055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang S. 2005. Modeling aging and cancer in the telomerase knockout mouse. Mutat Res 576: 39–53. 10.1016/j.mrfmmm.2004.08.020 [DOI] [PubMed] [Google Scholar]
- Chen J-H, Ozanne SE. 2006. Deep senescent human fibroblasts show diminished DNA damage foci but retain checkpoint capacity to oxidative stress. FEBS Lett 580: 6669–6673. 10.1016/j.febslet.2006.11.023 [DOI] [PubMed] [Google Scholar]
- Chen Z, Trotman LC, Shaffer D, Lin H-K, Dotan ZA, Niki M, Koutcher JA, Scher HI, Ludwig T, Gerald W, et al. 2005. Crucial role of p53-dependent cellular senescence in suppression of Pten-deficient tumorigenesis. Nature 436: 725–730. 10.1038/nature03918 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiche A, Le Roux I, Joest von M, Sakai H, Aguín SB, Cazin C, Salam R, Fiette L, Alegria O, Flamant P, et al. 2017. Injury-induced senescence enables in vivo reprogramming in skeletal muscle. Cell Stem Cell 20: 407–414.e4. 10.1016/j.stem.2016.11.020 [DOI] [PubMed] [Google Scholar]
- Chin L, Artandi SE, Shen Q, Tam A, Lee SL, Gottlieb GJ, Greider CW, DePinho RA. 1999. p53 deficiency rescues the adverse effects of telomere loss and cooperates with telomere dysfunction to accelerate carcinogenesis. Cell 97: 527–538. 10.1016/S0092-8674(00)80762-X [DOI] [PubMed] [Google Scholar]
- Chuprin A, Gal H, Biron-Shental T, Biran A, Amiel A, Rozenblatt S, Krizhanovsky V. 2013. Cell fusion induced by ERVWE1 or measles virus causes cellular senescence. Genes Dev 27: 2356–2366. 10.1101/gad.227512.113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciccia A, Elledge SJ. 2010. The DNA damage response: making it safe to play with knives. Mol Cell 40: 179–204. 10.1016/j.molcel.2010.09.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collado M, Serrano M. 2010. Senescence in tumours: evidence from mice and humans. Nat Rev Cancer 10: 51–57. 10.1038/nrc2772 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collado M, Gil J, Efeyan A, Guerra C, Schuhmacher AJ, Barradas M, Benguría A, Zaballos A, Flores JM, Barbacid M, et al. 2005. Tumour biology: senescence in premalignant tumours. Nature 436: 642 10.1038/436642a [DOI] [PubMed] [Google Scholar]
- Coppé J-P, Kauser K, Campisi J, Beauséjour CM. 2006. Secretion of vascular endothelial growth factor by primary human fibroblasts at senescence. J Biol Chem 281: 29568–29574. 10.1074/jbc.M603307200 [DOI] [PubMed] [Google Scholar]
- Coppé J-P, Patil CK, Rodier F, Sun Y, Muñoz DP, Goldstein J, Nelson PS, Desprez P-Y, Campisi J. 2008. Senescence-associated secretory phenotypes reveal cell-nonautonomous functions of oncogenic RAS and the p53 tumor suppressor. PLoS Biol 6: 2853–2868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Courtois-Cox S, Genther Williams SM, Reczek EE, Johnson BW, McGillicuddy LT, Johannessen CM, Hollstein PE, MacCollin M, Cichowski K. 2006. A negative feedback signaling network underlies oncogene-induced senescence. Cancer Cell 10: 459–472. 10.1016/j.ccr.2006.10.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Courtois-Cox S, Jones SL, Cichowski K. 2008. Many roads lead to oncogene-induced senescence. Oncogene 27: 2801–2809. 10.1038/sj.onc.1210950 [DOI] [PubMed] [Google Scholar]
- Criscione SW, Teo YV, Neretti N. 2016. The chromatin landscape of cellular senescence. Trends Genet 32: 751–761. 10.1016/j.tig.2016.09.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cruickshanks HA, McBryan T, Nelson DM, Vanderkraats ND, Shah PP, van Tuyn J, Singh Rai T, Brock C, Donahue G, Dunican DS, et al. 2013. Senescent cells harbour features of the cancer epigenome. Nat Cell Biol 15: 1495–1506. 10.1038/ncb2879 [DOI] [PMC free article] [PubMed] [Google Scholar]
- d'Adda di Fagagna F, Reaper PM, Clay-Farrace L, Fiegler H, Carr P, von Zglinicki T, Saretzki G, Carter NP, Jackson SP. 2003. A DNA damage checkpoint response in telomere-initiated senescence. Nature 426: 194–198. 10.1038/nature02118 [DOI] [PubMed] [Google Scholar]
- Deaton AM, Bird A. 2011. CpG islands and the regulation of transcription. Genes Dev 25: 1010–1022. 10.1101/gad.2037511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Cecco M, Criscione SW, Peckham EJ, Hillenmeyer S, Hamm EA, Manivannan J, Peterson AL, Kreiling JA, Neretti N, Sedivy JM. 2013a. Genomes of replicatively senescent cells undergo global epigenetic changes leading to gene silencing and activation of transposable elements. Aging Cell 12: 247–256. 10.1111/acel.12047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Cecco M, Criscione SW, Peterson AL, Neretti N, Sedivy JM, Kreiling JA. 2013b. Transposable elements become active and mobile in the genomes of aging mammalian somatic tissues. Aging (Albany NY) 5: 867–883. 10.18632/aging.100621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demaria M, Ohtani N, Youssef SA, Rodier F, Toussaint W, Mitchell JR, Laberge R-M, Vijg J, Van Steeg H, Dollé MET, et al. 2014. An essential role for senescent cells in optimal wound healing through secretion of PDGF-AA. Dev Cell 31: 722–733. 10.1016/j.devcel.2014.11.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demaria M, O'Leary MN, Chang J, Shao L, Liu S, Alimirah F, Koenig K, Le C, Mitin N, Deal AM, et al. 2017. Cellular senescence promotes adverse effects of chemotherapy and cancer relapse. Cancer Discov 7: 165–176. 10.1158/2159-8290.CD-16-0241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng Q, Liao R, Wu B-L, Sun P. 2004. High intensity ras signaling induces premature senescence by activating p38 pathway in primary human fibroblasts. J Biol Chem 279: 1050–1059. 10.1074/jbc.M308644200 [DOI] [PubMed] [Google Scholar]
