

SUMMARY
N6-Methyladenosine (m6A) represents the most prevalent internal modification on mRNA and requires a multicomponent m6A methyltransferase complex in mammals. How their plant counterparts determine the global m6A modification landscape and its molecular link to plant development remain unknown. Here we show that FKBP12 INTERACTING PROTEIN 37 KD (FIP37) is a core component of the m6A methyltransferase complex, which underlies control of shoot stem cell fate in Arabidopsis. The mutants lacking FIP37 exhibit massive overproliferation of shoot meristems and a transcriptome-wide loss of m6A RNA modifications. We further demonstrate that FIP37 mediates m6A RNA modification on key shoot meristem genes inversely correlated with their mRNA stability, thus confining their transcript levels to prevent shoot meristem overproliferation. Our results suggest an indispensable role of FIP37 in mediating m6A mRNA modification, which is required for maintaining the shoot meristem as a renewable source for continuously producing all aerial organs in plants.
In Brief
Shen et al. show that FIP37 is a core component of the m6A methyltransferase complex and determines Arabidopsis m6A mRNA modification pattern. FIP37 mediates m6A modification of key shoot meristem regulator mRNAs and, through the control of their stability, prevents overproliferation of the shoot apical meristem that produces all aerial organs.
Graphical Abstract

INTRODUCTION
m6A is the most abundant internal modification of eukaryotic mRNA, and has been found in yeast, plants, flies, and mammals (Clancy et al., 2002; Levis and Penman, 1978; Nichols, 1979; Wei et al., 1975; Zhong et al., 2008). The reversible m6A modification affects almost all aspects of RNA metabolism, including stability, translation efficiency, nuclear retention, and splicing (Fu et al., 2014; Fustin et al., 2013; Wang et al., 2014a, 2014b, 2015; Yue et al., 2015; Zhao et al., 2014; Zhou et al., 2015). In mammals, m6A mRNA methylation is catalyzed by the methyltransferase complex containing methyltransferase like 3 (METTL3), METTL14, and Wilms’ tumor 1-associating protein (WTAP), which are likely conserved among eukaryotic species (Liu et al., 2014; Ping et al., 2014; Schwartz et al., 2014; Wang et al., 2014b). The m6A modification is reversible and can be removed by m6A demethylases, such as fat-mass and obesity associated protein (FTO) and its homolog alkylated DNA repair protein AlkB homolog 5 (ALKBH5) (Jia et al., 2011, 2013; Zheng et al., 2013). m6A-modified transcripts are recognized by the “reader” proteins, such as YTH domain family proteins, which mediate the downstream effects of the m6A modification (Dominissini et al., 2012; Wang et al., 2014a, 2015; Xu et al., 2014). Despite the progress in elucidating m6A mRNA methylation machineries in mammals, the relevant knowledge on their plant counterparts is mostly unknown.
A recent comparison of m6A methylomes in two accessions of Arabidopsis thaliana (Can-0 and Hen-16) has suggested that m6A methylation is a highly conserved and abundant mRNA modification in Arabidopsis (Luo et al., 2014). The Arabidopsis ortholog of METTL3, MTA, which is essential for embryonic and post-embryonic development, is required for m6A mRNA methylation (Bodi et al., 2012; Zhong et al., 2008). FIP37, an Arabidopsis ortholog of mammalian WTAP, has been found to interact with MTA, and its loss-of-function mutants display premature arrest of seed development because of a strong delay in embryo and endosperm development (Vespa et al., 2004; Zhong et al., 2008). Although these observations imply a conserved nature of the methyltransferase complex in plants, so far how plant counterparts sculpt the global m6A mRNA modification landscape and the molecular link to plant development are completely unknown.
In this study, we show that FIP37 is a core component of the plant m6A methyltransferase complex and plays an indispensable role in determining the m6A mRNA modification pattern in Arabidopsis. We further demonstrate that m6A RNA modification mediated by FIP37 confers a novel layer of gene regulation for temporally and spatially confining the expression of key shoot meristem genes in the shoot apical meristem (SAM). This determines proliferation of SAM stem cells that continuously generate all aerial parts of a plant throughout its lifetime. Our findings reveal a hitherto unknown global m6A mRNA modification pattern determined by the plant m6A methyltransferase complex, and establish an essential molecular link between m6A mRNA modification and determination of soot stem cell fate in plants.
RESULTS
Loss of Function of FIP37 Causes Massive Overproliferation of SAM
To study the biological role of FIP37, a highly conserved gene across plant species (Figure S1A), we identified two heterozygous transfer DNA insertion mutants, fip37–1S/+ and fip37–4/+, in the Columbia (Col) background (Figure 1A), both of which failed to produce viable homozygous seeds after self-pollination. About one-quarter of the immature seeds of fip37–1S/+ or fip37–4/+ showed abnormal white color and failed to develop further (Figures 1B, S1B, and S1C). To assess the post-embryonic function of FIP37, we partially complemented fip37–4 with the LEC1:FIP37 transgene, in which the FIP37 coding region was driven by the promoter of LEAFY COTYLEDON 1 (LEC1), which is specifically active in seeds during embryogenesis (Lotan et al., 1998). We obtained 27 independent transgenic lines, all of which exhibited similar phenotypes as discussed later. Here we present the characterization of one such fip37–4 LEC1:FIP37 line containing a single copy of the transgene. As expected, most of the seeds from fip37–4/+ harboring LEC1:FIP37 developed normally (Figures 1B and S1C), resulting in successful generation of homozygous fip37–4 seedlings in the LEC1:FIP37 background. FIP37 expression remained at very low levels in fip37–4 LEC1:FIP37 compared with that in wild-type seedlings (Figure 1C). These results suggest that LEC1:FIP37 specifically rescues embryonic lethality of fip37–4, but has minimal expression at the post-embryonic stage.
Figure 1. Loss of Function of FIP37 Causes Massive Overproliferation of the SAM.

(A) Schematic diagram shows the T-DNA insertion sites in fip37–1S and fip37–4. Exons and other sequences are represented by black boxes and lines, respectively.
(B) Dissected siliques from plants with various genetic backgrounds. Asterisks indicate fip37–4 homozygous seeds that appear in abnormal white color and fail to develop further. Scale bars, 0.5 mm.
(C) qPCR analysis of FIP37expression in 15-day-old seedlings with various genetic backgrounds. Error bars, mean ± SD; asterisks indicate statistically significant differences of FIP37 expression between mutants and wild-type plants (two-tailed paired Student’s t test, p < 0.001); n = 3 biological replicates.
(D) Loss of function of FIP37 at the post-embryonic stage results in enlarged SAMs. Upper panels show top views of 15-day-old seedlings, while lower panels show the corresponding SAMs examined by scanning electron microscopy (SEM). Red asterisks indicate SAMs. Double-headed arrows indicate the parameters for comparing meristem size in (E). Scale bars, 1 mm (upper panels) and 50 μm (lower panels).
(E) Statistically significant differences in the SAM size between 15-day-old wild-type (WT) and fip37–4 LEC1:FIP37 seedlings. Error bars, mean ± SD; **p < 0.001, two-tailed paired Student’s t test; n = 15.
(F) Median longitudinal sections of the SAMs of wild-type (upper panels) and fip37–4 LEC1:FIP37 (lower panels) seedlings at 5 (D5), 10 (D10), and 20 days (D20) after germination. Asterisks indicate SAMs. Scale bars, 50 μm.
See also Figures S1 and S2.
Notably, fip37–4 LEC1:FIP37 mainly exhibited enlarged SAMs compared with wild-type plants during seedling development (Figures 1D–1F). A close examination of fip37–4 LEC1:FIP37 revealed enlargement of vegetative SAMs and a visible delay in the generation of leaves early in 5-day-old seedlings (Figures 1F and S1F). Progressive SAM enlargement and a dramatic arrest in leaf production were continuously observed in developing mutants (Figures 1F and S1F). In 15-day-old seedlings, fasciated fip37–4 LEC1:FIP37 SAMs were significantly larger than symmetrical dome-shaped wild-type SAMs (Figures 1D and 1E). Instead of continuously producing leaves, the flattened mutant SAMs generated only two small lobed leaves before producing many finger-like tissues from the peripheral regions (Figures 1D and S1F). In 20-day-old seedlings, wild-type SAMs were transformed into inflorescence SAMs that further generated floral meristems, whereas fip37–4 LEC1:FIP37 developed massively overproliferated SAMs with cytoplasmically dense cells, but without any observable reproductive tissue (Figures 1F and S1F). At a later stage, fip37–4 LEC1:FIP37 plants produced extremely large SAMs bearing compact dome-shaped meristematic tissues even with the outgrowth of meristems on lobed leaves (Figures S1G–S1I), and eventually died.
