

Abstract
Fibroblast growth factor (FGF)-21, a member of the family of FGFs, exhibits protective effects against myocardial ischemia and ischemia/reperfusion injury; it is also an enhancer of autophagy. However, the mechanisms underlying the protective role of FGF-21 against cardiomyocyte hypoxia/reoxygenation (H/R) injury remain unclear. The present study aimed to investigate the effect of FGF-21 on H9c2 cardiomyocyte injury induced by H/R and the mechanism associated with changes in autophagy. Cultured H9c2 cardiomyocytes subjected to hypoxia were treated with a vehicle or FGF-21 during reoxygenation. The viability of H9c2 rat cardiomyocytes was measured using Cell Counting Kit-8 and trypan blue exclusion assays. The contents of creatine kinase (CK) and creatine kinase isoenzymes (CK-MB), cardiac troponin I (cTnT), cardiac troponin T (cTnI) and lactate dehydrogenase (LDH) in culture medium were detected with a CK, CK-MB, cTnT, cTnI and LDH assay kits. The protein levels were examined by western blot analysis. Autophagic flux was detected by Ad-mCherry-GFP-LC3B autophagy fluorescent adenovirus reagent. The results indicated that FGF-21 alleviated H/R-induced H9c2 myocardial cell injury and enhanced autophagic flux during H/R, and that this effect was antagonized by co-treatment with 3-methyladenine, an autophagy inhibitor. Furthermore, FGF-21 increased the expression levels of Beclin-1 and Vps34 proteins, but not of mechanistic target of rapamycin. These data indicate that FGF-21 treatment limited H/R injury in H9c2 cardiomyocytes by promoting autophagic flux through upregulation of the expression levels of Beclin-1 and Vps34 proteins.
Keywords: FGF-21, autophagy, hypoxia/reoxygenation, cardiomyocytes
Introduction
Cardiovascular disease is a significant threat to human health and is the second leading cause of mortality following malignant tumors (1). Acute myocardial infarction is one of the major causes of morbidity and mortality in coronary artery cardiovascular disease, which may lead to myocardial cell damage and heart dysfunction. Timely restoration of coronary artery blood perfusion by antiplatelet and anticoagulant therapy, percutaneous coronary intervention, and angioplasty treatment technology are the most effective treatments for myocardial ischemia at present (2-4). However, the accompanying ischemia/reperfusion (I/R) injury, which is associated with poor prognosis, may cause cell death and aggravate myocardial injury. Therefore, methods for effectively preventing and mitigating I/R injury have been a major topic of discussion in the field of myocardial protection studies.
Fibroblast growth factor (FGF-21) is a member of the FGF family (5). It regulates the metabolism of glucose and lipids and serves important roles in obesity, diabetes mellitus, metabolic syndrome and nonalcoholic fatty liver disease (6,7). It may also antagonize the development of a number of cardiovascular diseases, including atherosclerosis, coronary heart disease, myocardial ischemia, myocardial hypertrophy, diabetes, cardiomyopathy and hypertension (8-11). A previous study indicated that FGF-21 may protect the heart from ischemia (12). Other studies have suggested that FGF21 protects the heart from myocardial ischemia and I/R injury by activating the protein kinase B (AKT) and 5-adenosine monophosphate-activated protein kinase (AMPK) signaling pathway through its binding with the FGF receptor 1, and that the underlying mechanisms are associated with anti-apoptotic and anti-inflammatory effects, antioxidant stress, and myocardial cell energy metabolism and mitochondrial function regulation, among others (13). However, the mechanisms underlying the protective effect of FGF-21 are not completely explained by these aforementioned interactions.
Autophagy is a process that involves the degradation and digestion of mature proteins and cell structures through lysosomal degradation pathways, to maintain cellular homeostasis and organelle recycling (13,14). In selective autophagy, targets are recognized by autophagy receptors, including Sequestosome 1 (p62), that initiate membrane recruitment through interaction with microtubule-associated proteins 1A/1B light chain 3A (LC3) and other autophagy-associated proteins. Zhao et al (15) demonstrated that acetylcholine may increase the tolerance of the myocardium to hypoxia/reoxygenation (H/R) injury by activating autophagy though the AMPK-mechanistic target of rapamycin (mTOR) pathway. Previous studies have indicated that, by inducing autophagy, FGF-21 may ameliorate nonalcoholic fatty liver disease and alleviate inflammation and fibrosis in type 1 diabetic mouse heart (7,16). Rühlmann et al (17) suggested that FGF-21 exerts neuroprotective effects on apolipoprotein E-knockout (ApoE-KO) mice with long-term restricted caloric intake by prolonged activation of the AMPK-mTOR signaling pathway. However, the role of FGF-21 in autophagy during H9c2 cardiomyocyte H/R remains unclear. The present study aimed to investigate the effect of FGF-21 on H9c2 cardiomyocyte injury induced by H/R and the mechanism associated with changes in autophagy. Cultured H9c2 cardiomyocytes subjected to hypoxia were treated with a vehicle or FGF-21 during reoxygenation. The viability of H9c2 rat cardiomyocytes was measured using Cell Counting Kit-8 (CCK-8) and trypan blue exclusion assays. The contents of creatine kinase (CK) and CK isoenzymes (CK-MB), cardiac troponin I (cTnT), cardiac troponin T (cTnI) and lactate dehydrogenase (LDH) in culture medium were detected with a CK, CK-MB, cTnT, cTnI and LDH assay kits. The protein levels were examined by western blot analysis. Autophagic flux was detected by Ad-mCherry-GFP-LC3B autophagy fluorescent adenovirus reagent.