- Deng Y, Chan SS, Chang S. 2008. Telomere dysfunction and tumour suppression: the senescence connection. Nat Rev Cancer 8: 450–458. 10.1038/nrc2393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng L, Liang H, Xu M, Yang X, Burnette B, Arina A, Li X-D, Mauceri H, Beckett M, Darga T, et al. 2014. STING-dependent cytosolic DNA sensing promotes radiation-induced type I interferon-dependent antitumor immunity in immunogenic tumors. Immunity 41: 843–852. 10.1016/j.immuni.2014.10.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dillinger S, Straub T, Németh A. 2017. Nucleolus association of chromosomal domains is largely maintained in cellular senescence despite massive nuclear reorganisation. PLoS ONE 12: e0178821 10.1371/journal.pone.0178821 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dörr JR, Yu Y, Milanovic M, Beuster G, Zasada C, Däbritz JHM, Lisec J, Lenze D, Gerhardt A, Schleicher K, et al. 2013. Synthetic lethal metabolic targeting of cellular senescence in cancer therapy. Nature 501: 421–425. 10.1038/nature12437 [DOI] [PubMed] [Google Scholar]
- Dou Z, Xu C, Donahue G, Shimi T, Pan J-A, Zhu J, Ivanov A, Capell BC, Drake AM, Shah PP, et al. 2015. Autophagy mediates degradation of nuclear lamina. Nature 527: 105–109. 10.1038/nature15548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dou Z, Ghosh K, Vizioli MG, Zhu J, Sen P, Wangensteen KJ, Simithy J, Lan Y, Lin Y, Zhou Z, et al. 2017. Cytoplasmic chromatin triggers inflammation in senescence and cancer. Nature 550: 402–406. 10.1038/nature24050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Downward J. 2003. Targeting RAS signalling pathways in cancer therapy. Nat Rev Cancer 3: 11–22. 10.1038/nrc969 [DOI] [PubMed] [Google Scholar]
- DuPage M, Dooley AL, Jacks T. 2009. Conditional mouse lung cancer models using adenoviral or lentiviral delivery of Cre recombinase. Nat Protoc 4: 1064–1072. 10.1038/nprot.2009.95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dvorak HF. 1986. Tumors: wounds that do not heal. Similarities between tumor stroma generation and wound healing. N Engl J Med 315: 1650–1659. 10.1056/NEJM198612253152606 [DOI] [PubMed] [Google Scholar]
- Dvorak HF. 2015. Tumors: wounds that do not heal—redux. Cancer Immunol Res 3: 1–11. 10.1158/2326-6066.CIR-14-0209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Efeyan A, Zoncu R, Sabatini DM. 2012. Amino acids and mTORC1: from lysosomes to disease. Trends Mol Med 18: 524–533. 10.1016/j.molmed.2012.05.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feldser DM, Greider CW. 2007. Short telomeres limit tumor progression in vivo by inducing senescence. Cancer Cell 11: 461–469. 10.1016/j.ccr.2007.02.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finney R, Bishop J. 1993. Predisposition to neoplastic transformation caused by gene replacement of H-ras1. Science 260: 1524–1527. 10.1126/science.8502998 [DOI] [PubMed] [Google Scholar]
- Freund A, Patil CK, Campisi J. 2011. p38MAPK is a novel DNA damage response-independent regulator of the senescence-associated secretory phenotype. EMBO J 30: 1536–1548. 10.1038/emboj.2011.69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freund A, Laberge R-M, Demaria M, Campisi J. 2012. Lamin B1 loss is a senescence-associated biomarker. Mol Biol Cell 23: 2066–2075. 10.1091/mbc.e11-10-0884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fumagalli M, Rossiello F, Clerici M, Barozzi S, Cittaro D, Kaplunov JM, Bucci G, Dobreva M, Matti V, Beauséjour CM, et al. 2012. Telomeric DNA damage is irreparable and causes persistent DNA-damage-response activation. Nat Cell Biol 14: 355–365. 10.1038/ncb2466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Funayama R, Saito M, Tanobe H, Ishikawa F. 2006. Loss of linker histone H1 in cellular senescence. J Cell Biol 175: 869–880. 10.1083/jcb.200604005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- García-Prat L, Martínez-Vicente M, Perdiguero E, Ortet L, Rodríguez-Ubreva J, Rebollo E, Ruiz-Bonilla V, Gutarra S, Ballestar E, Serrano AL, et al. 2016. Autophagy maintains stemness by preventing senescence. Nature 529: 37–42. 10.1038/nature16187 [DOI] [PubMed] [Google Scholar]
- Georgilis A, Klotz S, Hanley CJ, Herranz N, Weirich B, Morancho B, Leote AC, D'Artista L, Gallage S, Seehawer M, et al. 2018. PTBP1-mediated alternative splicing regulates the inflammatory secretome and the pro-tumorigenic effects of senescent cells. Cancer Cell 34: 85–102.e9. 10.1016/j.ccell.2018.06.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerland L-M, Peyrol S, Lallemand C, Branche R, Magaud J-P, Ffrench M. 2003. Association of increased autophagic inclusions labeled for β-galactosidase with fibroblastic aging. Exp Gerontol 38: 887–895. 10.1016/S0531-5565(03)00132-3 [DOI] [PubMed] [Google Scholar]
- Glück S, Guey B, Gulen MF, Wolter K, Kang T-W, Schmacke NA, Bridgeman A, Rehwinkel J, Zender L, Ablasser A. 2017. Innate immune sensing of cytosolic chromatin fragments through cGAS promotes senescence. Nat Cell Biol 19: 1061–1070. 10.1038/ncb3586 [DOI] [PMC free article] [PubMed] [Google Scholar]
- González-Suárez E, Samper E, Flores JM, Blasco MA. 2000. Telomerase-deficient mice with short telomeres are resistant to skin tumorigenesis. Nat Genet 26: 114–117. 10.1038/79089 [DOI] [PubMed] [Google Scholar]
- Guerra C, Mijimolle N, Dhawahir A, Dubus P, Barradas M, Serrano M, Campuzano V, Barbacid M. 2003. Tumor induction by an endogenous K-ras oncogene is highly dependent on cellular context. Cancer Cell 4: 111–120. 10.1016/S1535-6108(03)00191-0 [DOI] [PubMed] [Google Scholar]
- Hayflick L. 1965. The limited in vitro lifetime of human diploid cell strains. Exp Cell Res 37: 614–636. 10.1016/0014-4827(65)90211-9 [DOI] [PubMed] [Google Scholar]
- He S, Sharpless NE. 2017. Senescence in health and disease. Cell 169: 1000–1011. 10.1016/j.cell.2017.05.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- He Q, Au B, Kulkarni M, Shen Y, Lim KJ, Maimaiti J, Wong CK, Luijten MNH, Chong HC, Lim EH, et al. 2018. Chromosomal instability-induced senescence potentiates cell non-autonomous tumourigenic effects. Oncogenesis 7: 62 10.1038/s41389-018-0072-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helman A, Avrahami D, Klochendler A, Glaser B, Kaestner KH, Ben-Porath I, Dor Y. 2016. Effects of ageing and senescence on pancreatic β-cell function. Diabetes Obes Metab 18: 58–62. 10.1111/dom.12719 [DOI] [PubMed] [Google Scholar]