To confirm the phenotypes of fip37–4 LEC1:FIP37, we also complemented fip37–4 with the ABI3:FIP37 transgene, in which FIP37 was driven by another embryo-specific promoter of ABA INSENSITIVE3 (ABI3) (Parcy et al., 1994).Among 22 independent transgenic lines generated, 20 lines exhibited similar phenotypes to fip37–4 LEC1:FIP37. In addition, we introduced the LEC1:FIP37 transgene into fip37–1S through crossing fip37–4/+ LEC1:FIP37 with fip37–1S/+. Examination of fip37–1S LEC1:FIP37 and fip37–4 ABI3:FIP37 revealed a SAM defect comparable with that of fip37–4 LEC1:FIP37 (Figures 1C, 1D, S1B, and S1C). Because LEC1:FIP37 and ABI3:FIP37 in the wild-type background grew normally (Figure S1D), the SAM defect observed in fip37–4 LEC1:FIP37, fip37–1S LEC1:FIP37, and fip37–4 ABI3:FIP37 should be attributable to very low expression of FIP37 in these mutants (Figure 1C). We further created over 80 FIP37 knockdown transgenic plants (AmiR-fip37) by artificial microRNA (AmiR) interference (Schwab etal., 2006). Sixty-six independent lines exhibited similar growth defects, albeit at different levels, to fip37–4 LEC1:FIP37, including enlarged SAM and abnormal leaf development (Figures S2A–S2G). Downregulation of FIP37 in selected AmiR-fip37 lines was correlated with the severity of the defects (Figure S2H). In contrast, overexpression of FIP37 did not show obvious growth defects (Figures S2I and S2J), implying that an excessive amount of FIP37 might not affect plant development. Taken together, our results suggest that FIP37 is essential for preventing SAM overproliferation during post-embryonic development.
FIP37 Is Highly Expressed in Actively Proliferating Tissues
Quantitative real-time PCR analysis revealed that FIP37 was expressed in various plant tissues examined (Figure S3A) (Vespa et al., 2004). We also monitored the detailed expression of FIP37 using an FIP37:GUS reporter line. GUS signals were consistently strong in actively proliferating tissues, such as shoot apices, young leaves, and developing floral organs and seeds (Figures 2A and S3C–S3H). In situ hybridization also revealed strong FIP37 expression in the wild-type SAM, but not in the fip37–4 LEC1:FIP37 SAM (Figures 2B and S3B). Confocal analysis of fip37–4 gFIP37-GFP, in which the FIP37 genomic fragment fused with the GFP gene fully rescued fip37–4 (Figure S1C), showed FIP37-GFP localization in the nuclei of the cells in the whole SAM (Figure 2C, upper panels), root tip (Figure 2C, lower panels), and other actively proliferating tissues (Figure S3I). Similar localization was also observed in fip37–4 35S:FIP37-GFP (Figures S3J and S3K). Notably, FIP37-GFP was localized throughout the nucleoplasm excluding nucleoli that accommodate ribosomal biogenesis or immobilizes proteins (Figures 2C and S3K; Audas et al., 2012). Further immunostaining of a functional fip37–4 gFIP37–4HA line, in which gFIP37–4HA fully rescued the embryo-lethal phenotype of fip37–4 (Figures 1B, 1C, and S1C), and its derived protoplast cells confirmed the localization of FIP37–4HA throughout the nucleoplasm excluding nucleoli (Figures 2D, S3L, and S3M).
Figure 2. Gene Expression Patterns and Protein Localization of FIP37.

(A) GUS staining of 5-day-old and 10-day-old FIP37:GUS seedlings. Cotyledons (c) are labeled. Scale bars, 1 mm.
(B) In situ localization of FIP37 in the SAM of a 10-day-old wild-type seedling. The inset shows a section of a 10-day-old fip37–4 LEC1:FIP37 SAM hybridized with the FIP37 antisense probe as a control. Scale bars, 50 μm.
(C) Confocal analysis of FIP37-GFP in the SAM (upper panel) and root tip (lower panel) of a 10-day-old fip37–4 gFIP37-GFP seedling. Inset shows an enlarged view of the nucleus of a root tip cell. GFP, GFP fluorescence; BF, bright-field image; Merge, merge of GFP and bright-field images. Scale bars, 20 μm.
(D) Immunolocalization of FIP37–4HA in the nucleus of a fip37–4 gFIP37–4HA root tip cell. BF, bright-field image; Merge, merge of anti-HA and bright-field images. Scale bars, 10 μm.
See also Figure S3.
FIP37 Is Indispensable for m6A mRNA Modification
To investigate the underlying mechanisms of FIP37-mediated SAM development, we then explored the molecular nature of FIP37 in Arabidopsis. The finding that FIP37 is orthologous to WTAP, a key component of the m6A mRNA methyltransferase complex in mammals (Liu et al., 2014; Ping et al., 2014; Schwartz et al., 2014), led us to test whether FIP37 is involved in regulating m6A mRNA modification in Arabidopsis. To this end, we first compared the total m6A levels in mRNAs extracted from 5-day-old wild-type and fip37–4 LEC1:FIP37 seedlings by dot blot analysis using m6A antibody, and found a dramatic reduction of m6A levels in fip37–4 LEC1:FIP37 (Figure 3A). Quantitative measurement of m6A levels by liquid chromatography-tandem mass spectrometry (LC-MS/MS) further revealed that total m6A levels in fip37–4 LEC1:FIP37 were significantly decreased to only ~20% of those in wild-type and LEC1:FIP37 seedlings (Figures 3B, S4A and S4B), demonstrating that FIP37 is indispensable for global m6A RNA methylation in Arabidopsis.
Figure 3. Effect of Loss of FIP37 on the Arabidopsis m6A RNA Methylome and Transcriptome.

(A) Dot blot analysis of m6A levels in mRNA purified from 5-day-old wild-type and fip37–4 LEC1:FIP37 seedlings.
(B) m6A percentage relative to adenosine (m6A/A ratio) determined by LC-MS/MS in mRNA purified from 5-day-old wild-type, LEC1:FIP37, and fip37–4 LEC1:FIP37 seedlings. LC-MS/MS was repeated with three biological replicates. Statistical analysis was performed using two-tailed paired Student’s t test. The results are considered statistically significant at p < 0.05. Error bars, mean ± SD.
(C) Heatmap representing IP enrichment values for m6A peaks with statistically significant difference between fip37–4 LEC1:FIP37 and wild-type seedlings.
(D) Cumulative distribution function of log2 peak intensity of m6A peaks in fip37–4 LEC1:FIP37 and wild-type seedlings.
(E) Comparison of distribution of m6A peaks in different segments of wild-type (upper panel) and fip37–4 LEC1:FIP37 (lower panel) transcripts. Left panels show pie charts presenting the percentages of m6A peaks in different transcript segments, while right panels show relative enrichment of m6A peaks in different transcript segments.
(F) Distribution of m6A peaks in transcript segments divided into 5′ UTR, CDS, and 3′ UTR in wild-type and fip37–4 LEC1:FIP37 seedlings.
(G and H) Boxplot comparison of enrichment fold of m6A peaks in different transcript segments (G) or shared m6A peaks (H) in wild-type and fip37–4 LEC1:FIP37 seedlings.
(I) Heatmap showing differentially expressed genes in wild-type and fip37–4 LEC1:FIP37 seedlings.
(J) Cumulative distribution of changes in gene expression between fip37–4 LEC1:FIP37 and wild-type seedlings for non-m6A and m6A targeted genes. p < 10 × 10−16, two-sided Mann-Whitney test.
(K) Boxplot comparison of expression levels of non-m6A genes and genes bearing m6A in different transcript segments between fip37–4 LEC1:FIP37 and wild-type seedlings.
See also Figures S4 and S5, and Table S1, S2, and S3.
To understand how FIP37 contributes to the global m6A mRNA modification landscape, we compared transcriptome-wide m6A methylomes between 5-day-old Col-0 wild-type and fip37–4 LEC1:FIP37 seedlings through applying m6A-seq (Dominissini et al., 2013) to two biological replicates for each genotype with high Pearson correlation coefficient (Figure S4C). Approximately 24–54 and 26–65 million reads for wild-type and fip37–4 LEC1:FIP37 seedlings, respectively, were generated for each library. There were 2.4–4.7 and 2.3–5.6 million distinct reads for wild-type and fip37–4 LEC1:FIP37 seedlings, respectively, uniquely aligned to the Arabidopsis TAIR 10 genome (MAPQ ≥ 13; 95% mapping to unique loci). We used MACS to scale down the larger datasets, and m6A peaks were identified using a peak-detection algorithm with an estimated false discovery rate (FDR) <0.05. After combining the libraries of two replicates, our analysis revealed 4,276 putative high-confidence m6A peaks within 3,970 coding gene transcripts and 30 non-coding RNAs in wild-type seedlings (Table S1), whereas only 1,028 peaks within 686 coding genes and 35 non-coding RNAs were identified in fip37–4 LEC1:FIP37 (Table S2), demonstrating a global loss of m6A peaks in fip37–4 LEC1:FIP37 (Figures 3C and 3D). Subsequently, we validated m6A-seq results with independent m6A-immunoprecipitation (IP)-qPCR on randomly selected m6A targets (Figures S4D–S4N), suggesting that our m6A-seq data was accurate and robust.