Materials and methods
Cell culture
H9c2 rat cardiomyocyte cells were purchased from the Shanghai Institutes for Biological Sciences, Chinese Academy of Sciences (Shanghai, China). They were cultured in Dulbecco's modified Eagle's medium (DMEM; HyClone, Thermo Fisher Scientific, Inc., Wilmington, DE, USA) supplemented with 10% fetal bovine serum (FBS; HyClone; Thermo Fisher Scientific, Inc.) at 37°C in a humidified incubator supplied with 5% CO2. When the cells grew to 70-80% confluence, they were treated with serum-free DMEM for 12 h for synchronization, following which the experiments were conducted.
H/R cell model and drug administration
Serum medium was removed from the synchronized cardiomyocytes, they were rinsed three times with PBS, deoxygenated Hanks' balanced salt solution was added (69 mM NaCl, 5 mM KCl, 0.3 mM KH2PO4, 4 mM NaHCO3, 0.3 mM Na2HPO4, 0.5 mM MgCl2, 0.4 mM MgSO4 and 1.3 mM CaCl2) and the culture flask or plate (6-, 24- or 96-well plate) was maintained for 120 min at 37°C in an oxygen-deficient container (95% N2 and 5% CO2, O2 concentration ≤1%). Then, the solution was replaced with DMEM supplemented with 10% FBS in a suitable environment (5% CO2, 37°C) supplemented with 0, 12.5, 25, 50 or 100 ng/ml FGF-21 (ProteinTech Group, Inc., Chicago, IL USA) with or without 5 mmol/l 3-methyladenine (3-MA; Sigma-Aldrich; Merck KGaA, Darmstadt, Germany) and incubated at 37°C for 60 min for completion of the H/R model.
Viability assays
The viability of H9c2 rat cardiomyocytes was measured using CCK-8 (Vazyme Biotech Co., Ltd., Nanjing, China) and trypan blue exclusion assays (Beyotime Institute of Biotechnology, Haimen, China). A total of 2×103 cells were seeded into each well of a 96-well plate following H/R treatment. In accordance with the protocol of the manufacturer, 10 µl CCK-8 solution was added and the cells were incubated at 37°C for 4 h, following which the optical density (OD) was measured using a microplate reader (λ=450 nm). The average OD of 5 wells was recorded, and cell viability was calculated. The procedure was repeated at least three times, and cells in the control group were considered to be 100% viable. The trypan blue exclusion assay was performed as follows: 1×106 cells were cultured in each well of a 6-well plate. Following the H/R experiment, the culture medium was discarded and cells were washed three times with PBS, followed by dilution with trypan blue stock in PBS (4% final concentration). Then, 0.04% diluted trypan blue solution was added to the 6-well plate (500 µl/well). After 3 min, the stained cells were observed under a light microscope at ×10 magnification, the dead cells were dyed blue and the live cells were colorless and transparent.
Measurement of LDH, CK and CK-MB
Following the H/R experiment, the cell culture medium was collected at the end of the reoxygenation stage, and levels of LDH, CK and CK-MB in the cell culture were measured by LDH, CK and CK-MB assay kits (all from Nanjing Jiancheng Bioengineering Institute, Nanjing, China), in accordance with the manufacturer's protocol.
Measurement of cTnI and cTnT
Following the H/R experiment, the cell culture medium was collected at the end of the reoxygenation stage, and levels of cTnT and cTnI were measured with a cTnT assay kit (Roche Diagnostics GmbH, Mannheim, Germany) and a cTnI assay kit (Roche Diagnostics GmbH) with Roche Elecsys Analyser.
Ad-mCherry-green fluorescent protein (GFP)-microtubule- associated proteins 1A/1B light chain 3β (LC3B) autophagy fluorescence double labeling adenovirus autophagy assay
Ad-mCherry-GFP-LC3B autophagy fluorescent adenovirus reagent (Beyotime Institute of Biotechnology) at a multiplicity of infection of 80 was added to H9c2 cardiomyocytes that were cultured in 24-well plates (50 µl/well). After 12 h transfection of 8×106 pfu (Plaque forming units) adenovirus, H/R was performed. Following establishment of the H/R model, the expression of mCherry and GFP was visualized with confocal fluorescence microscopy at ×100 magnification. Autophagy flux was evaluated by calculating the number of yellow and red puncta (18) with Image J (version 1.8.0; National Institutes of Health, Bethesda, MD, USA).