- Herkel J, Jagemann B, Wiegard C, Lazaro JFG, Lueth S, Kanzler S, Blessing M, Schmitt E, Lohse AW. 2003. MHC class II-expressing hepatocytes function as antigen-presenting cells and activate specific CD4 T lymphocyutes. Hepatology 37: 1079–1085. 10.1053/jhep.2003.50191 [DOI] [PubMed] [Google Scholar]
- Hernandez-Segura A, de Jong TV, Melov S, Guryev V, Campisi J, Demaria M. 2017. Unmasking transcriptional heterogeneity in senescent cells. Curr Biol 27: 2652–2660.e4. 10.1016/j.cub.2017.07.033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herranz N, Gallage S, Mellone M, Wuestefeld T, Klotz S, Hanley CJ, Raguz S, Acosta JC, Innes AJ, Banito A, et al. 2015. mTOR regulates MAPKAPK2 translation to control the senescence-associated secretory phenotype. Nat Cell Biol 17: 1205–1217. 10.1038/ncb3225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hewitt G, Jurk D, Marques FDM, Correia-Melo C, Hardy T, Gackowska A, Anderson R, Taschuk M, Mann J, Passos JF. 2012. Telomeres are favoured targets of a persistent DNA damage response in ageing and stress-induced senescence. Nat Commun 3: 708 10.1038/ncomms1708 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hingorani SR, Petricoin EF, Maitra A, Rajapakse V, King C, Jacobetz MA, Ross S, Conrads TP, Veenstra TD, Hitt BA, et al. 2003. Preinvasive and invasive ductal pancreatic cancer and its early detection in the mouse. Cancer Cell 4: 437–450. 10.1016/S1535-6108(03)00309-X [DOI] [PubMed] [Google Scholar]
- Hoare MW, Narita M. 2018. The power behind the throne: senescence and the hallmarks of cancer. Annu Rev Cancer Biol 2: 175–194. 10.1146/annurev-cancerbio-030617-050352 [DOI] [Google Scholar]
- Hoare M, Young ARJ, Narita M. 2011. Autophagy in cancer: having your cake and eating it. Semin Cancer Biol 21: 397–404. [DOI] [PubMed] [Google Scholar]
- Hoare M, Ito Y, Kang T-W, Weekes MP, Matheson NJ, Patten DA, Shetty S, Parry AJ, Menon S, Salama R, et al. 2016. NOTCH1 mediates a switch between two distinct secretomes during senescence. Nat Cell Biol 18: 979–992. 10.1038/ncb3397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hobbs GA, Der CJ, Rossman KL. 2016. RAS isoforms and mutations in cancer at a glance. J Cell Sci 129: 1287–1292. 10.1242/jcs.182873 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong H, Takahashi K, Ichisaka T, Aoi T, Kanagawa O, Nakagawa M, Okita K, Yamanaka S. 2009. Suppression of induced pluripotent stem cell generation by the p53–p21 pathway. Nature 460: 1132–1135. 10.1038/nature08235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iannello A, Thompson TW, Ardolino M, Lowe SW, Raulet DH. 2013. p53-dependent chemokine production by senescent tumor cells supports NKG2D-dependent tumor elimination by natural killer cells. J Exp Med 210: 2057–2069. 10.1084/jem.20130783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito Y, Hoare M, Narita M. 2017. Spatial and temporal control of senescence. Trends Cell Biol 27: 820–832. 10.1016/j.tcb.2017.07.004 [DOI] [PubMed] [Google Scholar]
- Ivanov A, Pawlikowski J, Manoharan I, van Tuyn J, Nelson DM, Rai TS, Shah PP, Hewitt G, Korolchuk VI, Passos JF, et al. 2013. Lysosome-mediated processing of chromatin in senescence. J Cell Biol 202: 129–143. 10.1083/jcb.201212110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson EL, Willis N, Mercer K, Bronson RT, Crowley D, Montoya R, Jacks T, Tuveson DA. 2001. Analysis of lung tumor initiation and progression using conditional expression of oncogenic K-ras. Genes Dev 15: 3243–3248. 10.1101/gad.943001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jafri MA, Ansari SA, Alqahtani MH, Shay JW. 2016. Roles of telomeres and telomerase in cancer, and advances in telomerase-targeted therapies. Genome Med 8: 69 10.1186/s13073-016-0324-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jun J-I, Lau LF. 2010a. Cellular senescence controls fibrosis in wound healing. Aging (Albany NY) 2: 627–631. 10.18632/aging.100201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jun J-I, Lau LF. 2010b. The matricellular protein CCN1 induces fibroblast senescence and restricts fibrosis in cutaneous wound healing. Nat Cell Biol 12: 676–685. 10.1038/ncb2070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Junttila MR, Karnezis AN, Garcia D, Madriles F, Kortlever RM, Rostker F, Brown-Swigart L, Pham DM, Seo Y, Evan GI, et al. 2010. Selective activation of p53-mediated tumour suppression in high-grade tumours. Nature 468: 567–571. 10.1038/nature09526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang T-W, Yevsa T, Woller N, Hoenicke L, Wuestefeld T, Dauch D, Hohmeyer A, Gereke M, Rudalska R, Potapova A, et al. 2011. Senescence surveillance of pre-malignant hepatocytes limits liver cancer development. Nature 479: 547–551. 10.1038/nature10599 [DOI] [PubMed] [Google Scholar]
- Kang C, Xu Q, Martin TD, Li MZ, Demaria M, Aron L, Lu T, Yankner BA, Campisi J, Elledge SJ. 2015. The DNA damage response induces inflammation and senescence by inhibiting autophagy of GATA4. Science 349: aaa5612 10.1126/science.aaa5612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawamura T, Suzuki J, Wang YV, Menendez S, Morera LB, Raya A, Wahl GM, Izpisua Belmonte JC. 2009. Linking the p53 tumour suppressor pathway to somatic cell reprogramming. Nature 460: 1140–1144. 10.1038/nature08311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim NW, Piatyszek MA, Prowse KR, Harley CB, West MD, Ho PL, Coviello GM, Wright WE, Weinrich SL, Shay JW. 1994. Specific association of human telomerase activity with immortal cells and cancer. Science 266: 2011–2015. 10.1126/science.7605428 [DOI] [PubMed] [Google Scholar]