We next compared the distribution of m6A peaks along wild-type and fip37–4 LEC1:FIP37 transcripts relative to landmarks in their architecture, based on the single-transcript, intron-free transcriptome dataset. Wild-type m6A peaks were greatly enriched near the stop codon and 3′ UTR of coding genes (Figures 3E and 3F). The stop codon segment (200-nucleotide window centered on the stop codon) was the most enriched in m6A peaks, with 66.72% of the peaks, representing a 9.4-fold enrichment over the distribution expected by chance (Figure 3E). In coding sequences (CDSs), m6A peaks were mainly enriched near the start codon (Figure 3F). In contrast, loss of FIP37 leads to a dramatic change in the topology of m6A methylome. The enrichment near the stop codon and 3′ UTR in wild-type were largely abolished in fip37–4 LEC1:FIP37 (Figures 3E–3G), while the m6A modification in the CDS and 5′ UTR were less affected (Figure 3G), resulting in the change of percentage and relative enrichment of the m6A peak in different regions of expressed mRNAs (Figures 3E–3G). Most m6A peaks (3,778/4276, 88%) found in Col-0 were FIP37 dependent, and were present in wild-type but totally lost in fip37–4 LEC1:FIP37. Gene ontology analysis indicated that these genes regulate multiple biological processes (Figure S5A). Although other 498 m6A peaks were present in both wild-type and fip37–4 LEC1:FIP37, the enrichment fold of these shared peaks was also reduced in fip37–4 LEC1:FIP37 (Figures 3H, S5B, and S5C). Thus, almost all m6A peaks identified in wild-type seedlings were either fully or partially dependent on FIP37, suggesting that FIP37 is essential for sculpting the global m6A mRNA modification landscape in Arabidopsis. In addition, over 95% of the m6A sites contained an RRACH motif (R = G or A; H = A, C, or U) (Figure S5D), which is similar to that found in various organisms (Dominissini et al., 2012; Luo et al., 2014; Zhao et al., 2014).
To test whether there is a potential correlation between m6A mRNA methylation and gene expression levels mediated by FIP37, we performed RNA-seq on 5-day-old wild-type versus fip37–4 LEC1:FIP37 seedlings with three highly reproducible biological replicates (Figures 3I and S5E). Plotting the genes with m6A peaks in different segments against their gene expression levels in wild-type seedlings found that transcripts of highly expressed genes were more likely to be methylated at the stop codon (Figure S5F). There were 3,116 upregulated and 2,943 downregulated transcripts, respectively, in fip37–4 LEC1:FIP37 (fold change >1.5, p < 0.05) (Table S3). Comparison of differentially expressed genes with our list of m6A peaks revealed that the transcript abundance for 874 or 193 out of 3,970 m6A-modified genes was decreased or increased in fip37–4 LEC1:FIP37, respectively. Genes bearing m6A modifications at the CDS, stop codon, and 3′ UTR tended to be downregulated, while those with m6A modifications at the 5′ UTR were more likely upregulated in fip37–4 LEC1:FIP37 (Figures 3J and 3K). These observations implicate a complex relationship between FIP37-mediated m6A methylation and gene expression level in plants.
FIP37-Mediated m6A Modification Confines STM and WUS Expression
In line with massive overproliferation of SAM in fip37–4 LEC1:FIP37, our RNA-seq data revealed that the expression levels of two key SAM regulators, WUSCHEL (WUS) and SHOOTMERISTEMLESS (STM) (Barton and Poethig, 1993; Endrizzi et al., 1996; Laux et al., 1996; Lenhard et al., 2002; Long et al., 1996; Mayer et al., 1998), were significantly increased in fip37–4 LEC1:FIP37 (Table S3). We then examined whether the effect of FIP37 on the expression levels of WUS and STM is associated with FIP37-mediated m6A methylation of these two genes. As the m6A-seq approach likely underestimates the fraction of m6A modified genes with low expression levels (Batista et al., 2014; Dominissini et al., 2013; Fray and Simpson, 2015), our m6A-seq analyses using the stringent mapping criterion (MAPQ ≥ 13) did not reveal any known SAM regulators whose transcripts only occupied a very small percentage of mRNAs extracted from 5-day-old seedlings (Table S1). Thus, we applied a loose mapping criterion (MAPQ ≥ 1) to identify m6A peaks in genes expressed at lower levels. Notably, we found that there was an m6A peak within the stop codon region of STM mRNA in wild-type seedlings, which was greatly diminished in fip37–4 LEC1:FIP37 (Figure 4A, upper panel). The change in this m6A peak in STM was further confirmed by independent m6A-IP-qPCR using mRNAs extracted from only shoot apices (Figure 4A, lower panel). As WUS is only specifically expressed within the organizing center of the SAM (Figure 4F; Long et al., 1996; Mayer et al., 1998), its abundance in mRNAs extracted from whole seedlings could be too low to be detected by m6A-seq. Similarly, we performed m6A-IP-qPCR using mRNAs extracted from shoot apices, and found that WUS mRNA from wild-type shoot apices was also modified with m6A at the region near its stop codon, which was completely abolished in fip37–4 LEC1:FIP37 (Figure 4B). Furthermore, RNA immunoprecipitation (RIP)-qPCR assays using shoot apices of fip37–4 gFIP37–4HA (Figures 4C and S1C) revealed a direct binding of FIP37–4HA to both STM and WUS transcripts in vivo (Figure 4D). These results suggest that FIP37 is directly associated with STM and WUS transcripts and mediates m6A methylation of both transcripts in SAMs.
Figure 4. Upregulation of STM and WUS Is Coupled with Their Loss of m6A in fip37–4 LEC1:FIP37.

(A) m6A modification in STM mRNA is abolished in fip37–4 LEC1:FIP37. Upper panel shows m6A peaks revealed by m6A-seq In STM mRNA from wild-type and fip37–4 LEC1:FIP37. The STM transcript structure is shown beneath, with thick boxes and lines representing exons and introns, respectively. Lower panel shows m6A-IP-qPCR results, in which cDNA fragments amplified are indicated below the STM transcript structure. Shoot apices of 5-day-old wild-type and fip37–4 LEC1:FIP37 seedlings were harvested for m6A-IP-qPCR in (A and B). Error bars, mean ± SD; *p < 0.05 in (A and B), two-tailed paired Student’s t test; n = 3 biological replicates.
(B) m6A modification in WUS mRNA is abolished in fip37–4 LEC1:FIP37. Upper panel shows the WUS transcript structure labeled with cDNA fragments amplified in m6A-IP-qPCR assays shown in the lower panel. Error bars, mean ± SD.
(C) Western blot analysis using anti-HA antibody shows the expression of FIP37–4HA in protein extracts (Input) or immunoprecipitated fractions (Eluate) from shoot apices of 5-day-old fip37–4 gFIP37–4HA seedlings. The arrowhead indicates the FIP37–4HA band.
(D) RIP assay shows direct binding of FIP37–4HAto STM and WUS transcripts. Shoot apices of 5-day-old fip37–4 gFIP37–4HA seedlings were harvested for RIP. Enrichment of ACTIN2 (ACT) serves as a negative control. Error bars, mean ± SD. Asterisks indicate statistically significant differences in FIP37–4HA enrichment with STM or WUS transcripts compared with that with ACTIN2 (two-tailed paired Student’s t test, p < 0.05); n = 3 biological replicates.
(E) qPCR analysis of mRNA expression of STM and WUS in fip37–4 LEC1:FIP37 versus wild-type seedlings. Gene expression levels in wild-type seedlings are set as 1, which is indicated by a red dotted line. Error bars, mean ± SD; asterisks indicate statistically significant differences in gene expression between fip37–4 LEC1:FIP37 and wild-type seedlings (two-tailed paired Student’s t test, p < 0.05). n = 3 biological replicates.
(F) In situ localization of STM and WUS in wild-type (WT) and fip37–4 LEC1:FIP37 SAMs. Arrow indicates an incipient leaf primordium in a wild-type SAM. Arrowheads indicate STM expression in the regions where lateral organs emerge in a fip37–4 LEC1:FIP37 SAM. Scale bars, 50 μm.
(G and H) The FIP37-GR fusion protein is biologically functional. One-day-old fip37–4gFIP37-GR seedlings were mock treated (0.03% ethanol and 0.012% Silwet L-77) (Mock) or treated with 10 μM dexamethasone (Dex) and 0.012% Silwet L-77 once a week. At 3 weeks after the first treatment, a mock-treated plant shows similar phenotypes to fip37–4 LEC1:FIP37 (G) (upper panel, a top view of the seedling; lower panel, an enlarged view of the SAM shown in the upper panel), whereas a Dex-treated plant exhibits a rescued SAM phenotype with continuous generation of leaves (H). Scale bars, 1 mm.