Western blot analysis
Cells were rinsed with cold PBS twice and lysed in radioimmunoprecipitation buffer containing phenylmethylsulfonyl fluoride (all from Beyotime Institute of Biotechnology). Cell protein concentrations were measured using the BCA method (Beyotime Institute of Biotechnology). A total of 20 µg of protein was loaded on 10% SDS-PAGE gels and then transferred to polyvinylidene fluoride membranes (Roche Diagnostics Co., Ltd., Shanghai, China). Following blocking in 5% non-fat milk for 1 h at room temperature, the membranes were probed with primary antibodies (all dilutions were 1:1,000) at 4°C overnight; the following primary antibodies were used: Anti-Beclin-1 (11306-1-AP; ProteinTech Group, Inc.), anti-LC3 (14600-1-AP; ProteinTech Group, Inc.), anti-P62 (ab5641; Epitomics; Abcam, Cambridge, UK), anti-phosphatidylinositol 3-kinase (PI3K) catalytic subunit type 3 (Vps34; 20584-1-AP; ProteinTech Group, Inc.), Phospho-mTOR (Ser2448) antibody (2971; Cell Signaling Technology, Danvers, MA, USA), mTOR antibody (2972; Cell Signaling Technology) and anti-GAPDH (13937-1-AP; ProteinTech Group, Inc.). The membranes were then incubated for 2 h with secondary antibodies at 37°C (HRP-conjugated Affinipure goat anti-rabbit IgG; 1:7,000; BA1056; Wuhan Boster Biological Technology Ltd., Wuhan, China). Bands were visualized using an ECL chemiluminescent agent (CW0048; CWBio, Beijing, China). An automatic chemical luminous imaging analysis system was used for capturing images. The image analysis software ImageJ (version 1.8.0; National Institutes of Health) was used for analyzing the results, and expression of the protein was determined relative to that of GAPDH.
Statistical analysis
Statistical analysis was performed using GraphPad Prism 5.0 software (GraphPad Software, Inc., La Jolla, CA, USA). All experimental data are expressed as mean ± standard deviation. P<0.05 was considered to indicate a statistically significant difference, as determined using one-way analysis of variance with Tukey's range test.
Results
Cytoprotective effects of FGF-21 in cells during H/R injury
After 2 h of hypoxia treatment, H9c2 cells were incubated in 0, 12.5, 25, 50, and 100 ng/ml FGF-21 for 1 h at the time of reoxygenation, and the viability of the cells was measured using the CCK-8 assay. The results demonstrated that, compared with the control group, cell viability was significantly decreased in the H/R group; treatment with 25 or 50 ng/ml FGF-21 increased the viability of cells with H/R [H/R vehicle group = 35.77±6.514%; H/R + FGF-21 (25 ng/ml) group = 52.77±6.394%; and H/R + FGF-21 (50 ng/ml) group = 77.30±4.899%; n=3; P<0.05; Fig. 1A]. These data suggest that an appropriate concentration of FGF-21 may improve cell survival. Subsequent experiments were conducted with 50 ng/ml FGF-21.
Figure 1.

Effects of FGF-21 on cell survival. (A) Effects of different doses of FGF-21 (0, 12.5, 25, 50 and 100 ng/ml) on the viability of cultured H9c2 cardiomyocytes, as determined by the Cell Counting Kit-8 assay. (B) Quantitatively analysis of dead cells and (C) dead cells were visualized using trypan blue exclusion assay (blue cells). Scale bar, 100 µm. *P<0.05 vs. control group and #P<0.05 vs. H/R group. Values are presented as mean ± standard deviation. Experiments were repeated in triplicate. FGF-21, fibroblast growth factor-21; H/R, hypoxia/reoxygenation.
The trypan blue exclusion assay indicated that, compared with the control group, the rate of damaged cells was significantly increased in the H/R group; however, FGF-21 (50 ng/ml) significantly decreased the number of dead cells following exposure to H/R (Fig. 1B and C).
H/R increased levels of LDH, CK, CK-MB, cTnT and cTnI in the cell culture medium at the end of reoxygenation, which was rescued by FGF-21 (Fig. 2). These results suggest that FGF-21 significantly alleviates H/R-induced H9c2 myocardial cell injury.
Figure 2.

Effects of FGF-21 on signatures of cell injury. Cell injury was measured by (A) LDH, (B) CK, (C) CK-MB, (D) cTnT and (E) cTnI release. *P<0.05 vs. control group and #P<0.05 vs. H/R group. Values are expressed as mean ± standard deviation. Experiments were repeated in triplicate. FGF-21, fibroblast growth factor-21; H/R, hypoxia/reoxygenation; LDH, lactate dehydrogenase; CK, creatine kinase; CK-MB, creatine kinase isoenzymes; cTnT, cardiac troponin T; cTnI, cardiac troponin I.
FGF-21 enhances autophagic flux of cardiomyocytes during H/R
The western blot analysis results demonstrated that, compared with the control group, the formation of lipid modified microtubule-associated proteins 1A/1B light chain (LC3-II) and expression levels of the Beclin-1 protein in cardiomyocytes were significantly increased in the H/R group. The expression of p62 was significantly decreased, compared with the control group. p62 is a receptor that facilitates selective autophagy by interacting simultaneously with cargoes and the LC3 protein on the autophagosome to maintain cellular homeostasis. The formation of LC3-II and expression levels of Beclin-1 were additionally increased and the expression of p62 was also additionally decreased in the FGF21-treated group compared with those in the H/R group. However, this effect was partly abolished by 3-MA. Following addition of the autophagic inhibitor 3-MA, the formation of LC3-II and expression levels of Beclin-1 were downregulated and the expression of p62 was upregulated compared with those in the FGF-21-treated group (Fig. 3).
Figure 3.

Effects of FGF-21 on autophagy makers in H9c2 cardiomyocytes. (A) Western blot analysis of LC3-II protein levels. (B) Densitometric analysis of LC3-II levels. (C) Western blot analysis of Beclin-1 protein levels. (D) Densitometric analysis of Beclin-1 levels. (E) Western blot analysis of p62 protein levels. (F) Densitometric analysis of p62 levels. *P<0.05 vs. control group, #P<0.05 vs. H/R group and &P<0.05 vs. H/R + FGF-21 group. Values are presented as mean ± standard deviation. Experiments were repeated in triplicate. FGF-21, fibroblast growth factor-21; H/R, hypoxia/reoxygenation; 3-MA, 3-methylad-enine; LC3-II, lipid modified microtubule-associated proteins 1A/1B light chain; p62, sequestosome 1.