- Kim K-H, Chen C-C, Monzon RI, Lau LF. 2013. Matricellular protein CCN1 promotes regression of liver fibrosis through induction of cellular senescence in hepatic myofibroblasts. Mol Cell Biol 33: 2078–2090. 10.1128/MCB.00049-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim YH, Choi YW, Lee J, Soh EY, Kim J-H, Park TJ. 2017. Senescent tumor cells lead the collective invasion in thyroid cancer. Nat Commun 8: 15208 10.1038/ncomms15208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirkland JL, Tchkonia T. 2017. Cellular senescence: a translational perspective. EBioMedicine 21: 21–28. 10.1016/j.ebiom.2017.04.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirkwood TB, Austad SN. 2000. Why do we age? Nature 408: 233–238. 10.1038/35041682 [DOI] [PubMed] [Google Scholar]
- Komatsu M, Ichimura Y. 2010. Selective autophagy regulates various cellular functions. Genes Cells 15: 923–933. 10.1111/j.1365-2443.2010.01433.x [DOI] [PubMed] [Google Scholar]
- Kong X, Feng D, Wang H, Hong F, Bertola A, Wang F-S, Gao B. 2012. Interleukin-22 induces hepatic stellate cell senescence and restricts liver fibrosis in mice. Hepatology 56: 1150–1159. 10.1002/hep.25744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kortlever RM, Higgins PJ, Bernards R. 2006. Plasminogen activator inhibitor-1 is a critical downstream target of p53 in the induction of replicative senescence. Nat Cell Biol 8: 877–884. 10.1038/ncb1448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krizhanovsky V, Lowe SW. 2009. Stem cells: the promises and perils of p53. Nature 460: 1085–1086. 10.1038/4601085a [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krizhanovsky V, Yon M, Dickins RA, Hearn S, Simon J, Miething C, Yee H, Zender L, Lowe SW. 2008. Senescence of activated stellate cells limits liver fibrosis. Cell 134: 657–667. 10.1016/j.cell.2008.06.049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krtolica A, Parrinello S, Lockett S, Desprez PY, Campisi J. 2001. Senescent fibroblasts promote epithelial cell growth and tumorigenesis: a link between cancer and aging. Proc Natl Acad Sci 98: 12072–12077. 10.1073/pnas.211053698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuilman T, Peeper DS. 2009. Senescence-messaging secretome: SMS-ing cellular stress. Nat Rev Cancer 9: 81–94. 10.1038/nrc2560 [DOI] [PubMed] [Google Scholar]
- Kuilman T, Michaloglou C, Vredeveld LCW, Douma S, van Doorn R, Desmet CJ, Aarden LA, Mooi WJ, Peeper DS. 2008. Oncogene-induced senescence relayed by an interleukin-dependent inflammatory network. Cell 133: 1019–1031. 10.1016/j.cell.2008.03.039 [DOI] [PubMed] [Google Scholar]
- Kuilman T, Michaloglou C, Mooi WJ, Peeper DS. 2010. The essence of senescence. Genes Dev 24: 2463–2479. 10.1101/gad.1971610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurz DJ, Decary S, Hong Y, Erusalimsky JD. 2000. Senescence-associated β-galactosidase reflects an increase in lysosomal mass during replicative ageing of human endothelial cells. J Cell Sci 113: 3613–3622. [DOI] [PubMed] [Google Scholar]
- Kwon Y, Kim JW, Jeoung JA, Kim M-S, Kang C. 2017. Autophagy is pro-senescence when seen in close-up, but anti-senescence in long-shot. Mol Cells 40: 607–612. 10.14348/molcells.2017.0151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laberge R-M, Sun Y, Orjalo AV, Patil CK, Freund A, Zhou L, Curran SC, Davalos AR, Wilson-Edell KA, Liu S, et al. 2015. MTOR regulates the pro-tumorigenic senescence-associated secretory phenotype by promoting IL1A translation. Nat Cell Biol 17: 1049–1061. 10.1038/ncb3195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazzerini Denchi E, Attwooll C, Pasini D, Helin K. 2005. Deregulated E2F activity induces hyperplasia and senescence-like features in the mouse pituitary gland. Mol Cell Biol 25: 2660–2672. 10.1128/MCB.25.7.2660-2672.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lecot P, Alimirah F, Desprez P-Y, Campisi J, Wiley C. 2016. Context-dependent effects of cellular senescence in cancer development. Br J Cancer 114: 1180–1184. 10.1038/bjc.2016.115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee YS, Bae S-C. 2016. How do K-RAS-activated cells evade cellular defense mechanisms? Oncogene 35: 827–832. 10.1038/onc.2015.153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee BY, Han JA, Im JS, Morrone A, Johung K, Goodwin EC, Kleijer WJ, DiMaio D, Hwang ES. 2006. Senescence-associated β-galactosidase is lysosomal β-galactosidase. Aging Cell 5: 187–195. 10.1111/j.1474-9726.2006.00199.x [DOI] [PubMed] [Google Scholar]
- Lenain C, Gusyatiner O, Douma S, van den Broek B, Peeper DS. 2015. Autophagy-mediated degradation of nuclear envelope proteins during oncogene-induced senescence. Carcinogenesis 36: 1263–1274. 10.1093/carcin/bgv124 [DOI] [PubMed] [Google Scholar]
- Li T, Chen ZJ. 2018. The cGAS–cGAMP–STING pathway connects DNA damage to inflammation, senescence, and cancer. J Exp Med 215: 1287–1299. 10.1084/jem.20180139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Collado M, Villasante A, Strati K, Ortega S, Cañamero M, Blasco MA, Serrano M. 2009. The Ink4/Arf locus is a barrier for iPS cell reprogramming. Nature 460: 1136–1139. 10.1038/nature08290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin AW, Barradas M, Stone JC, van Aelst L, Serrano M, Lowe SW. 1998. Premature senescence involving p53 and p16 is activated in response to constitutive MEK/MAPK mitogenic signaling. Genes Dev 12: 3008–3019. 10.1101/gad.12.19.3008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Johnson SM, Fedoriw Y, Rogers AB, Yuan H, Krishnamurthy J, Sharpless NE. 2011. Expression of p16INK4a prevents cancer and promotes aging in lymphocytes. Blood 117: 3257–3267. 10.1182/blood-2010-09-304402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- López-Otín C, Blasco MA, Partridge L, Serrano M, Kroemer G. 2013. The hallmarks of aging. Cell 153: 1194–1217. 10.1016/j.cell.2013.05.039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowe SW, Cepero E, Evan G. 2004. Intrinsic tumour suppression. Nature 432: 307–315. 10.1038/nature03098 [DOI] [PubMed] [Google Scholar]