(I) Single Dex treatment of fip37–4 gFIP37-GR is able to rescue the developmental defects of fip37–4. Upper panel, a 2-week-old fip37–4 gFIP37-GR plant before Dex treatment shows an overproliferated SAM and a dramatic arrest in leaf production. Lower panel, a 4-week-old fip37–4 gFIP37-GR plant shows continuous generation of leaves from the SAM after a single Dex treatment at 2 weeks old. Scale bars, 1 mm.
(J) Induced FIP37 activity upregulates m6A levels in STM and WUS mRNA. Time-course m6A-IP-qPCR was performed using shoot apices of fip37–4 gFIP37-GR mock treated (Mock) or treated with 10 μM Dex for 0, 4, and 8 hr. Error bars, mean ± SD; asterisks indicate statistically significant differences in m6A enrichment levels between mock- and Dex-treated seedlings (two-tailed paired Student’s t test, p < 0.05); n = 3 biological replicates.
(K) Induced FIP37 activity does not affect m6A levels in CLV3 and KNAT1 mRNA. m6A-IP-qPCR was performed using shoot apices of fip37–4 gFIP37-GR mock treated (Mock) or treated with 10 μM Dex for 4 hr. Error bars, mean ± SD; n = 3 biological replicates.
(L) Induced FIP37 activity reduces the mRNA levels of WUS and STM. Time-course expression of WUS and STM in 5-day-old fip37–4 gFIP37-GR shoot apices mock treated (Mock) or treated with 10 μM Dex for 0, 4, and 8 hr. The relative expression of each gene at different time points was calculated by normalizing the gene expression in Dex-treated samples against that in mock-treated samples. Error bars, mean ± SD; n = 3 biological replicates.
(M) Induced FIP37 activity does not affect CLV3 and KNAT1 expression. Expression of CLV3 and KNAT1 was examined in 5-day-old fip37–4 gFIP37-GR shoot apices mock treated (Mock) or treated with 10 μM Dex for 4 hr. Error bars, mean ± SD; n = 3 biological replicates.
See also Figures S6.
Besides STM and WUS, we also screened out other candidate genes that could be regulated by FIP37 and relevant to SAM development from our RNA-seq data (Table S3). These genes included those involved in meristem function or leaf initiation, such as ASYMMETRIC LEAVES 2 (AS2), CLAVATA3 (CLV3), and KNOTTED-LIKE FROM ARABIDOPSIS THALIANA 1 (KNAT1) (Fletcher et al., 1999; Lincoln et al., 1994; Semiarti et al., 2001), and those involved in hormone metabolism, perception, or signaling that may be related to SAM function, such as GA REQUIRING 1 (GA1), GIBBERELLIN 2-OXIDASE 1 (GA2OX1), YUCCA5 (YUC5), PIN-FORMED 4 (PIN4), and CYTOKININ OXIDASE 5 (CKX5) (Jasinski et al., 2005; Vernoux et al., 2010). qPCR analysis confirmed a change in the expression of these genes in 5-day-old fip37–4 LEC1:FIP37 versus wild-type seedlings (Figure S6A). However, m6A-IP-qPCR analysis of mRNAs extracted from shoot apices revealed that none of these genes was m6A-modified in wild-type and fip37–4 LEC1:FIP37 seedlings (Figure S6B). Consistently, RIP did not detect direct binding of FIP37–4HA to these transcripts in shoot apices of 5-day-old fip37–4 gFIP37–4HA (Figure S6C). These observations exclude the transcripts of these genes as direct targets of FIP37-mediated m6A methylation.
We further quantitatively monitored the effect of loss of FIP37 on STM and WUS expression in developing seedlings. In 5-day-old fip37–4 LEC1:FIP37 that exhibited only slight defects compared with wild-type plants (Figures 1F and S1F), WUS and STM expression was obviously upregulated (Figure 4E). In 15-day-old mutants that exhibited significantly enlarged SAMs (Figures 1D–1F), WUS was dramatically upregulated over 50-fold compared with that in wild-type plants, while STM was also increased around 10-fold (Figure 4E). In situ hybridization further revealed that in contrast to its expression confined to the organizing center of a wild-type SAM, WUS expression was expanded to a larger region including the overlying layers of the central zone early in a 5-day-old mutant (Figures 4F and S6D). The domain expressing WUS was dramatically enlarged concomitantly with the SAM overproliferation in a 15-day-old mutant (Figures 4F and S6D). In line with the change in WUS expression, the expression of another important meristem regulator CLV3, which is activated by WUS, but in turn restricts WUS expression (Brand et al., 2000; Fletcher et al., 1999; Schoof et al., 2000; Yadav et al., 2011), only appeared in the adjacent layers above the WUS-expressing domains in both SAMs of 5- and 15-day-old mutants (Figure S6D). In wild-type plants, STM expression was clearly downregulated in incipient leaf primordia (Figures 4F and S6D). However, its expression was expanded in the regions where lateral organs emerged early in the SAM of a 5-day-old mutant, and was later very strong in the entire overproliferated SAM of a 15-day-old mutant except the peripheral region (Figures 4F and S6D). These expression patterns, together with an apparent correlation of phenotypic severity with the upregulated levels of WUS and STM in AmiR-fip37 lines (Figure S2H), indicate that massive SAM overproliferation in FIP37 knockdown seedlings might be attributable to an expanded expression of WUS and STM in SAMs associated with loss of m6A on both transcripts.
To test whether FIP37 quickly affects m6A and mRNA levels of STM and WUS in vivo, we created a steroid-inducible version of FIP37 (fip37–4 gFIP37-GR), in which fip37–4 was transformed with the FIP37 genomic fragment fused to the steroid-binding domain of the rat glucocorticoid receptor (GR). Induction of FIP37 activity could be achieved by dexamethasone treatment of fip37–4 gFIP37-GR plants, which releases the FIP37-GR fusion protein in the cytoplasm to the nucleus (Dalman et al., 1991). Dexamethasone was applied to floral buds and siliques of fip37–4/+ gFIP37-GR to allow the setting of seeds homozygous for fip37–4. In the absence of applied dexamethasone after seed germination, fip37–4 gFIP37-GR exhibited the defects in SAM development and generation of aerial organs similarly to fip37–4 LEC1:FIP37 (Figures 1 and 4G). In contrast, dexamethasone treatment almost fully rescued these developmental defects in fip37–4 gFIP37-GR (Figure 4H). Notably, a single dexamethasone treatment was able to significantly rescue the defects (Figure 4I). These observations suggest that the FIP37-GR fusion protein is biologically functional in a hormone-dependent manner. Dexamethasone treatment of shoot apices of 5-day-old fip37–4 gFIP37-GR for 4 hr or longer resulted in a significant increase in m6A levels in WUS and STM mRNA (Figure 4J), which was associated with a reduction of WUS and STM transcript levels (Figure 4L). In contrast, dexamethasone treatment did not affect the m6A status and expression levels of CLV3 and KNAT1 in fip37–4 gFIP37-GR shoot apices (Figures 4K and 4M). These results substantiate that FIP37-mediated m6A RNA modification mainly targets to WUS and STM in the SAM.
As FIP37 has been reported to interact with the m6A methyl-transferase MTA (Zhong et al., 2008), we further tested whether MTA plays a similar role to FIP37 in controlling SAM development. Downregulation of MTA by AmiR interference (AmiR-mta) exhibited similar growth defects to AmiR-fip37 (Figures 5A and S2). Like AmiR-fip37 (Figure S2G), strong AmiR-mta lines developed meristem-like tissues on the leaf surface (Figure 5B). As expected, m6A levels was significantly reduced on a global scale (Figure 5C) and also in several tested mRNAs, including WUS and STM (Figure 5D), in AmiR-mta. Consequently, WUS and STM expression was also unregulated in AmiR-mta (Figure 5E). These results indicate that MTA and FIP37 could similarly regulate SAM development through mediating m6A modification on WUS and STM mRNAs.
Figure 5. Downregulation of MTA Causes Various Developmental Defects and a Reduction in m6A Levels.

(A) Phenotypic comparison of developing wild-type and AmiR-mta seedlings. Scale bars, 0.1 cm.
(B) A strong AmiR-mta line at 1 month old exhibits overproliferation of the SAM and abnormal leaf development. Red arrowheads indicate the formation of meristematic-like structure. Scale bars, 0.1 cm.
(C) Dot blot analysis shows a reduction of m6A levels in 10-day-old AmiR-mta transgenic plants and fip37–4 LEC1:FIP37.
(D) m6A levels of selected genes are reduced in AmiR-mta. Shoot apices of 10-day-old wild-type and AmiR-mta seedlings were harvested for m6A-IP-qPCR. Error bars, mean ± SD; *p < 0.05, two-tailed paired Student’s t test; n = 3 biological replicates.
(E) qPCR analysis of expression of MTA, FIP37, WUS, and STM in 10-day-old wild-type (WT) and AmiR-mta with strong or weak phenotypes. The gene expression level in wild-type seedlings is set as 1. Error bars, mean ± SD; n = 3 biological replicates.
