

ABSTRACT
The Discoidin Domain Receptor 1 (DDR1) receptor tyrosine kinase performs pleiotropic functions in the control of cell adhesion, proliferation, survival, migration, and invasion. Aberrant DDR1 function as a consequence of either mutations or increased expression has been associated with various human diseases including cancer. Pharmacological inhibition of DDR1 results in significant therapeutic benefit in several pre-clinical cancer models. Here, we discuss the potential implication of DDR1-dependent pro-survival functions in the development of cancer resistance to chemotherapeutic regimens and speculate on the molecular mechanisms that might mediate such important feature.
KEYWORDS: cancer, chemoresistance, Discoidin Domain Receptor 1, RTK signalling, receptor crosstalk
In recent years, DDR1 function has been increasingly associated to the development of a variety of cancers, including lung, breast, brain, prostate, liver, head and neck and pancreas among others [1]. Although DDR1 shows a pleiotropic nature including context or cell type dependent antiproliferative functions [1], its expression is often elevated in solid malignant tumours compared to neighbouring normal tissue, and high DDR1 expression has been correlated with poor prognosis in several tumour types [2–4]. Furthermore, DDR1 was reported as the most highly phosphorylated Receptor Tyrosine Kinase (RTK) in Non-Small Cell Lung Cancer, a strong indication of its functional implication in this disease [5]. Finally, DDR1 has been very recently discovered to be involved in metastatic dissemination phenomenon [6]. Yet, in spite of these evidences, the exact DDR1-dependent molecular mechanisms implicated in cancer progression are still incompletely understood. This may be in part due to the complex signalling downstream of DDR1 as well as to the fact that it can potentially regulate different and essential features of tumour biology including proliferation, survival, differentiation, migration and invasion [1]. The predominant contribution of DDR1 is most likely tumour-context dependent and has been excellently reviewed elsewhere [1]. Evidences from in vitro studies support the implication of DDR1 in the maintenance of important cancer features [7]. Furthermore, DDR1 knockdown in xenograft models of colorectal, lung and pancreatic cancer resulted in a significant therapeutic benefit [3,8,9]. Even more interesting from a pre-clinical perspective, DDR1 inhibition with small molecule compounds suppressed the tumour growth of gastric cancer xenografts [10], aggressive KRas-mutant lung adenocarcinoma (LUAD), both of murine and human origin [11] and of KRas-driven pancreatic ductal adenocarcinoma (PDAC) [12].
The findings listed above strongly support the implication of DDR1 function in cancer development. Yet, we would like to propose that the pro-survival function of DDR1 might also contribute to an essential cancer feature that is cell persistence after treatment. This pro-survival role may be particularly relevant in the context of genotoxic treatments, which, in spite of the increasing implementation of targeted therapies, still remain the standard of care for a substantial number of cancer patients. In this context, resistance to cancer chemotherapy is unfortunately the most common clinical output. Chemoresistance is a truly complex and multifaceted phenomenon [13] and the discussion of such intricate scenario is beyond the scope of this article. It is anyway clear that the deregulation of programmed cell death is an essential component of this response. As such, DDR1-mediated activation of pro-survival pathways resulting in ineffective induction of cell death may contribute to the onset of a chemoresistant phenotype [8]. Elevated DDR1 expression is associated with particularly aggressive cancer types and shows a clear correlation with unfavourable disease prognosis [2–4]. It is therefore possible that such high DDR1 activity may contribute to the intrinsic chemoresistant phenotype that often accompanies poor cancer outcome.
Recently, various pre-clinical studies have provided suggestive evidences of DDR1-dependent functions cooperating in the onset of the chemoresistant phenotype. One of the earliest evidences indicated that DDR1 activation induced NFKB-mediated cyclooxygenase 2 expression resulting in increased chemoresistance of breast cancer cells [14]. Later on, DDR1 knockdown was shown to significantly increase the sensitivity of ovarian cancer cell lines to cisplatin treatment resulting in elevated apoptosis [15]. A similar phenotype was observed in Hodgkin lymphoma [16]. Likewise, pharmacological inhibition of DDR1 resulted in increased therapeutic response in PDAC when administered concomitantly with cytotoxic chemotherapy [12]. Finally, perhaps the most compelling evidence was recently reported for gastric cancer. In this clinical setting, DDR1 expression was a prognostic marker only in patients receiving adjuvant treatment and was significantly correlated with poor survival [10].