- Lujambio A, Akkari L, Simon J, Grace D, Tschaharganeh DF, Bolden JE, Zhao Z, Thapar V, Joyce JA, Krizhanovsky V, et al. 2013. Non-cell-autonomous tumor suppression by p53. Cell 153: 449–460. 10.1016/j.cell.2013.03.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malaquin N, Martinez A, Rodier F. 2016. Keeping the senescence secretome under control: molecular reins on the senescence-associated secretory phenotype. Exp Gerontol 82: 39–49. 10.1016/j.exger.2016.05.010 [DOI] [PubMed] [Google Scholar]
- Mallette FA, Gaumont-Leclerc M-F, Ferbeyre G. 2007. The DNA damage signaling pathway is a critical mediator of oncogene-induced senescence. Genes Dev 21: 43–48. 10.1101/gad.1487307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marión RM, Strati K, Li H, Murga M, Blanco R, Ortega S, Fernandez-Capetillo O, Serrano M, Blasco MA. 2009. A p53-mediated DNA damage response limits reprogramming to ensure iPS cell genomic integrity. Nature 460: 1149–1153. 10.1038/nature08287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marthandan S, Menzel U, Priebe S, Groth M, Guthke R, Platzer M, Hemmerich P, Kaether C, Diekmann S. 2016. Conserved genes and pathways in primary human fibroblast strains undergoing replicative and radiation induced senescence. Biol Res 49: 34 10.1186/s40659-016-0095-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michaloglou C, Vredeveld LCW, Soengas MS, Denoyelle C, Kuilman T, van der Horst CMAM, Majoor DM, Shay JW, Mooi WJ, Peeper DS. 2005. BRAFE600-associated senescence-like cell cycle arrest of human naevi. Nature 436: 720–724. 10.1038/nature03890 [DOI] [PubMed] [Google Scholar]
- Milanovic M, Fan DNY, Belenki D, Däbritz JHM, Zhao Z, Yu Y, Dörr JR, Dimitrova L, Lenze D, Monteiro Barbosa IA, et al. 2018. Senescence-associated reprogramming promotes cancer stemness. Nature 553: 96–100. 10.1038/nature25167 [DOI] [PubMed] [Google Scholar]
- Mosteiro L, Pantoja C, Alcazar N, Marión RM, Chondronasiou D, Rovira M, Fernandez-Marcos PJ, Muñoz-Martin M, Blanco-Aparicio C, Pastor J, et al. 2016. Tissue damage and senescence provide critical signals for cellular reprogramming in vivo. Science 354: aaf4445 10.1126/science.aaf4445 [DOI] [PubMed] [Google Scholar]
- Muñoz-Espín D, Serrano M. 2014. Cellular senescence: from physiology to pathology. Nat Rev Mol Cell Biol 15: 482–496. 10.1038/nrm3823 [DOI] [PubMed] [Google Scholar]
- Muñoz-Espín D, Cañamero M, Maraver A, Gómez-López G, Contreras J, Murillo-Cuesta S, Rodríguez-Baeza A, Varela-Nieto I, Ruberte J, Collado M, et al. 2013. Programmed cell senescence during mammalian embryonic development. Cell 155: 1104–1118. 10.1016/j.cell.2013.10.019 [DOI] [PubMed] [Google Scholar]
- Muñoz-Espín D, Rovira M, Galiana I, Giménez C, Lozano-Torres B, Paez-Ribes M, Llanos S, Chaib S, Muñoz-Martin M, Ucero AC, et al. 2018. A versatile drug delivery system targeting senescent cells. EMBO Mol Med 10: e9355 10.15252/emmm.201809355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nacarelli T, Liu P, Zhang R. 2017. Epigenetic basis of cellular senescence and its implications in aging. Genes 8: E343 10.3390/genes8120343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nardella C, Clohessy JG, Alimonti A, Pandolfi PP. 2011. Pro-senescence therapy for cancer treatment. Nat Rev Cancer 11: 503–511. 10.1038/nrc3057 [DOI] [PubMed] [Google Scholar]
- Narita M, Lowe SW. 2005. Senescence comes of age. Nat Med 11: 920–922. 10.1038/nm0905-920 [DOI] [PubMed] [Google Scholar]
- Narita M, Nuñez S, Heard E, Narita M, Lin AW, Hearn SA, Spector DL, Hannon GJ, Lowe SW. 2003. Rb-mediated heterochromatin formation and silencing of E2F target genes during cellular senescence. Cell 113: 703–716. 10.1016/S0092-8674(03)00401-X [DOI] [PubMed] [Google Scholar]
- Narita M, Narita M, Krizhanovsky V, Nuñez S, Chicas A, Hearn SA, Myers MP, Lowe SW. 2006. A novel role for high-mobility group a proteins in cellular senescence and heterochromatin formation. Cell 126: 503–514. 10.1016/j.cell.2006.05.052 [DOI] [PubMed] [Google Scholar]
- Narita M, Young ARJ, Arakawa S, Samarajiwa SA, Nakashima T, Yoshida S, Hong S, Berry LS, Reichelt S, Ferreira M, et al. 2011. Spatial coupling of mTOR and autophagy augments secretory phenotypes. Science 332: 966–970. 10.1126/science.1205407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson G, Wordsworth J, Wang C, Jurk D, Lawless C, Martin-Ruiz C, Zglinicki von T. 2012. A senescent cell bystander effect: senescence-induced senescence. Aging Cell 11: 345–349. 10.1111/j.1474-9726.2012.00795.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orjalo AV, Bhaumik D, Gengler BK, Scott GK, Campisi J. 2009. Cell surface-bound IL-1α is an upstream regulator of the senescence-associated IL-6/IL-8 cytokine network. Proc Natl Acad Sci 106: 17031–17036. 10.1073/pnas.0905299106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Sullivan RJ, Kubicek S, Schreiber SL, Karlseder J. 2010. Reduced histone biosynthesis and chromatin changes arising from a damage signal at telomeres. Nat Struct Mol Biol 17: 1218–1225. 10.1038/nsmb.1897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Padeken J, Heun P. 2014. Nucleolus and nuclear periphery: velcro for heterochromatin. Curr Opin Cell Biol 28: 54–60. 10.1016/j.ceb.2014.03.001 [DOI] [PubMed] [Google Scholar]
- Pal S, Tyler JK. 2016. Epigenetics and aging. Sci Adv 2: e1600584 10.1126/sciadv.1600584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmero I, Pantoja C, Serrano M. 1998. p19ARF links the tumour suppressor p53 to Ras. Nature 395: 125–126. 10.1038/25870 [DOI] [PubMed] [Google Scholar]