To monitor autophagic flux, tandem fluorescent mCherry-GFP-LC3B was performed on H9c2 cardiomyocytes (Ad-LC3-H9c2). Normal Ad-LC3-H9c2 cells exhibited basal autophagy with few autophagosomes and autolysosomes. Ad-LC3-H9c2 cells subjected to H/R exhibited increased autophagosomes and few autolysosomes, suggesting that autophagic flux was increased during myocardial H/R. In the FGF-21-treated group, Ad-LC3-H9c2 cells subjected to H/R possessed more autophagosomes and autolysosomes compared with those in the untreated group, indicating that FGF-21 treatment additionally enhanced autophagic flux. However, co-treatment with FGF-21 and the autophagic inhibitor 3-MA decreased autophagosomes and autolysosomes compared with those in the FGF-21-treated group. In addition, in the 3-MA-treated group, Ad-LC3-H9c2 cells subjected to H/R exhibited fewer autophagosomes and autolysosomes compared with that in the untreated group, indicating that FGF-21 treatment attenuated autophagic flux (Fig. 4). These data suggest that FGF-21 induced upregulation of autophagic flux during H/R injury.
Figure 4.
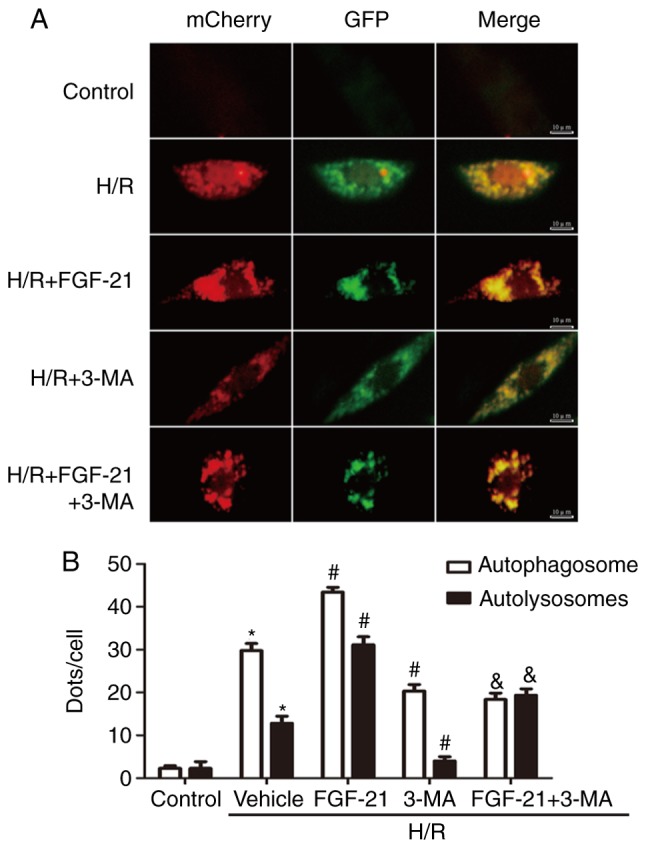
Effects of FGF-21 on autophagic flux in H9c2 cardiomyocytes. (A) H9c2 cardiomyocytes transfected with adenovirus harboring tandem fluorescent mCherry-GFP-LC3 (Ad-LC3-H9c2) for 24 h were subjected to different treatments. Representative images of immunofluorescent H9c2 cardiomyocytes expressing mRFP-GFP-LC3. Green fluorescence represents GFP and red fluorescence represents mRFP. (B) Semi-quantitative analysis of autophagosomes (yellow dots in merged images) and autolysosomes (red only dots in merged images). *P<0.05 vs. control group, #P<0.05 vs. H/R group and &P<0.05 vs. H/R + FGF-21 group. Values are expressed as mean ± standard deviation. Experiments were repeated in triplicate. FGF-21, fibroblast growth factor-21; H/R, hypoxia/reoxygenation; 3-MA, 3-methyladenine; GFP, green fluorescent protein; mRFP, monomeric red fluorescent protein.
FGF-21-mediated autophagy enhances cell survival
CCK-8 and trypan blue exclusion assays revealed that 3-MA partly abolished the effect of FGF-21 on the myocardial viability rate in the H/R + FGF-21 group (Fig. 5). These results suggest that 3-MA may reverse the protective effect of FGF-21 against H9c2 cardiomyocyte survival during H/R injury.
Figure 5.

Effects of autophagy on FGF-21-induced cell protection. (A) The Cell Counting Kit-8 assay determined cell viability in cultured H9c2 cardiomyocytes. (B) Quantitatively analysis of dead cells and (C) dead cells were visualized using trypan blue exclusion assay (blue cells). Scale bar, 100 µm. *P<0.05 vs. H/R group and #P<0.05 vs. H/R + FGF-21 group. Values are presented as mean ± standard deviation. Experiments were repeated in triplicate. FGF-21, fibroblast growth factor-21; H/R, hypoxia/reoxygenation; 3-MA, 3-methyladenine.