How this contribution to chemoresistance is brought about in molecular terms is unclear and, taking into account the pleiotropic DDR1 functions, most probably it will be tumour- and context-dependent. To add an additional level of complexity, DDR1 interacts with other signalling pathways with clear pro-survival implications. These include additional extracellular matrix receptors such as integrins [7] that not only contribute to the activation of pro-survival pathways but also to drug response and chemoresistance [17]. In addition, DDR1 displays a functional crosstalk to cytokines such as TGF-β, both in homeostasis and cancer [9,18]. Elevated TGF-β also correlates with poor cancer prognosis and chemoresistance and several combinatorial therapies combining chemo with TGF-β inhibitors have been recently proposed [19]. Furthermore, DDR1 association with the insulin growth factor receptor 1 (IGF-1R) regulates IGF-1R trafficking and expression levels resulting in collagen-dependent and independent phosphorylation of DDR1 [20]. Similarly, DDR1 and insulin receptor (IR) co-localize and interact upon stimulation with insulin or IGF-2 [21]. As above, the insulin/IGF-R axis has been linked to chemoresistance in several tumour types [22]. Indeed, the combination of IGF signalling blockade plus chemotherapy has been recently proposed as a treatment for PDAC and colorectal cancer (CRC) [23,24]. Finally, in CRC cell lines DDR1-dependent pro-survival effects are mediated through a direct interaction with Notch1 that is essential to suppress genotoxic-mediated cell death [8].
Although direct interaction, such as that observed for DDR1 and IR mentioned above, is not in principle mandatory for network co-activity, the formation of higher order clusters may facilitate co-regulation. De-regulation caused by RTK overexpression may subvert spatial compartmentalization control in cancer [25]. The recent finding that DDR1 forms clusters by lateral association upon ligand binding reinforces the importance of spatial regulation for DDR1 signalling [26]. Membrane clustering is also an important feature for RTK signalling together with some of the potential chemoresistant co-actors mentioned above like integrins and TGF-β [27,28]. Moreover, DDR1 has been described to physically interact with syntenin 2 and hence PKCa, thus activating JAK2/STAT3 pathway and sustaining pluripotency factors and self-renewal capacity of metastasis-initiating cells [6]. It is therefore tempting to speculate that DDR1 overexpression in cancer might facilitate the formation of higher order “signalosomes” that by membrane-proximal co-activation could enhance the pro-survival properties of cancer cells.
Finally, DDR1 may also be an important mediator in the acquired chemoresistance, either by the acquisition of mutations during treatment and/or by chemotherapy-induced selection of pre-existing mutant subclones. Indeed, DDR1 mutations can be found in datasets from several cancer projects [29] (Figure 1a). We have failed to identify hotspots within DDR1 when taking into account the distribution of mutations in several cancer types. Yet, the mutations so far identified display some selectivity towards specific protein domains in certain tumour types. For instance, while most DDR1 mutations in melanoma tend to cluster in the extracellular and transmembrane domains those observed in LUAD or stomach cancer are mainly located in intracellular regions including the kinase domain (Figure 1b). This finding could suggest that cancer context may impose different selective pressures for the accumulation of specific DDR1 alterations. Whether any of these alterations might provide a selective advantage during chemotherapy is unknown. The answer to this question is complicated by the lack of exhaustive clinical records for all patient data, that would otherwise facilitate the analysis of any putative correlation between the prevalence of certain DDR1 alterations and the onset of a chemoresistant phenotype. This is further challenged by the paucity of matched tumour biopsies prior to and following the chemotherapeutic regimens. Additional studies will be required to evaluate whether such mutations result in a relevant impact on DDR1 functions and eventually affect patient response and survival after chemotherapy. Yet, as indicated in the previous sections, increased DDR1 activity due to higher expression levels in cancer may suffice to convey a pro-survival function without the need of acquired mutations. Interestingly, DDR1 has been recently identified as a constituent of a small group of cancer-associated factors that is maintained after chemotherapy treatment, is essential for cell line survival and elevated in drug-resistant stem-like cancer cells [30].