- Parrinello S, Coppé J-P, Krtolica A, Campisi J. 2005. Stromal–epithelial interactions in aging and cancer: senescent fibroblasts alter epithelial cell differentiation. J Cell Sci 118: 485–496. 10.1242/jcs.01635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parry AJ, Narita M. 2016. Old cells, new tricks: chromatin structure in senescence. Mamm Genome 27: 320–331. 10.1007/s00335-016-9628-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parry AJ, Hoare M, Bihary D, Hänsel-Hertsch R, Smith S, Tomimatsu K, Mannion E, Smith A, D'Santos P, Russell IA, et al. 2018. NOTCH-mediated non-cell autonomous regulation of chromatin structure during senescence. Nat Commun 9: 1840 10.1038/s41467-018-04283-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Passos JF, Saretzki G, Ahmed S, Nelson G, Richter T, Peters H, Wappler I, Birket MJ, Harold G, Schaeuble K, et al. 2007. Mitochondrial dysfunction accounts for the stochastic heterogeneity in telomere-dependent senescence. PLoS Biol 5: e110 10.1371/journal.pbio.0050110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Passos JF, Nelson G, Wang C, Richter T, Simillion C, Proctor CJ, Miwa S, Olijslagers S, Hallinan J, Wipat A, et al. 2010. Feedback between p21 and reactive oxygen production is necessary for cell senescence. Mol Syst Biol 6: 347 10.1038/msb.2010.5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patschan S, Chen J, Polotskaia A, Mendelev N, Cheng J, Patschan D, Goligorsky MS. 2008. Lipid mediators of autophagy in stress-induced premature senescence of endothelial cells. Am J Physiol Heart Circ Physiol 294: H1119–H1129. 10.1152/ajpheart.00713.2007 [DOI] [PubMed] [Google Scholar]
- Pérez-Mancera PA, Young ARJ, Narita M. 2014. Inside and out: the activities of senescence in cancer. Nat Rev Cancer 14: 547–558. 10.1038/nrc3773 [DOI] [PubMed] [Google Scholar]
- Pitiyage GN, Slijepcevic P, Gabrani A, Chianea YG, Lim KP, Prime SS, Tilakaratne WM, Fortune F, Parkinson EK. 2011. Senescent mesenchymal cells accumulate in human fibrosis by a telomere-independent mechanism and ameliorate fibrosis through matrix metalloproteinases. J Pathol 223: 604–617. 10.1002/path.2839 [DOI] [PubMed] [Google Scholar]
- Quintanilla M, Brown K, Ramsden M, Balmain A. 1986. Carcinogen-specific mutation and amplification of Ha-ras during mouse skin carcinogenesis. Nature 322: 78–80. 10.1038/322078a0 [DOI] [PubMed] [Google Scholar]
- Risques RA, Kennedy SR. 2018. Aging and the rise of somatic cancer-associated mutations in normal tissues. PLoS Genet 14: e1007108 10.1371/journal.pgen.1007108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritschka B, Storer M, Mas A, Heinzmann F, Ortells MC, Morton JP, Sansom OJ, Zender L, Keyes WM. 2017. The senescence-associated secretory phenotype induces cellular plasticity and tissue regeneration. Genes Dev 31: 172–183. 10.1101/gad.290635.116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodier F, Coppé J-P, Patil CK, Hoeijmakers WAM, Muñoz DP, Raza SR, Freund A, Campeau E, Davalos AR, Campisi J. 2009. Persistent DNA damage signalling triggers senescence-associated inflammatory cytokine secretion. Nat Cell Biol 11: 973–979. 10.1038/ncb1909 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodier F, Muñoz DP, Teachenor R, Chu V, Le O, Bhaumik D, Coppé J-P, Campeau E, Beauséjour CM, Kim S-H, et al. 2011. DNA-SCARS: distinct nuclear structures that sustain damage-induced senescence growth arrest and inflammatory cytokine secretion. J Cell Sci 124: 68–81. 10.1242/jcs.071340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudolph KL, Chang S, Lee HW, Blasco M, Gottlieb GJ, Greider C, DePinho RA. 1999. Longevity, stress response, and cancer in aging telomerase-deficient mice. Cell 96: 701–712. 10.1016/S0092-8674(00)80580-2 [DOI] [PubMed] [Google Scholar]
- Sadaie M, Salama R, Carroll T, Tomimatsu K, Chandra T, Young ARJ, Narita M, Pérez-Mancera PA, Bennett DC, Chong H, et al. 2013. Redistribution of the Lamin B1 genomic binding profile affects rearrangement of heterochromatic domains and SAHF formation during senescence. Genes Dev 27: 1800–1808. 10.1101/gad.217281.113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakaki M, Ebihara Y, Okamura K, Nakabayashi K, Igarashi A, Matsumoto K, Hata K, Kobayashi Y, Maehara K. 2017. Potential roles of DNA methylation in the initiation and establishment of replicative senescence revealed by array-based methylome and transcriptome analyses. PLoS ONE 12: e0171431 10.1371/journal.pone.0171431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salama R, Sadaie M, Hoare M, Narita M. 2014. Cellular senescence and its effector programs. Genes Dev 28: 99–114. 10.1101/gad.235184.113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarkisian CJ, Keister BA, Stairs DB, Boxer RB, Moody SE, Chodosh LA. 2007. Dose-dependent oncogene-induced senescence in vivo and its evasion during mammary tumorigenesis. Nat Cell Biol 9: 493–505. 10.1038/ncb1567 [DOI] [PubMed] [Google Scholar]
- Schafer MJ, White TA, Iijima K, Haak AJ, Ligresti G, Atkinson EJ, Oberg AL, Birch J, Salmonowicz H, Zhu Y, et al. 2017. Cellular senescence mediates fibrotic pulmonary disease. Nat Commun 8: 14532 10.1038/ncomms14532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitt CA, Fridman JS, Yang M, Lee S, Baranov E, Hoffman RM, Lowe SW. 2002. A senescence program controlled by p53 and p16INK4a contributes to the outcome of cancer therapy. Cell 109: 335–346. 10.1016/S0092-8674(02)00734-1 [DOI] [PubMed] [Google Scholar]
- Serrano M, Lin AW, McCurrach ME, Beach D, Lowe SW. 1997. Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a. Cell 88: 593–602. 10.1016/S0092-8674(00)81902-9 [DOI] [PubMed] [Google Scholar]
- Sewing A, Wiseman B, Lloyd AC, Land H. 1997. High-intensity Raf signal causes cell cycle arrest mediated by p21Cip1. Mol Cell Biol 17: 5588–5597. 10.1128/MCB.17.9.5588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah PP, Donahue G, Otte GL, Capell BC, Nelson DM, Cao K, Aggarwala V, Cruickshanks HA, Rai TS, McBryan T, et al. 2013. Lamin B1 depletion in senescent cells triggers large-scale changes in gene expression and the chromatin landscape. Genes Dev 27: 1787–1799. 10.1101/gad.223834.113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharpless NE, DePinho RA. 2007. How stem cells age and why this makes us grow old. Nat Rev Mol Cell Biol 8: 703–713. 10.1038/nrm2241 [DOI] [PubMed] [Google Scholar]