STM and WUS Are Responsible for SAM Overproliferation in fip37–4 LEC1:FIP37
To genetically test whether WUS and STM contribute to SAM overproliferation in fip37–4 LEC1:FIP37, we crossed wus-8, a newly identified mutant (Figure S7A), and stm-10 (Kanrar et al., 2006) in the Col background with fip37–4 LEC1:FIP37. wus-8 resembled wus-1 in the Landsberg erecta (Ler) background (Laux et al., 1996) and repetitively initiated defective SAMs that terminated prematurely, while stm-10 failed to maintain the SAM after generating a few leaves (Kanrar et al., 2006) (Figures 6A and S7B). Both wus-8 and stm-10 substantially suppressed the enlargement of SAMs in 18-day-old fip37–4 LEC1:FIP37 seedlings, while wus-8 had a stronger effect on further delaying leaf generation (Figures 6A and S7B). One month after germination, 55% of wus-8 fip37–4 LEC1:FIP37 and 40% of stm-10 fip37–4 LEC1:FIP37 seedlings still developed enlarged SAMs and meristem-like tissues on leaves (Figure S7C), although these phenotypes were less pronounced than those in fip37–4 LEC1:FIP37, indicating that wus-8 or stm-10 only partially suppresses SAM overproliferation in fip37–4 LEC1:FIP37. Since both WUS and STM were targets of FIP37-mediated m6A modification and highly upregulated in fip37–4 LEC1:FIP37 (Figure 4), we further generated stm-10 wus-8 fip37–4 LEC1:FIP37. This mutant exhibited the same phenotype to stm-10 wus-8 and never produced any SAM and leaf before its death (Figures 6A and 6B), suggesting that both WUS and STM are responsible for SAM overproliferation in fip37–4 LEC1:FIP37. In contrast, clv3–2 fip37–4 LEC1:FIP37 developed the SAM of a size comparable with that of fip37–4 LEC1:FIP37 (Figure S7D), substantiating that CLV3 does not directly contribute to the SAM defect in fip37–4 LEC1:FIP37.
Figure 6. SAM Overproliferation in fip37–4 LEC1:FIP37 Is Attributable to WUS and STM Activity.
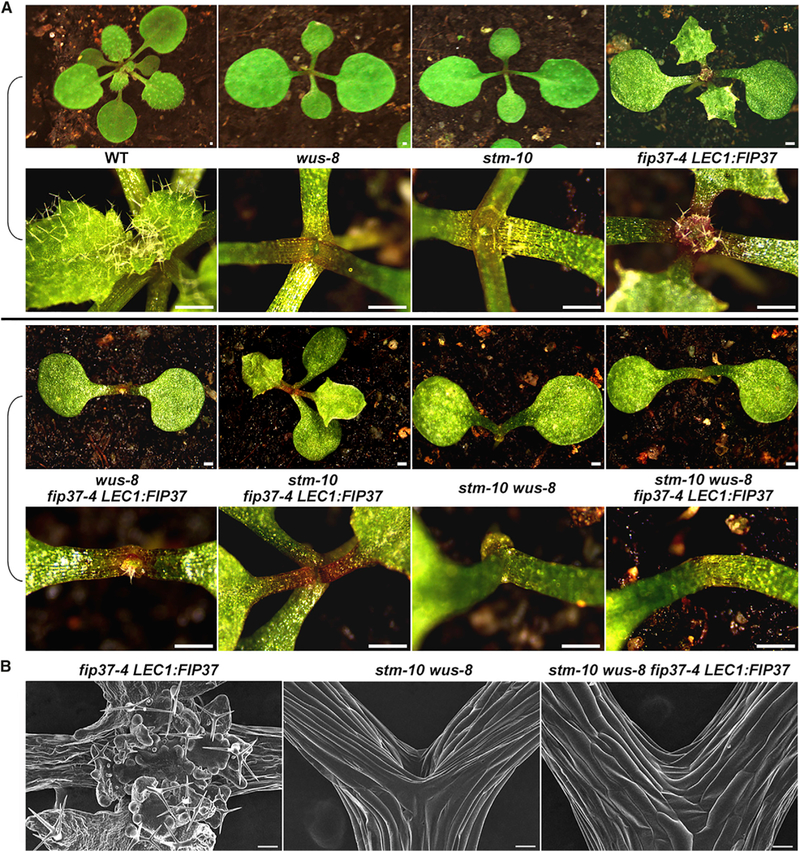
(A) SAM overproliferation in fip37–4 LEC1:FIP37 is partially suppressed by wus-8 or stm-10, and fully suppressed by stm-10 wus-8. Upper panels show top views of 18-day-old seedlings with various genetic backgrounds, while lower panels show close-up views of their SAM regions. Scale bars, 0.5 mm.
(B) SEM analysis of the SAMs of 18-day-old fip37–4 LEC1:FIP37, stm-10 wus-8, and stm-10 wus-8 fip37–4 LEC1:FIP37 seedlings. Scale bars, 100 μm.
See also Figure S7.
FIP37 Affects the Decay Rate of STM and WUS mRNAs
As FIP37 mediates m6A modification near the stop codon and 3′ UTR of STM and WUS mRNAs (Figures 4A and 4B), we sought to assess the correlation between this modification and the effect of FIP37 on STM and WUS expression levels. Although multiple cellular mechanisms contribute to changes in steady-state RNA levels, the role of m6A in mRNA decay has been suggested as one of the major effects in mammalian cells (Batista et al., 2014; Fustin et al., 2013; Wang et al., 2014a, 2014b). Thus, we investigated whether m6A modification by FIP37 affects the mRNA decay of STM and WUS by measuring their RNA levels in 5-day-old wild-type and fip37–4 LEC1:FIP37 seedlings treated with the transcription inhibitor actinomycin D. Both STM and WUS showed significant increases in their cumulative RNA stability in fip37–4 LEC1:FIP37 versus wild-type seedlings from 8 to 24 hr after actinomycin D treatment, whereas non-m6A targeted transcripts, such as β-tubulin (TUB8) and WRKY6, displayed unchanged RNA stability (Figure 7A). These results suggest that m6A methylation by FIP37 on STM and WUS accelerates decay of both transcripts.
Figure 7. FIP37-Mediated m6A Methylation Accelerates Decay of STM and WUS Transcripts.

(A) qPCR analyses of transcripts levels of STM, WUS, TUB8, and WRKY6 in 5-day-old wild-type and fip37–4 LEC:FIP37 seedlings treated by actinomycin D versus mock treated for 0, 8, and 24 hr. The relative expression of each gene at different time points was calculated by normalizing the gene expression in actinomycin D-treated samples against that in mock-treated samples. Error bars, mean ± SD; *p < 0.001, two-tailed paired Student’s t test; n = 3 biological replicates.
(B) A model describing regulation of shoot meristem proliferation by FIP37-mediated m6A modification in Arabidopsis. FIP37 determines the m6A mRNA modification landscape and acts as a core component of the plant methyltransferase complex including MTA. In the wild-type SAM (left panel), FIP37-mediated m6A modification confines WUS expression in the organizing center (blue dotted circle) and STM expression in the undifferentiated region of the SAM (green dotted lines). Loss of FIP37 abolishes m6A modification of WUS and STM (right panel). This delays decay of WUS and STM mRNAs, causing their expanded expression and a resulting overproliferated SAM. SAM, shoot apical meristem; LP, leaf primordium.
See also Table S4.
In mammals, WTAP and m6A play a role in mediating alternative splicing (Dominissini et al., 2012; Ping et al., 2014). To determine whether FIP37 also affects splicing, we calculated the number of alternative splicing events according to 12 basic types using our RNA-seq data. There were no significant differences in all alternative splicing events between wild-type and fip37–4 LEC1:FIP37 seedling (Figure S5G and Table S4), suggesting that FIP37 is not responsible for RNA splicing in Arabidopsis. In particular, the patterns of alternative splicing of STM and WUS remained the same in wild-type and fip37–4 LEC1:FIP37 seedlings (Table S4), demonstrating that FIP37 does not affect the RNA splicing of STM and WUS.
DISCUSSION
m6A is the conserved and most abundant mRNA modification that affects almost all aspects of RNA metabolism. Loss of function of core components of m6A methyltransferase complexes in mammals, including METTL3 and WTAP, leads to embryonic lethality (Fukusumi et al., 2008; Geula et al., 2015), which makes it technically difficult to study the developmental effects of m6Ain multicellular organisms. Thus, by far most of the studies on m6A have been restricted in cell lines, and the molecular mechanisms of the role of m6A in the development of multicellular organisms remain largely unknown. Through rescue of embryonic lethality of null mutants of FIP37, the Arabidopsis ortholog of WTAP, using embryo-specific expression of FIP37, here we have shown for the first time that a plant m6A methyltransferase component FIP37 governs the distribution pattern of m6A mRNA modification in the transcriptome and plays an indispensable role in regulating shoot stem cell fate in Arabidopsis (Figure 7B).