Figure 1.
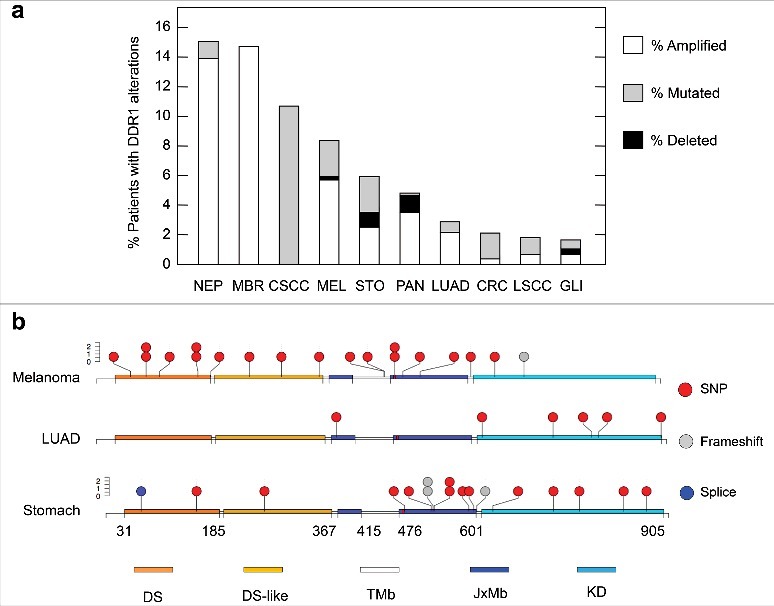
Schematic summary of DDR1 alterations found in human cancer. a) Frequency of patients showing DDR1 amplification (empty columns), mutation (grey columns) or deletion (black columns) in the indicated cancer studies. NEP (Neuroendocrine Prostate), MBR (Metastatic Breast), CSCC (Cutaneous Squamous Cell Carcinoma), MEL (Melanoma), STO (Stomach), PAN (Pancreas), LUAD (Lung Adenocarcinoma), CRC (Colorectal), LSCC (Lung Squamous Cell Carcinoma), GLI (Glioma). Data obtained from cBioPortal (www.cbioportal.org). b) Distribution of mutations with respect to DDR1 domains in patients from the indicated cancer studies. The figure represents single nucleotide polymorphisms (SNP, red circles), frameshift (grey circles) or mutations within splicing elements (blue circles). The indicated splice mutant is described as X_58 splice, potentially resulting in a truncated protein. DDR1 domains are indicated as DS (Discoidin Domain), DS-like (Discoidin Domain Like), TMb (Trans-membrane), JxMb (Juxta-membrane), KD (Kinase Domain). Numbering at the bottom indicates domain boundaries. Data obtained from cBioPortal (www.cbioportal.org).
In conclusion, further experimental work will be required to evaluate whether, as we hypothesize, the use of DDR1 inhibitors including orally available and more specific compounds developed recently [10,12], may provide a much-needed therapeutic benefit for chemoresistant cancer patients.