- Shaw AT, Meissner A, Dowdle JA, Crowley D, Magendantz M, Ouyang C, Parisi T, Rajagopal J, Blank LJ, Bronson RT, et al. 2007. Sprouty-2 regulates oncogenic K-ras in lung development and tumorigenesis. Genes Dev 21: 694–707. 10.1101/gad.1526207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shay JW, Bacchetti S. 1997. A survey of telomerase activity in human cancer. Eur J Cancer 33: 787–791. 10.1016/S0959-8049(97)00062-2 [DOI] [PubMed] [Google Scholar]
- Shay JW, Wright WE. 2000. Hayflick, his limit, and cellular ageing. Nat Rev Mol Cell Biol 1: 72–76. 10.1038/35036093 [DOI] [PubMed] [Google Scholar]
- Shay JW, Wright WE. 2005. Senescence and immortalization: role of telomeres and telomerase. Carcinogenesis 26: 867–874. 10.1093/carcin/bgh296 [DOI] [PubMed] [Google Scholar]
- Shen H-M, Mizushima N. 2014. At the end of the autophagic road: an emerging understanding of lysosomal functions in autophagy. Trends Biochem Sci 39: 61–71. 10.1016/j.tibs.2013.12.001 [DOI] [PubMed] [Google Scholar]
- Shimi T, Butin-Israeli V, Adam SA, Hamanaka RB, Goldman AE, Lucas CA, Shumaker DK, Kosak ST, Chandel NS, Goldman RD. 2011. The role of nuclear lamin B1 in cell proliferation and senescence. Genes Dev 25: 2579–2593. 10.1101/gad.179515.111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song S, Peng P, Tang Z, Zhao J, Wu W, Li H, Shao M, Li L, Yang C, Duan F, et al. 2017. Decreased expression of STING predicts poor prognosis in patients with gastric cancer. Sci Rep 7: 39858 10.1038/srep39858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stergachis AB, Neph S, Reynolds A, Humbert R, Miller B, Paige SL, Vernot B, Cheng JB, Thurman RE, Sandstrom R, et al. 2013. Developmental fate and cellular maturity encoded in human regulatory DNA landscapes. Cell 154: 888–903. 10.1016/j.cell.2013.07.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storer M, Mas A, Robert-Moreno A, Pecoraro M, Ortells MC, Di Giacomo V, Yosef R, Pilpel N, Krizhanovsky V, Sharpe J, et al. 2013. Senescence is a developmental mechanism that contributes to embryonic growth and patterning. Cell 155: 1119–1130. 10.1016/j.cell.2013.10.041 [DOI] [PubMed] [Google Scholar]
- Swanson EC, Manning B, Zhang H, Lawrence JB. 2013. Higher-order unfolding of satellite heterochromatin is a consistent and early event in cell senescence. J Cell Biol 203: 929–942. 10.1083/jcb.201306073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taguchi J, Yamada Y. 2017. Unveiling the role of senescence-induced cellular plasticity. Cell Stem Cell 20: 293–294. 10.1016/j.stem.2017.02.001 [DOI] [PubMed] [Google Scholar]
- Takahashi A, Imai Y, Yamakoshi K, Kuninaka S, Ohtani N, Yoshimoto S, Hori S, Tachibana M, Anderton E, Takeuchi T, et al. 2012. DNA damage signaling triggers degradation of histone methyltransferases through APC/CCdh1 in senescent cells. Mol Cell 45: 123–131. 10.1016/j.molcel.2011.10.018 [DOI] [PubMed] [Google Scholar]
- Takahashi A, Loo TM, Okada R, Kamachi F, Watanabe Y, Wakita M, Watanabe S, Kawamoto S, Miyata K, Barber GN, et al. 2018. Downregulation of cytoplasmic DNases is implicated in cytoplasmic DNA accumulation and SASP in senescent cells. Nat Commun 9: 1249 10.1038/s41467-018-03555-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takai H, Smogorzewska A, De Lange T. 2003. DNA damage foci at dysfunctional telomeres. Curr Biol 13: 1549–1556. 10.1016/S0960-9822(03)00542-6 [DOI] [PubMed] [Google Scholar]
- Takasugi M, Okada R, Takahashi A, Virya Chen D, Watanabe S, Hara E. 2017. Small extracellular vesicles secreted from senescent cells promote cancer cell proliferation through EphA2. Nat Commun 8: 15729 10.1038/ncomms15728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tasdemir N, Banito A, Roe J-S, Alonso-Curbelo D, Camiolo M, Tschaharganeh DF, Huang C-H, Aksoy O, Bolden JE, Chen C-C, et al. 2016. BRD4 connects enhancer remodeling to senescence immune surveillance. Cancer Discov 6: 612–629. 10.1158/2159-8290.CD-16-0217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tato-Costa J, Casimiro S, Pacheco T, Pires R, Fernandes A, Alho I, Pereira P, Costa P, Castelo HB, Ferreira J, et al. 2016. Therapy-induced cellular senescence induces epithelial-to-mesenchymal transition and increases invasiveness in rectal cancer. Clin Colorectal Cancer 15: 170–178.e3. 10.1016/j.clcc.2015.09.003 [DOI] [PubMed] [Google Scholar]
- Tomimatsu K, Narita M. 2015. Translating the effects of mTOR on secretory senescence. Nat Cell Biol 17: 1230–1232. 10.1038/ncb3244 [DOI] [PubMed] [Google Scholar]
- Tuveson DA, Shaw AT, Willis NA, Silver DP, Jackson EL, Chang S, Mercer KL, Grochow R, Hock H, Crowley D, et al. 2004. Endogenous oncogenic K-rasG12D stimulates proliferation and widespread neoplastic and developmental defects. Cancer Cell 5: 375–387. 10.1016/S1535-6108(04)00085-6 [DOI] [PubMed] [Google Scholar]
- Tyner SD, Venkatachalam S, Choi J, Jones S, Ghebranious N, Igelmann H, Lu X, Soron G, Cooper B, Brayton C, et al. 2002. p53 mutant mice that display early ageing-associated phenotypes. Nature 415: 45–53. 10.1038/415045a [DOI] [PubMed] [Google Scholar]
- Utikal J, Polo JM, Stadtfeld M, Maherali N, Kulalert W, Walsh RM, Khalil A, Rheinwald JG, Hochedlinger K. 2009. Immortalization eliminates a roadblock during cellular reprogramming into iPS cells. Nature 460: 1145–1148. 10.1038/nature08285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vandal G, Geiling B, Dankort D. 2014. Ras effector mutant expression suggest a negative regulator inhibits lung tumor formation. PLoS ONE 9: e84745 10.1371/journal.pone.0084745 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Deursen JM. 2014. The role of senescent cells in ageing. Nature 509: 439–446. 10.1038/nature13193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Tuyn J, Jaber-Hijazi F, MacKenzie D, Cole JJ, Mann E, Pawlikowski JS, Rai TS, Nelson DM, McBryan T, Ivanov A, et al. 2017. Oncogene-expressing senescent melanocytes up-regulate MHC class II, a candidate melanoma suppressor function. J Invest Dermatol 137: 2197–2207. 10.1016/j.jid.2017.05.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaziri H, Benchimol S. 1998. Reconstitution of telomerase activity in normal human cells leads to elongation of telomeres and extended replicative life span. Curr Biol 8: 279–282. 10.1016/S0960-9822(98)70109-5 [DOI] [PubMed] [Google Scholar]