Compared with the H/R + FGF-21 group, levels of LDH, CK, CK-MB, cTnT and cTnI in cell culture medium at the end of reoxygenation in the H/R + FGF-21+3-MA group were significantly increased (Fig. 6), which additionally supports the hypothesis that 3-MA may reverse the protective effect of FGF-21 against H9c2 cardiomyocyte H/R injury.
Figure 6.

Effects of autophagy on FGF-21-induced decrease of signatures of cell injury. Cell injury was measured by (A) LDH, (B) CK, (C) CK-MB, (D) cTnT and (E) cTnI release. *P<0.05 vs. H/R group and #P<0.05 vs. H/R + FGF-21 group. Values are expressed as mean ± standard deviation. Experiments were repeated in triplicate. FGF-21, fibroblast growth factor-21; H/R, hypoxia/reoxygenation; 3-MA, 3-methyladenine; LDH, lactate dehydrogenase; CK, creatine kinase; CK-MB, creatine kinase isoenzymes; cTnT, cardiac troponin T; cTnI, cardiac troponin I.
Effect of FGF-21 on H/R-induced changes in mTOR and Vps34 signaling
Acetylcholine may increase the tolerance of the myocardium to H/R injury by activating autophagy through the AMPK-mTOR pathway (15). A previous study demonstrated that FGF21 exerted neuroprotective effects on ApoE-KO mice with long-term restricted caloric intake by prolonging activation of the AMPK-mTOR signaling pathway (17). The present study examined the expression of the phosphorylated (p)-mTOR protein and total mTOR protein in each group of cells: No significant differences between the control, H/R and H/R + FGF-21 groups were observed (Fig. 7A-C), indicating that FGF-21-induced autophagy in H/R cardiomyocytes may not occur through the mTOR signaling pathway.
Figure 7.

Effects of FGF-21 on expression levels of mTOR and Vps34. (A) Western blot analysis of mTOR and p-mTOR in H9c2 cardiomyocytes. The quantification of (B) mTOR/GAPDH and (C) p-mTOR/GAPDH. (D) Western blot analysis of Vps-34 in H9c2 cardiomyocytes. (E) Quantification of Vps34/GAPDH. *P<0.05 vs. control group, #P<0.05 vs. H/R group and &P<0.05 vs. H/R + FGF-21 group. Values are presented as mean ± standard deviation. Experiments were repeated in triplicate. FGF-21, fibroblast growth factor-21; H/R, hypoxia/reoxygenation; 3-MA, 3-methyladenine; mTOR, mechanistic target of rapamycin; p, phosphorylated.
The Beclin-1/Vps34 complex formed by Vps34 and Beclin-1 may modulate the formation of the autophagic membrane. The present study suggested that FGF-21 increased the expression level of the Beclin-1 protein in H/R cardio-myocytes (Fig. 3). The autophagy inhibitor 3-MA used in the present study is an inhibitor of Vps34; therefore, expression levels of the Vps34 protein were examined in each group. The results demonstrated that, compared with the control group, the expression levels of the Vsp34 protein were significantly increased in the H/R group, and it was additionally increased in the H/R + FGF-21 group. However, co-treatment with FGF-21 and the autophagic inhibitor 3-MA decreased the expression level of the Vsp34 protein compared with that in the H/R + FGF-21 group. In addition, compared with the H/R group, the expression level of the Vsp34 protein was significantly attenuated in the H/R + 3-MA group (Fig. 7D and E). These results suggest that the Vps34 protein serves a role in FGF-21-enhanced autophagy in H/R cardiomyocytes.
Discussion
Cardiomyocyte H/R models are designed to mimic in vivo myocardial I/R. At present, a number of previous studies have used this cell model for studying the mechanism of H/R injury (19-21). In the present study, compared with the control group, the survival rate of cardiomyocytes in the H/R group was significantly decreased and levels of CK, CK-MB, cTnT, cTnI and LDH were significantly increased in the culture medium, indicating that the H/R model was successfully established.
Numerous studies have demonstrated that levels of autophagy increase in the heart during I/R in animal models and in isolated cardiomyocytes during H/R (22,23). Whether autophagy has a protective or deleterious role in the I/R injury process in the heart is unclear at present. Low levels of autophagy triggered by mild-to-moderate hypoxia or ischemia are protective and appear to prevent activation of apoptosis by degrading and removing damaged mitochondria (24,25). It has also been suggested that autophagy enhances and exacerbates myocardial injury during reperfusion, indicating that excessive autophagy is detrimental to the heart (14). Ling et al (26) identified that H/R blocked autophagic flux in cardiomyocytes, causing accumulation of autophagosomes. However, the experiments in the present study indicated that autophagic flux was improved in cardiomyocytes during H/R, which may be associated with the duration of the H/R stages. In previous studies, the time intervals selected for establishing the H/R models were varied (23,27,28); for example, each stage of the hypoxia and reoxygenation model described by Ling et al (26) was 3 h. In our preliminary experiment, the highest survival rate was observed in the H9c2 cardiomyocytes treated with hypoxia for 2 h and reoxygenation for 1 h; therefore, the 2/1 h model was selected. The results from this model indicated that prolonged H/R may inhibit cardiomyocyte autophagic flux. Besides, in the model in the present study, the application of 3-MA during reoxygenation inhibited autophagic flux, aggravated cell damage and increased cell death, indicating that reoxygenation-induced autophagic flux in cardiomyocytes may occur as a compensatory response to H/R injury and serve a protective role in cells.