FIP37 affects almost all m6A peaks identified in wild-type seedlings, most of which are fully dependent on FIP37. This suggests that FIP37 acts as a fundamental component of the plant methyltransferase complex including the Arabidopsis ortholog of METTL3, MTA (Bodi et al., 2012; Zhong et al., 2008), whose role in sculpting the m6A mRNA modification landscape is still unknown. Notably, FIP37-mediated m6A exhibits two unique features that could be specific in plants. First, the mammal counterpart of FIP37, WTAP, is localized in nuclear speckles where it interacts with METTL3 and METTL14, possibly facilitating m6A methylation associated with alternative splicing (Liu et al., 2014; Ping et al., 2014). Consistently, there is a correlation of m6A methylation with multi-isoform genes and differentially spliced exons and introns in mammals (Dominissini et al., 2012), indicating a role of m6A in mRNA splicing. In contrast, FIP37 islocalized throughout the nucleoplasm excluding nucleoli in living plant cells, and does not obviously affect the patterns of alternative splicing in the transcriptome, suggesting that m6A mediated by FIP37 does not mainly contribute to alternative splicing in plants. Second, in mammals, m6A is preferentially associated with transcripts expressed at medium levels (Batista et al., 2014; Dominissini et al., 2012), and WTAP-dependent methylation is usually not associated with abundant transcripts (Schwartz et al., 2014). However, FIP37-mediated m6A is more likely to mark highly expressed transcripts in plants. Thus, despite the known conservation of m6A RNA methylation in eukaryotes, plants may evolve with different strategies in m6A methylation machinery and its effect on RNA metabolism.
Our results have also revealed a new layer of gene regulation underlying control of shoot stem cell fate in plants. The SAM containing pluripotent stem cells is an ultimate source of all aerial parts of a plant. A balance between proliferation and differentiation of the SAM allows the plant to keep the regenerative power while producing other specialized cell types to support its continuous growth. Downregulation of FIP37 results in extremely enlarged SAMs and an arrest in generating aerial tissues, which considerably shortens the lifespan of Arabidopsis plants. These dramatic phenotypes are specifically associated with loss of m6A modification and significantly decreased mRNA decay of WUS and STM transcripts. Our results suggest that dynamic restriction of both WUS and STM expression in the SAM is directly regulated by FIP37-mediated m6A modification, which is crucial for sustainable post-embryonic development. While WUS and STM have been suggested to act in independent pathways (Lenhard et al., 2002; Mayer et al., 1998), regulation of WUS and STM by FIP37-mediated m6A modification permits a coordinated control of their respective roles in promoting the specification of stem cells and antagonizing cell differentiation, thus achieving an appropriate control of SAM proliferation and differentiation.
EXPERIMENTAL PROCEDURES
Plant Material and Growth Conditions
Arabidopsis thaliana plants were grown on soil or Murashige and Skoog (MS) plates under long-day conditions (16 hr light/8 hr dark) at 23°C ± 2°C. The mutants fip37–1S, fip37–4, wus-8, and stm-10 were in the Col background. Seeds of fip37–1S (CS16192), fip37–4 (SALK_018636), wus-8 (CS349353), and stm-10 (CS3781) were obtained from the Arabidopsis Biological Resource Center (http://www.arabidopsis.org/).
Expression Analysis
Total RNA was extracted using the RNeasy Plus Mini kit (QIAGEN) and reverse transcribed using the M-MLV Reverse Transcriptase (Promega) according to the manufacturer’s instructions. Quantitative real-time PCR was performed on three biological replicates using 7900HT Fast Real-Time PCR systems (Applied Biosystems) with Maxima SYBR Green/ROX qPCR Master Mix (Fermentas). The expression of TUB2 was used as an internal control. The relative expression levels of various genes were calculated as previously described (Liu et al., 2007). The difference between the cycle threshold (Ct) of target genes and the Ct of control primers (ΔCt = Cttarget gene – CtControl) was used to calculate the normalized expression of target genes. The primers used for gene expression analysis are listed in Supplemental Experimental Procedures.
Measurement of the m6A/A Ratio by Liquid Chromatography-Tandem Mass Spectrometry
mRNA samples were digested into single ribonucleosides as previously described (Su et al., 2014), followed by a spin-filter cleanup. Samples were subjected to LC-MS/MS analysis on an Agilent 6490 Triple Quadrupole mass spectrometer.
Dot Blot Analysis of m6A Levels
Purified mRNA was first denatured by heating at 95°C for 3 min, followed by chilling on ice directly. mRNA was spotted on a Hybond-N+ membrane (Amersham) optimized for nucleic acid transfer. After UV crosslinking in a Stratalinker 2400 UV Crosslinker (Stratagene), the membrane was washed by 1 × PBST buffer (PBS with Tween-20), blocked with 5% of non-fat milk in PBST, and incubated with anti-m6A antibody (1:250; Synaptic Systems) overnight at 4°C. After incubating with horseradish-peroxidase-conjugated anti-rabbit IgG secondary antibody (Santa Cruz), the membrane was visualized by an ECL Western Blotting Detection Kit (Thermo).
m6A Sequencing and Validation
Five-day-old wild-type and fip37–4 LEC1:FIP37 seedlings grown on MS plates were collected. fip37–4 LEC1:FIP37 were identified in the segregated progenies from fip37–4/+ LEC1:FIP37 under a microscope. Total RNA was extracted using the RNeasy Plus Mini kit (QIAGEN) according to the manufacturer’s instructions. m6A sequencing was performed as previously described (Dominissini et al., 2013). RNA samples were fragmented into ~100-nucleotide-long fragments by RNA fragmentation buffer (10 mM Tris-HCl [pH 7.0], 10 mM ZnCl2). Fragmented RNA was incubated for 2 hr at 4°C with 0.5 mg/ml anti-m6A polyclonal antibody (202–003; Synaptic Systems) in IP buffer (150 mM NaCl, 0.1% Igepal CA-630, 10 mM Tris-HCl [pH 7.4]) supplemented with RNasin Plus RNase inhibitor (Promega). The mixture was then immunoprecipitated by incubation with Protein A/G Plus-Agarose (Santa Cruz) that was pre-bound with BSA at 4°C for an additional 2 hr. After extensive washing, bound RNA was eluted from the beads with 6.7 mM N6-methyladenosine (Sigma-Aldrich) in IP buffer, and precipitated by ethanol. NEBNext Ultra Directional RNA Library Prep Kit for Illumina (NEB) was used to construct the libraries from immunoprecipitated RNA and input RNA. Sequencing was performed on the Illumina Hiseq 2500 platform.
For validation of m6A sequencing results, m6A immunoprecipitation was performed using either whole seedlings or SAMs of 5-day-old wild-type and fip37–4 LEC1:FIP37 plants grown on MS plates. RNA samples were fragmented into ~200-nucleotide-long fragments using RNA fragmentation buffer (Ambion). The input RNA and immunoprecipitated RNA were reverse transcribed with random hexamers (Invitrogen) using M-MLV Reverse Transcriptase (Promega). Relative enrichment of each gene was determined by quantitative real-time PCR and calculated first by normalizing the amount of a target cDNA fragment against that of TUB2 as an internal control, and then by normalizing the value for the immunoprecipitated sample against that for the input. The primers used for real-time PCR are listed in Supplemental Experimental Procedures.
Peak Calling and Analysis
Sequence data were analyzed according to the procedure previously described (Dominissini et al., 2013). Briefly, the BWA with MEN algorithm was run to align read sequences of the input and IP samples to the Columbia reference genome and annotation file (TAIR10). Non-unique reads with MAPQ less than 13 were discarded from downstream analysis. The peak-caller MACS (Dominissini et al., 2013; Feng et al., 2011) was used for peak detection with default settings, and the parameter of effective genome size was adjusted to the calculated transcriptome size (65084215). Peaks were considered if their MACS-assigned FDR value was less than 0.05 and fold change more than 1.3.
Motif Analysis
The consensus m6A motif sequences were analyzed as previously described (Luo et al., 2014). The 100 nucleotide sequences centered on all the summits of m6A peaks were used for the analysis.
RNA-Seq
Five-day-old wild-type and fip37–4 LEC1:FIP37 seedlings grown on MS plates were harvested for total RNA extraction using the RNeasy Plus Mini kit (QIAGEN) according to the manufacturer’s instructions. The amount and quality of RNA were tested by Qubit RNA Assay Kit (Life Technologies), gel electrophoresis, and an Agilent Bioanalyzer 2100 system. rRNA was removed by a Ribo-Zero rRNA Removal Kit (Epicentre). A NEBNext Ultra Directional RNA Library Prep Kit for Illumina (NEB) was used to construct the library, and sequencing was performed on the Illumina Hiseq 2500 platform.