Funding Statement
This work was supported by Fondation ARC pour la Recherche sur le Cancer (PJA2016120478). This work was supported by project PJA20161204784 from Fondation ARC pour la recherche sur le cancer to D.S.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
References
- 1.Valiathan RR, Marco M, Leitinger B, et al.. Discoidin domain receptor tyrosine kinases: new players in cancer progression. Cancer Metastasis Rev. 2012;31:295–321. doi: 10.1007/s10555-012-9346-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Yang SH, Baek HA, Lee HJ, et al.. Discoidin domain receptor 1 is associated with poor prognosis of non-small cell lung carcinomas. Oncol Rep. 2010;24:311–9. [DOI] [PubMed] [Google Scholar]
- 3.Valencia K, Ormazabal C, Zandueta C, et al.. Inhibition of Collagen Receptor Discoidin Domain Receptor-1 (DDR1) reduces cell survival, homing, and colonization in lung cancer bone metastasis. Clin Cancer Res. 2012;18:969–80. doi: 10.1158/1078-0432.CCR-11-1686. [DOI] [PubMed] [Google Scholar]
- 4.Heinzelmann-Schwarz VA, Gardiner-Garden M, Henshall SM, et al.. Overexpression of the cell adhesion molecules DDR1, claudin 3, and Ep-CAM in metaplastic ovarian epithelium and ovarian cancer. Clin Cancer Res. 2004;10:4427–36. doi: 10.1158/1078-0432.CCR-04-0073. [DOI] [PubMed] [Google Scholar]
- 5.Rikova K, Guo A, Zeng Q, et al.. Global survey of phosphotyrosine signaling identifies oncogenic kinases in lung cancer. Cell. 2007;131:1190–203. doi: 10.1016/j.cell.2007.11.025. [DOI] [PubMed] [Google Scholar]
- 6.Gao H, Chakraborty G, Zhang Z, et al.. Multi-organ site metastatic reactivation mediated by non-canonical discoidin domain receptor 1 signaling. Cell. 2016;166:47–62. doi: 10.1016/j.cell.2016.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Leitinger B. Discoidin domain receptor functions in physiological and pathological conditions. Int Rev Cell Mol Biol. 2014;310:39–87. doi: 10.1016/B978-0-12-800180-6.00002-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kim H-G, Hwang S-Y, Aaronson SA, et al.. DDR1 receptor tyrosine kinase promotes prosurvival pathway through Notch1 activation. J Biol Chem. 2011;286:17672–81. doi: 10.1074/jbc.M111.236612. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 9.Rudra-Ganguly N, Lowe C, Mattie M, et al.. Discoidin domain receptor 1 contributes to tumorigenesis through modulation of TGFBI expression. PLoS One. 2014;9:e111515. doi: 10.1371/journal.pone.0111515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hur H, Ham I-H, Lee D, et al.. Discoidin domain receptor 1 activity drives an aggressive phenotype in gastric carcinoma. BMC Cancer. 2017;17:87. doi: 10.1186/s12885-017-3051-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ambrogio C, López-Gómez G, Falcone M, et al.. Combined inhibition of DDR1 and Notch signaling is a therapeutic strategy for KRAS-driven lung adenocarcinoma. Nat Med. 2016;22:270–7. doi: 10.1038/nm.4041. [DOI] [PubMed] [Google Scholar]
- 12.Aguilera KY, Huang H, Du W, et al.. Inhibition of discoidin domain receptor 1 reduces collagen-mediated tumorigenicity in pancreatic ductal adenocarcinoma. Mol Cancer Ther. 2017;16(11):2473–2485. doi: 10.1158/1535-7163.MCT-16-0834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Holohan C, Van Schaeybroeck S, Longley DB, et al.. Cancer drug resistance: an evolving paradigm. Nat Rev Cancer. 2013;13:714–26. doi: 10.1038/nrc3599. [DOI] [PubMed] [Google Scholar]
- 14.Das S, Ongusaha PP, Yang YS, et al.. Discoidin domain receptor 1 receptor tyrosine kinase induces cyclooxygenase-2 and promotes chemoresistance through nuclear factor-kappaB pathway activation. Cancer Res. 2006;66:8123–30. doi: 10.1158/0008-5472.CAN-06-1215. [DOI] [PubMed] [Google Scholar]