Previous studies have suggested that several FGFs may protect the myocardium from ischemic and/or I/R injury: Palmen et al (29) identified that FGF-1 may enhance ischemic tolerance of the myocardium, promote recovery of cardiac function following I/R injury and decrease the level of myocardial cell death; in addition, earlier studies by our group have detected that FGF-2 may antagonize myocardial apoptosis, decrease the area of infarction, ameliorate impaired heart function, improve arrhythmia and protect cardiomyocytes against myocardial ischemia and I/R injury (30-32). Singla et al (33) revealed that FGF-9 may protect mice with diabetes from myocardial infarction. The cardioprotective effects of FGF-21 have recently attracted considerable attention: A previous study has identified that the myocardial infarct size is significantly increased in FGF-21-KO mice compared with that in wild-type mice (34). In addition, injection of the FGF-21 adenovirus expression vector (Ad-FGF-21) into wild-type mice skeletal muscle with myocardial infarction increased the density of capillaries in the infarct zone, decreased myocardial apoptosis and improved the ventricular systolic and diastolic function (35). Upon injection of FGF-21 small interfering RNA, the study identified that the aforementioned myocardial protective effect of FGF-21 was reversed (12). In addition, intravenous injection of FGF-21 in FGF-21-KO mice antagonized myocardial apoptosis induced by I/R, decreased the infarct size and improved cardiac function (34). The effect of exogenous FGF-21 on infarct size and cardiac function was also demonstrated in an isolated heart perfusion model (36). Cell experiments have also suggested that exogenous FGF-21 may alleviate morphological changes in H9c2 cardiomyocytes, depress nuclear fragmentation and decrease the rate of apoptosis (37). Underlying mechanisms include anti-apoptosis activities, regulation of energy metabolism, antioxidant stress and regulation of mitochondrial function (34-39). Consistent with the results of a previous study (11), the present study identified that FGF-21 attenuated cardiomyocyte H/R injury and that co-treatment with FGF-21 and 3-MA during reoxygenation significantly attenuated autophagic flux and the protective effect on cardiomyocytes, indicating that the protective effect of FGF-21 may be associated with enhanced autophagy.
Autophagy may be regulated by the Beclin-1/Vps34 complex, AMPK/mTOR, and PI3K/AKT/mTOR signaling pathways. Rühlmann et al (17) demonstrated that FGF-21 may serve a neuroprotective role by activating the AMPK/mTOR signaling pathway in ApoE-KO mice with long-term restricted caloric intake. In addition, Zhao et al (15) identified that acetylcholine increased the tolerance of the myocardium to H/R injury, which was activated through autophagy by the AMPK-mTOR pathway. However, in the experimental results of the present study, no significant change occurred in p-mTOR and mTOR proteins following the application of FGF-21 during reoxygenation, suggesting that FGF-21-enhanced autophagy and protection of the myocardium from H/R injury does not occur through the mTOR pathway.
Beclin-1 was the earliest mammalian autophagy gene identified, which is localized on human chromosome 17q21 (40). Beclin-1 is expressed in the Golgi apparatus and primarily regulates autophagy-associated proteins through the formation of the Vps34 complex, which localizes these autophagy-associated proteins into the structure of precursor precursors, thereby regulating autophagy (41). Ling et al (26) identified that the application of 3-MA or short hairpin-Beclin-1 decreased the expression of the autophagy-associated gene Beclin-1, partly abolishing the protective effect of polydatin against cardiomyocyte death during H/R, and myocardial infarct size during I/R. Similar results were also observed in the present study using 3-MA to inhibit the expression of the autophagy-associated genes Beclin-1 and Vps34. The results revealed that H9c2 cardiomyocyte injury was significantly increased in the H/R + FGF-21 + 3-MA group compared with that in the H/R + FGF-21 treatment group, suggesting that FGF-21-associated cardioprotection occurs through the Beclin-1/Vps34 complex pathway to enhance autophagy. However, mechanisms of cross-talk between autophagy and Vps34 remain unclear and should be explored in future studies.
Initiation of autophagy causes the conversion of LC3-I to LC3-II. An increase in LC3-II band intensity and a decrease in LC3-I expression are considered to be hallmarks of autophagy. In the present study, the LC3-I levels were not examined, which was a limitation. 3-MA is an autophagy inhibitor and used widely, but Bafilomycin A may also stop autophagosomes degradation; this reagent will be included in future studies. The PI3K-AKT pathway has been indicated to serve a critical role in autophagy. Previous studies have suggested that FGFs activates AKT and increases the level of p-AKT (31,42). As AKT is a regulator of autophagy, how FGF-21 affects p-AKT will be observed in future studies. The aim of the present study was to investigate the protective effect of exogenous FGF21 on the myocardium; as no specific agonists or inhibitors of FGF21 were identified, the protein and mRNA expression levels of FGF21 in H/R-treated H9c2 cardiomyocytes were not examined. The present study also lacks in vivo experiments to improve understanding of the role of FGF21 in heart disease; this will be explored in future studies.
Acknowledgments
Not applicable.
Funding
The present study was supported by the National Natural Scientific Foundation of China (81670429 and 81470435), the Educational Department of Hunan Province Foundation (13C795), the Innovation team of Basic Medicine of University of South China and Aid Program for Science and Technology Innovative Research Team in Higher Educational Institutions of Human Province (2008-244).