- Villeponteau B. 1997. The heterochromatin loss model of aging. Exp Gerontol 32: 383–394. 10.1016/S0531-5565(96)00155-6 [DOI] [PubMed] [Google Scholar]
- Wajapeyee N, Serra RW, Zhu X, Mahalingam M, Green MR. 2008. Oncogenic BRAF induces senescence and apoptosis through pathways mediated by the secreted protein IGFBP7. Cell 132: 363–374. 10.1016/j.cell.2007.12.032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiley CD, Flynn JM, Morrissey C, Lebofsky R, Shuga J, Dong X, Unger MA, Vijg J, Melov S, Campisi J. 2017. Analysis of individual cells identifies cell-to-cell variability following induction of cellular senescence. Aging Cell 16: 1043–1050. 10.1111/acel.12632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolstein JM, Lee DH, Michaud J, Buot V, Stefanchik B, Plotkin MD. 2010. INK4a knockout mice exhibit increased fibrosis under normal conditions and in response to unilateral ureteral obstruction. Am J Physiol Renal Physiol 299: F1486–F1495. 10.1152/ajprenal.00378.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woo S-R, Fuertes MB, Corrales L, Spranger S, Furdyna MJ, Leung MYK, Duggan R, Wang Y, Barber GN, Fitzgerald KA, et al. 2014. STING-dependent cytosolic DNA sensing mediates innate immune recognition of immunogenic tumors. Immunity 41: 830–842. 10.1016/j.immuni.2014.10.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia T, Konno H, Ahn J, Barber GN. 2016. Deregulation of STING signaling in colorectal carcinoma constrains DNA damage responses and correlates with tumorigenesis. Cell Rep 14: 282–297. 10.1016/j.celrep.2015.12.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie W, Kagiampakis I, Pan L, Zhang YW, Murphy L, Tao Y, Kong X, Kang B, Xia L, Carvalho FLF, et al. 2018. DNA methylation patterns separate senescence from transformation potential and indicate cancer risk. Cancer Cell 33: 309–321.e5. 10.1016/j.ccell.2018.01.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu M, Pirtskhalava T, Farr JN, Weigand BM, Palmer AK, Weivoda MM, Inman CL, Ogrodnik MB, Hachfeld CM, Fraser DG, et al. 2018. Senolytics improve physical function and increase lifespan in old age. Nat Med 24: 1246–1256. 10.1038/s41591-018-0092-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue W, Zender L, Miething C, Dickins RA, Hernando E, Krizhanovsky V, Cordon-Cardo C, Lowe SW. 2007. Senescence and tumour clearance is triggered by p53 restoration in murine liver carcinomas. Nature 445: 656–660. 10.1038/nature05529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang H, Wang H, Ren J, Chen Q, Chen ZJ. 2017a. cGAS is essential for cellular senescence. Proc Natl Acad Sci 114: E4612–E4620. 10.1073/pnas.1705499114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang L, Fang J, Chen J. 2017b. Tumor cell senescence response produces aggressive variants. Cell Death Discov 3: 17049 10.1038/cddiscovery.2017.49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yosef R, Pilpel N, Tokarsky-Amiel R, Biran A, Ovadya Y, Cohen S, Vadai E, Dassa L, Shahar E, Condiotti R, et al. 2016. Directed elimination of senescent cells by inhibition of BCL-W and BCL-XL. Nat Commun 7: 11190 10.1038/ncomms11190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshimoto S, Loo TM, Atarashi K, Kanda H, Sato S, Oyadomari S, Iwakura Y, Oshima K, Morita H, Hattori M, et al. 2013. Obesity-induced gut microbial metabolite promotes liver cancer through senescence secretome. Nature 499: 97–101. 10.1038/nature12347 [DOI] [PubMed] [Google Scholar]
- Young ARJ, Narita M, Ferreira M, Kirschner K, Sadaie M, Darot JFJ, Tavaré S, Arakawa S, Shimizu S, Watt FM, et al. 2009. Autophagy mediates the mitotic senescence transition. Genes Dev 23: 798–803. 10.1101/gad.519709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu L, McPhee CK, Zheng L, Mardones GA, Rong Y, Peng J, Mi N, Zhao Y, Liu Z, Wan F, et al. 2010. Termination of autophagy and reformation of lysosomes regulated by mTOR. Nature 465: 942–946. 10.1038/nature09076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang R, Poustovoitov MV, Ye X, Santos HA, Chen W, Daganzo SM, Erzberger JP, Serebriiskii IG, Canutescu AA, Dunbrack RL, et al. 2005. Formation of MacroH2A-containing senescence-associated heterochromatin foci and senescence driven by ASF1a and HIRA. Dev Cell 8: 19–30. 10.1016/j.devcel.2004.10.019 [DOI] [PubMed] [Google Scholar]
- Zhang R, Chen W, Adams PD. 2007. Molecular dissection of formation of senescence-associated heterochromatin foci. Mol Cell Biol 27: 2343–2358. 10.1128/MCB.02019-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu J, Woods D, McMahon M, Bishop JM. 1998. Senescence of human fibroblasts induced by oncogenic Raf. Genes Dev 12: 2997–3007. 10.1101/gad.12.19.2997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu F, Li Y, Zhang J, Piao C, Liu T, Li H-H, Du J. 2013. Senescent cardiac fibroblast is critical for cardiac fibrosis after myocardial infarction. PLoS ONE 8: e74535 10.1371/journal.pone.0074535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Y, Tchkonia T, Pirtskhalava T, Gower AC, Ding H, Giorgadze N, Palmer AK, Ikeno Y, Hubbard GB, Lenburg M, et al. 2015. The Achilles’ heel of senescent cells: from transcriptome to senolytic drugs. Aging Cell 14: 644–658. 10.1111/acel.12344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Y, Tchkonia T, Fuhrmann-Stroissnigg H, Dai HM, Ling YY, Stout MB, Pirtskhalava T, Giorgadze N, Johnson KO, Giles CB, et al. 2016. Identification of a novel senolytic agent, navitoclax, targeting the Bcl-2 family of anti-apoptotic factors. Aging Cell 15: 428–435. 10.1111/acel.12445 [DOI] [PMC free article] [PubMed] [Google Scholar]