Quality Control of the Raw Read Library
Raw data (raw reads) of fastq format were first processed using in-house Perl scripts. We further obtained clean data (clean reads) by removing reads containing adapter, reads containing ploy-N, and other low quality reads from the raw data. Q20, Q30, and GC content of the clean data were simultaneously calculated. All downstream analyses were based on the resulting clean data with high quality.
Mapping and Transcriptome Assembly
Clean reads were aligned to the Arabidopsis reference TAIR10 using TopHat v2.0.9. The mapped reads of each sample were assembled by Cufflinks v2.1.1 (Trapnell et al., 2010).
Quantification of Gene Expression Levels
Cuffdiff was used to calculate FPKMs of coding genes in each sample (Trapnell et al., 2010). FPKMs indicate fragments per kilobase of exon per million fragments mapped, which are calculated based on the length of the fragments and read counts mapped to these fragments. Gene FPKMs were computed by summing the FPKMs of transcripts in each gene group.
Differential Expression Analysis
Cuffdiff provides statistical routines for determining differential gene expression using a model based on the negative binomial distribution (Trapnell et al., 2010). Transcripts that displayed a significant difference (adjusted p value <0.05 and fold change >1.5) in expression levels in fip37–4 LEC1: FIP37 versus wild-type seedlings were assigned as differentially expressed transcripts.
RNA Immunoprecipitation
RIP was performed as previously described (Koster and Staiger, 2014) with minor modifications. Briefly, 5-day-old fip37–4 gFIP37–4HA seedlings were fixed on ice for 30 min in 1% formaldehyde supplemented with RNasin Plus RNase inhibitor (Promega) and phenylmethylsulfonyl fluoride under a vacuum. Fixed tissues were homogenized, and the extract was subjected to immunoprecipitation with HA antibody (sc-7392, Santa Cruz) bound to Dynabeads Protein G (Invitrogen). The input RNA and immunoprecipitated RNA were reverse transcribed with random hexamers (Invitrogen) using M-MLV Reverse Transcriptase (Promega). Relative enrichment of each gene was determined by quantitative real-time PCR and calculated first by normalizing the amount of a target cDNA fragment against that of TUB2 as an internal control, and then by normalizing the value for immunoprecipitated samples against that for the input.
Analysis of mRNA Stability
Five-day-old wild-type and fip37–4 LEC1:FIP37 seedlings grown on MS plates were harvested and incubated in liquid MS medium containing 10 μM actinomycin D (Sigma) or DMSO for different time periods. Total RNA was then extracted for reverse transcription and subsequent quantification of expression levels of various genes.
Supplementary Material
Highlights.
FIP37 is a core component of the plant m6A methyltransferase complex
FIP37 is essential for sculpting the global m6A Mrna modification landscape
RNA modification mediated by FIP37 confines expression of key stem cell regulators
fip37 mutants exhibit massive overproliferation of SAMs without aerial organs
ACKNOWLEDGMENTS
We thank the Arabidopsis Biological Resource Centre, J.C. Fletcher, and T. Ito for providing seeds and plasmids, Y. Wang for technical assistance, and members of the Yu lab for discussion and comments on the manuscript. This work was supported by an Academic Research Fund (MOE2015-T2–1-002) from the Ministry of Education-Singapore, the Singapore National Research Foundation Investigatorship Programme (NRF-NRFI2016–02), and intramural research support from National University of Singapore and Temasek Life Sciences Laboratory.
Footnotes
ACCESSION NUMBERS
The high-throughput sequencing data performed in this study were deposited in the NCBI Gene Expression Omnibus (GEO) database with the accession number GEO: GSE75508.
SUPPLEMENTAL INFORMATION
Supplemental Information includes Supplemental Experimental Procedures, seven figures, and four tables and can be found with this article online at http://dx.doi.org/10.1016/j.devcel.2016.06.008.
REFERENCES
- Audas TE, Jacob MD, and Lee S (2012). Immobilization of proteins in the nucleolus by ribosomal intergenic spacer noncoding RNA. Mol. Cell 45, 147–157. [DOI] [PubMed] [Google Scholar]
- Barton MK, and Poethig RS (1993). Formation of the shoot apical meristem in Arabidopsis thaliana: an analysis of development in the wild type and in the shoot meristemless mutant. Development 119, 823–831. [Google Scholar]
- Batista PJ, Molinie B, Wang J, Qu K, Zhang J, Li L, Bouley DM, Lujan E, Haddad B, Daneshvar K, et al. (2014). m6A RNA modification controls cell fate transition in mammalian embryonic stem cells. Cell Stem Cell 15, 707–719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bodi Z, Zhong S, Mehra S, Song J, Graham N, Li H, May S, and Fray RG (2012). Adenosine methylation in Arabidopsis mRNA is associated with the 3’ end and reduced levels cause developmental defects. Front. Plant Sci. 3, 48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brand U, Fletcher JC, Hobe M, Meyerowitz EM, and Simon R (2000). Dependence of stem cell fate in Arabidopsis on a feedback loop regulated by CLV3 activity. Science 289, 617–619. [DOI] [PubMed] [Google Scholar]
- Clancy MJ, Shambaugh ME, Timpte CS, and Bokar JA (2002). Induction of sporulation in Saccharomyces cerevisiae leads to the formation of N6-methyladenosine in mRNA: a potential mechanism for the activity of the IME4 gene. Nucleic Acids Res. 30, 4509–4518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalman FC, Scherrer LC, Taylor LP, Akil H, and Pratt WB (1991). Localization of the 90-kDa heat shock protein-binding site within the hormone-binding domain of the glucocorticoid receptor by peptide competition. J. Biol. Chem. 266, 3482–3490. [PubMed] [Google Scholar]
- Dominissini D, Moshitch-Moshkovitz S, Schwartz S, Salmon-Divon M, Ungar L, Osenberg S, Cesarkas K, Jacob-Hirsch J, Amariglio N, Kupiec M, et al. (2012). Topology of the human and mouse m6A RNA methylomes revealed by m6A-seq. Nature 485, 201–206. [DOI] [PubMed] [Google Scholar]
- Dominissini D, Moshitch-Moshkovitz S, Salmon-Divon M, Amariglio N, and Rechavi G (2013). Transcriptome-wide mapping of N6-methyladenosine by m6A-seq based on immunocapturing and massively parallel sequencing. Nat. Protoc. 8, 176–189. [DOI] [PubMed] [Google Scholar]
- Endrizzi K, Moussian B, Haecker A, Levin JZ, and Laux T (1996). The SHOOT MERISTEMLESS gene is required for maintenance of undifferentiated cells in Arabidopsis shoot and floral meristems and acts at a different regulatory level than the meristem genes WUSCHEL and ZWILLE. Plant J. 10, 967–979. [DOI] [PubMed] [Google Scholar]
- Feng J, Liu T, and Zhang Y (2011). Using MACS to identify peaks from ChlP-Seq data. Curr. Protoc. Bioinformatics Chapter 2, Unit 2 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fletcher JC, Brand U, Running MP, Simon R, and Meyerowitz EM (1999). Signaling of cell fate decisions by CLAVATA3 in Arabidopsis shoot meristems. Science 283, 1911–1914. [DOI] [PubMed] [Google Scholar]
- Fray RG, and Simpson GG (2015). The Arabidopsis epitranscriptome. Curr. Opin. Plant Biol. 27, 17–21. [DOI] [PubMed] [Google Scholar]
- Fu Y, Dominissini D, Rechavi G, and He C (2014). Gene expression regulation mediated through reversible m6A RNA methylation. Nat. Rev. Genet. 15, 293–306. [DOI] [PubMed] [Google Scholar]
- Fukusumi Y, Naruse C, and Asano M (2008). Wtap is required for differentiation of endoderm and mesoderm in the mouse embryo. Dev. Dyn. 237, 618–629. [DOI] [PubMed] [Google Scholar]
- Fustin JM, Doi M, Yamaguchi Y, Hida H, Nishimura S, Yoshida M, Isagawa T, Morioka MS, Kakeya H, Manabe I, et al. (2013). RNA-methylation-dependent RNA processing controls the speed of the circadian clock. Cell 155, 793–806. [DOI] [PubMed] [Google Scholar]
- Geula S, Moshitch-Moshkovitz S, Dominissini D, Mansour AA, Kol N, Salmon-Divon M, Hershkovitz V, Peer E, Mor N, Manor YS, et al. (2015). m6A mRNA methylation facilitates resolution of naive pluripotency toward differentiation. Science 347, 1002–1006. [DOI] [PubMed] [Google Scholar]
- Jasinski S, Piazza P, Craft J, Hay A, Woolley L, Rieu I, Phillips A, Hedden P, and Tsiantis M (2005). KNOX action in Arabidopsis is mediated by coordinate regulation of cytokinin and gibberellin activities. Curr. Biol. 15, 1560–1565. [DOI] [PubMed] [Google Scholar]