Availability of data and materials
The datasets used or analyzed during the current study are available from the corresponding author on reasonable request.
Authors' contributions
ZSJ, ZR, WX, ZHT and GHL conceived and designed the study. WX, ZR, YZ, MHL, ZR, HQY and YMH performed the experiments. WX, YZ and ZR wrote the paper. SLQ and ZSJ reviewed and edited the manuscript. All authors read and approved the manuscript.
Ethics approval and consent to participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
References
- 1.World Health Organization (WHO) World health statistics 2007. WHO; Geneva: 2007. [Google Scholar]
- 2.Przyklenk K, Dong Y, Undyala VV, Whittaker P. Autophagy as a therapeutic target for ischaemia/reperfusion injury? Concepts, controversies, and challenges. Cardiovasc Res. 2012;94:197–205. doi: 10.1093/cvr/cvr358. [DOI] [PubMed] [Google Scholar]
- 3.Jennings RB. Historical perspective on the pathology of myocardial ischemia/reperfusion injury. Circ Res. 2013;113:428–438. doi: 10.1161/CIRCRESAHA.113.300987. [DOI] [PubMed] [Google Scholar]
- 4.Zhang Y, Ren J. Targeting autophagy for the therapeutic application of histone deacetylase inhibitors in ischemia/reper-fusion heart injury. Circulation. 2014;129:1088–1091. doi: 10.1161/CIRCULATIONAHA.113.008115. [DOI] [PubMed] [Google Scholar]
- 5.Planavila A, Redondo-Angulo I, Villarroya F. FGF21 and cardiac physiopathology. Front Endocrinol (Lausanne) 2015;6:133. doi: 10.3389/fendo.2015.00133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cheung BM, Deng HB. Fibroblast growth factor 21: A promising therapeutic target in obesity-related diseases. Expert Rev Cardiovasc Ther. 2014;12:659–666. doi: 10.1586/14779072.2014.904745. [DOI] [PubMed] [Google Scholar]
- 7.Zhu S, Wu Y, Ye X, Ma L, Qi J, Yu D, Wei Y, Lin G, Ren G, Li D. FGF21 ameliorates nonalcoholic fatty liver disease by inducing autophagy. Mol Cell Biochem. 2016;420:107–119. doi: 10.1007/s11010-016-2774-2. [DOI] [PubMed] [Google Scholar]
- 8.Tanajak P, Sa-Nguanmoo P, Wang X, Liang G, Li X, Jiang C, Chattipakorn SC, Chattipakorn N. Fibroblast growth factor 21 (FGF21) therapy attenuates left ventricular dysfunction and metabolic disturbance by improving FGF21 sensitivity, cardiac mitochondrial redox homoeostasis and structural changes in pre-diabetic rats. Acta Physiol (Oxf) 2016;217:287–299. doi: 10.1111/apha.12698. [DOI] [PubMed] [Google Scholar]
- 9.Luo F, Guo Y, Ruan G, Li X. Metformin promotes cholesterol efflux in macrophages by up-regulating FGF21 expression: A novel anti-atherosclerotic mechanism. Lipids Health Dis. 2016;15:109. doi: 10.1186/s12944-016-0281-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cheng P, Zhang F, Yu L, Lin X, He L, Li X, Lu X, Yan X, Tan Y, Zhang C. Physiological and pharmacological roles of FGF21 in cardiovascular diseases. J Diabetes Res. 2016;2016:1540267. doi: 10.1155/2016/1540267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Planavila A, Redondo-Angulo I, Ribas F, Garrabou G, Casademont J, Giralt M, Villarroya F. Fibroblast growth factor 21 protects the heart from oxidative stress. Cardiovasc Res. 2015;106:19–31. doi: 10.1093/cvr/cvu263. [DOI] [PubMed] [Google Scholar]
- 12.Liu SQ, Tefft BJ, Roberts DT, Zhang LQ, Ren Y, Li YC, Huang Y, Zhang D, Phillips HR, Wu YH. Cardioprotective proteins upregulated in the liver in response to experimental myocardial ischemia. Am J Physiol Heart Circ Physiol. 2012;303:H1446–H1458. doi: 10.1152/ajpheart.00362.2012. [DOI] [PubMed] [Google Scholar]
- 13.Choi AM, Ryter SW, Levine B. Autophagy in human health and disease. N Engl J Med. 2013;368:651–662. doi: 10.1056/NEJMra1205406. [DOI] [PubMed] [Google Scholar]
- 14.Lavandero S, Troncoso R, Rothermel BA, Martinet W, Sadoshima J, Hill JA. Cardiovascular autophagy: Concepts, controversies, and perspectives. Autophagy. 2013;9:1455–1466. doi: 10.4161/auto.25969. [DOI] [PubMed] [Google Scholar]
- 15.Zhao M, Sun L, Yu XJ, Miao Y, Liu JJ, Wang H, Ren J, Zang WJ. Acetylcholine mediates AMPK-dependent autophagic cytoprotection in H9c2 cells during hypoxia/reoxygenation injury. Cell Physiol Biochem. 2013;32:601–613. doi: 10.1159/000354464. [DOI] [PubMed] [Google Scholar]