- 15.Deng Y, Zhao F, Hui L, et al.. Suppressing miR-199a-3p by promoter methylation contributes to tumor aggressiveness and cisplatin resistance of ovarian cancer through promoting DDR1 expression. J Ovarian Res. 2017;10:50. doi: 10.1186/s13048-017-0333-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cader FZ, Vockerodt M, Bose S, et al.. Lymphoid neoplasia: The EBV oncogene LMP1 protects lymphoma cells from cell death through the collagen-mediated activation of DDR1. Blood. 2013;122:4237–45. doi: 10.1182/blood-2013-04-499004. [DOI] [PubMed] [Google Scholar]
- 17.Seguin L, Desgrosellier JS, Weis SM, et al.. Integrins and cancer: Regulators of cancer stemness, metastasis, and drug resistance. Trends Cell Biol. 2015;25:234–40. doi: 10.1016/j.tcb.2014.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Roarty K, Serra R. Wnt5a is required for proper mammary gland development and TGF- -mediated inhibition of ductal growth. Development. 2007;134:3929–39. doi: 10.1242/dev.008250. [DOI] [PubMed] [Google Scholar]
- 19.Colak S, ten Dijke P. Targeting TGF-β signaling in cancer. Trends in Cancer. 2017;3:56–71. doi: 10.1016/j.trecan.2016.11.008. [DOI] [PubMed] [Google Scholar]
- 20.Malaguarnera R, Nicolosi ML, Sacco A, et al.. Novel cross talk between IGF-IR and DDR1 regulates IGF-IR trafficking, signaling and biological responses. Oncotarget. 2015;6:16084–105. doi: 10.18632/oncotarget.3177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Vella V, Malaguarnera R, Luisa Nicolosi M, et al.. Discoidin domain receptor 1 modulates insulin receptor signaling and biological responses in breast cancer cells. Oncotarget. 2017;5:43248–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Denduluri SK, Idowu O, Wang Z, et al.. Insulin-like growth factor (IGF) signaling in tumorigenesis and the development of cancer drug resistance. Genes Dis. 2015;2:14–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ireland L, Santos A, Ahmed MS, et al.. Chemoresistance in pancreatic cancer is driven by stroma-derived insulin-like growth factors. Cancer Res. 2016;76:6851–63. doi: 10.1158/0008-5472.CAN-16-1201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Codony-Servat J, Cuatrecasas M, Asensio E, et al.. Nuclear IGF-1R predicts chemotherapy and targeted therapy resistance in metastatic colorectal cancer. Br J Cancer. 2017;117(12):1777–1786. doi: 10.1038/bjc.2017.279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Casaletto JB, McClatchey AI. Spatial regulation of receptor tyrosine kinases in development and cancer. Nat Rev Cancer. 2012;12:387–400. doi: 10.1038/nrc3277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Juskaite V, Corcoran DS, Leitinger B. Collagen induces activation of DDR1 through lateral dimer association and phosphorylation between dimers. Elife. 2017;6:e25716. doi: 10.7554/eLife.25716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang SE, Xiang B, Zent R, et al.. Transforming growth factor β induces clustering of HER2 and integrins by activating Src-focal adhesion kinase and receptor association to the cytoskeleton. Cancer Res. 2009;69:475–82. doi: 10.1158/0008-5472.CAN-08-2649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Munger JS, Sheppard D. Cross talk among TGF-β signaling pathways, integrins, and the extracellular matrix. Cold Spring Harb Perspect Biol. 2011;3(11):a005017. doi: 10.1101/cshperspect.a005017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gao J, Aksoy BA, Dogrusoz U, et al.. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal. 2013;6:pl1. doi: 10.1126/scisignal.2004088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Selfors LM, Stover DG, Harris IS, et al.. Identification of cancer genes that are independent of dominant proliferation and lineage programs. Proc Natl Acad Sci [Internet]. 2017;114:E11276–84. Available from: http://www.pnas.org/lookup/doi/10.1073/pnas.1714877115 doi: 10.1073/pnas.1714877115. [DOI] [PMC free article] [PubMed] [Google Scholar]