- Jia GF, Fu Y, Zhao X, Dai Q, Zheng GQ, Yang Y, Yi CQ, Lindahl T, Pan T, Yang YG, et al. (2011). N6-Methyladenosine in nuclear RNA is a major substrate of the obesity-associated FTO. Nat. Chem. Biol. 7, 885–887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia G, Fu Y, and He C (2013). Reversible RNA adenosine methylation in biological regulation. Trends Genet. 29, 108–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanrar S, Onguka O, and Smith HM (2006). Arabidopsis inflorescence architecture requires the activities of KNOX-BELL homeodomain heterodimers. Planta 224, 1163–1173. [DOI] [PubMed] [Google Scholar]
- Koster T, and Staiger D (2014). RNA-binding protein immunoprecipitation from whole-cell extracts. Methods Mol. Biol. 1062, 679–695. [DOI] [PubMed] [Google Scholar]
- Laux T, Mayer KF, Berger J, and Jurgens G (1996). The WUSCHEL gene is required for shoot and floral meristem integrity in Arabidopsis. Development 122, 87–96. [DOI] [PubMed] [Google Scholar]
- Lenhard M, Jurgens G, and Laux T (2002). The WUSCHEL and SHOOT MERISTEMLESS genes fulfil complementary roles in Arabidopsis shoot meristem regulation. Development 129, 3195–3206. [DOI] [PubMed] [Google Scholar]
- Levis R, and Penman S (1978). 5’-terminal structures of poly(A)+ cytoplasmic messenger RNA and of poly(A)+ and poly(A)-heterogeneous nuclear RNA of cells of the dipteran Drosophila melanogaster. J. Mol. Biol. 120, 487–515. [DOI] [PubMed] [Google Scholar]
- Lincoln C, Long J, Yamaguchi J, Serikawa K, and Hake S (1994). A knotted1-like homeobox gene in Arabidopsis is expressed in the vegetative meristem and dramatically alters leaf morphology when overexpressed in transgenic plants. Plant Cell 6, 1859–1876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu C, Zhou J, Bracha-Drori K, Yalovsky S, Ito T, and Yu H (2007). Specification of Arabidopsis floral meristem identity by repression of flowering time genes. Development 134, 1901–1910. [DOI] [PubMed] [Google Scholar]
- Liu J, Yue Y, Han D, Wang X, Fu Y, Zhang L, Jia G, Yu M, Lu Z, Deng X, et al. (2014). A METTL3-METTL14 complex mediates mammalian nuclear RNA N6-adenosine methylation. Nat. Chem. Biol. 10, 93–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long JA, Moan EI, Medford JI, and Barton MK (1996). A member of the KNOTTED class of homeodomain proteins encoded by the STM gene of Arabidopsis. Nature 379, 66–69. [DOI] [PubMed] [Google Scholar]
- Lotan T, Ohto M,Yee KM, West MA, Lo R, Kwong RW,Yamagishi K, Fischer RL, Goldberg RB, and Harada JJ (1998). Arabidopsis LEAFY COTYLEDON1 is sufficient to induce embryo development in vegetative cells. Cell 93, 1195–1205. [DOI] [PubMed] [Google Scholar]
- Luo GZ, MacQueen A, Zheng G, Duan H, Dore LC, Lu Z, Liu J, Chen K, Jia G, Bergelson J, et al. (2014). Uniquefeatures of the m6A methylome in Arabidopsis thaliana. Nat. Commun. 5, 5630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayer KF, Schoof H, Haecker A, Lenhard M, Jurgens G, and Laux T (1998). Role of WUSCHEL in regulating stem cell fate in the Arabidopsis shoot meristem. Cell 95, 805–815. [DOI] [PubMed] [Google Scholar]
- Nichols JL (1979). ‘Cap’ structures in maize poly(A)-containing RNA. Biochim. Biophys. Acta 563, 490–495. [DOI] [PubMed] [Google Scholar]
- Parcy F,Valon C, Raynal M, Gaubier-Comella P, Delseny M, and Giraudat J (1994). Regulation of gene expression programs during Arabidopsis seed development: roles of the ABI3 locus and of endogenous abscisic acid. Plant Cell 6, 1567–1582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ping XL, Sun BF, Wang L, Xiao W, Yang X, Wang WJ, Adhikari S, Shi Y, Lv Y, Chen YS, et al. (2014). Mammalian WTAP is a regulatory subunit of the RNA N6-methyladenosine methyltransferase. Cell Res. 24, 177–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoof H, Lenhard M, Haecker A, Mayer KF, Jurgens G, and Laux T (2000). The stem cell population of Arabidopsis shoot meristems in maintained by a regulatory loop between the CLAVATA and WUSCHEL genes. Cell 100, 635–644. [DOI] [PubMed] [Google Scholar]
- Schwab R, Ossowski S, Riester M, Warthmann N, and Weigel D (2006). Highly specific gene silencing by artificial microRNAs in Arabidopsis. Plant Cell 18, 1121–1133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz S, Mumbach MR, Jovanovic M, Wang T, Maciag K, Bushkin GG, Mertins P, Ter-Ovanesyan D, Habib N, Cacchiarelli D, et al. (2014). Perturbation of m6A writers reveals two distinct classes of mRNA methylation at internal and 5’ sites. Cell Rep. 8, 284–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Semiarti E, Ueno Y,Tsukaya H, Iwakawa H, Machida C, and Machida Y (2001).The ASYMMETRIC LEAVES2 gene of Arabidopsis thaliana regulates formation of a symmetric lamina, establishment of venation and repression of meristem-related homeobox genes in leaves. Development 128, 1771–1783. [DOI] [PubMed] [Google Scholar]
- Su D, Chan CT, Gu C, Lim KS, Chionh YH, McBee ME, Russell BS, Babu IR, Begley TJ, and Dedon PC (2014). Quantitative analysis of ribo-nucleoside modifications in tRNA by HPLC-coupled mass spectrometry. Nat. Protoc. 9, 828–841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C, Williams BA, Pertea G, Mortazavi A, Kwan G, van Baren MJ, Salzberg SL, Wold BJ, and Pachter L (2010). Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 28, 511–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vespa L, Vachon G, Berger F, Perazza D, Faure JD, and Herzog M (2004). The immunophilin-interacting protein AtFIP37 from Arabidopsis is essential for plant development and is involved in trichome endoreduplication. Plant Physiol. 134, 1283–1292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vernoux T, Besnard F, and Traas J (2010). Auxin at the shoot apical meristem. Cold Spring Harb. Perspect. Biol. 2, a001487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Lu Z, Gomez A, Hon GC, Yue Y, Han D, Fu Y, Parisien M, Dai Q, Jia G, et al. (2014a). N6-methyladenosine-dependent regulation of messenger RNA stability. Nature 505, 117–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Li Y, Toth JI, Petroski MD, Zhang Z, and Zhao JC (2014b). N6-methyladenosine modification destabilizes developmental regulators in embryonic stem cells. Nat. Cell Biol. 16, 191–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Zhao BS, Roundtree IA, Lu Z, Han D, Ma H, Weng X, Chen K, Shi H, and He C (2015). N6-methyladenosine modulates messenger RNA translation efficiency. Cell 161, 1388–1399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei CM, Gershowitz A, and Moss B (1975). Methylated nucleotides block 5’ terminus of HeLa cell messenger RNA. Cell 4, 379–386. [DOI] [PubMed] [Google Scholar]
- Xu C, Wang X, Liu K, Roundtree IA, Tempel W, Li Y, Lu Z, He C, and Min J (2014). Structural basis for selective binding of m6A RNA by the YTHDC1 YTH domain. Nat. Chem. Biol. 10, 927–929. [DOI] [PubMed] [Google Scholar]
- Yadav RK, Perales M, Gruel J, Girke T, Jonsson H, and Reddy GV (2011). WUSCHEL protein movement mediates stem cell homeostasis in the Arabidopsis shoot apex. Genes Dev. 25, 2025–2030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yue Y, Liu J, and He C (2015). RNA N6-methyladenosine methylation in post-transcriptional gene expression regulation. Genes Dev. 29, 1343–1355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao X, Yang Y, Sun BF, Shi Y, Yang X, Xiao W, Hao YJ, Ping XL, Chen YS, Wang WJ, et al. (2014). FTO-dependent demethylation of N6-methyladenosine regulates mRNA splicing and is required for adipo- genesis. Cell Res. 24, 1403–1419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng GQ, Dahl JA, Niu YM, Fedorcsak P, Huang CM, Li CJ, Vagbo CB, Shi Y, Wang WL, Song SH, et al. (2013). ALKBH5 is a mammalian RNA demethylase that impacts RNA metabolism and mouse fertility. Mol.Cell 49,18–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong S, Li H, Bodi Z, Button J, Vespa L, Herzog M, and Fray RG (2008). MTA is an Arabidopsis messenger RNA adenosine methylase and interacts with a homolog of a sex-specific splicing factor. Plant Cell 20, 1278–1288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou J, Wan J, Gao X, Zhang X, Jaffrey SR, and Qian SB (2015). Dynamic mA mRNA methylation directs translational control of heat shock response. Nature 526, 591–594. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.