- 16.Zhang J, Cheng Y, Gu J, Wang S, Zhou S, Wang Y, Tan Y, Feng W, Fu Y, Mellen N, et al. Fenofibrate increases cardiac autophagy via FGF21/SIRT1 and prevents fibrosis and inflammation in the hearts of Type 1 diabetic mice. Clin Sci (Lond) 2016;130:625–641. doi: 10.1042/CS20150623. [DOI] [PubMed] [Google Scholar]
- 17.Rühlmann C, Wölk T, Blümel T, Stahn L, Vollmar B, Kuhla A. Long-term caloric restriction in ApoE-deficient mice results in neuroprotection via Fgf21-induced AMPK/mTOR pathway. Aging. 2016;8:2777–2789. doi: 10.18632/aging.101086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yu P, Zhang C, Gao CY, Ma T, Zhang H, Zhou MM, Yang YW, Yang L, Kong LY. Anti-proliferation of triple-negative breast cancer cells with physagulide P: ROS/JNK signaling pathway induces apoptosis and autophagic cell death. Oncotarget. 2017;8:64032–64049. doi: 10.18632/oncotarget.19299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gurusamy N, Lekli I, Mukherjee S, Ray D, Ahsan MK, Gherghiceanu M, Popescu LM, Das DK. Cardioprotection by resveratrol: A novel mechanism via autophagy involving the mTORC2 pathway. Cardiovasc Res. 2010;86:103–112. doi: 10.1093/cvr/cvp384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lin CH, Wu WS, Lin MT, Liu WP, Hsu RB, Chang CP. Attenuating ischemia-induced H9c2 myoblasts apoptosis by therapeutic hypothermia. Am J Med Sci. 2010;339:258–265. doi: 10.1097/MAJ.0b013e3181ce507f. [DOI] [PubMed] [Google Scholar]
- 21.Au KW, Kou CY, Woo AY, Chim SS, Fung KP, Cheng CH, Waye MM, Tsui SK. Calcyclin binding protein promotes DNA synthesis and differentiation in rat neonatal cardiomyocytes. J Cell Biochem. 2006;98:555–566. doi: 10.1002/jcb.20710. [DOI] [PubMed] [Google Scholar]
- 22.Matsui Y, Takagi H, Qu X, Abdellatif M, Sakoda H, Asano T, Levine B, Sadoshima J. Distinct roles of autophagy in the heart during ischemia and reperfusion: Roles of AMP-activated protein kinase and Beclin 1 in mediating autophagy. Circ Res. 2007;100:914–922. doi: 10.1161/01.RES.0000261924.76669.36. [DOI] [PubMed] [Google Scholar]
- 23.Hamacher-Brady A, Brady NR, Gottlieb RA. Enhancing macroautophagy protects against ischemia/reperfusion injury in cardiac myocytes. J Biol Chem. 2006;281:29776–29787. doi: 10.1074/jbc.M603783200. [DOI] [PubMed] [Google Scholar]
- 24.Decker RS, Wildenthal K. Lysosomal alterations in hypoxic and reoxygenated hearts. I. Ultrastructural and cytochemical changes. Am J Pathol. 1980;98:425–444. [PMC free article] [PubMed] [Google Scholar]
- 25.Hamacher-Brady A, Brady NR, Gottlieb RA. The interplay between pro-death and pro-survival signaling pathways in myocardial ischemia/reperfusion injury: Apoptosis meets autophagy. Cardiovasc Drugs Ther. 2006;20:445–462. doi: 10.1007/s10557-006-0583-7. [DOI] [PubMed] [Google Scholar]
- 26.Ling Y, Chen G, Deng Y, Tang H, Ling L, Zhou X, Song X, Yang P, Liu Y, Li Z, et al. Polydatin post-treatment alleviates myocardial ischaemia/reperfusion injury by promoting autophagic flux. Clin Sci (Lond) 2016;130:1641–1653. doi: 10.1042/CS20160082. [DOI] [PubMed] [Google Scholar]
- 27.Sagrillo-Fagundes L, Assunção Salustiano EM, Ruano R, Markus RP, Vaillancourt C. Melatonin modulates autophagy and inflammation protecting human placental trophoblast from hypoxia/reoxygenation. J Pineal Res. 2018;4:e12520. doi: 10.1111/jpi.12520. [DOI] [PubMed] [Google Scholar]
- 28.Liu F, Zhang J, Qian J, Wu G, Ma Z. Baicalin attenuates liverhypoxia/reoxygenation injury by inducing autophagy. Exp Ther Med. 2018;2:657–664. doi: 10.3892/etm.2018.6284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Palmen M, Daemen MJ, De Windt LJ, Willems J, Dassen WR, Heeneman S, Zimmermann R, Van Bilsen M, Doevendans PA. Fibroblast growth factor-1 improves cardiac functional recovery and enhances cell survival after ischemia and reperfusion: A fibroblast growth factor receptor, protein kinase C, and tyrosine kinase-dependent mechanism. J Am Coll Cardiol. 2004;44:1113–1123. doi: 10.1016/j.jacc.2004.05.067. [DOI] [PubMed] [Google Scholar]
- 30.Jiang ZS, Padua RR, Ju H, Doble BW, Jin Y, Hao J, Cattini PA, Dixon IM, Kardami E. Acute protection of ischemic heart by FGF-2: Involvement of FGF-2 receptors and protein kinase C. Am J Physiol Heart Circ Physiol. 2002;282:H1071–H1080. doi: 10.1152/ajpheart.00290.2001. [DOI] [PubMed] [Google Scholar]
- 31.Jiang ZS, Srisakuldee W, Soulet F, Bouche G, Kardami E. Non-angiogenic FGF-2 protects the ischemic heart from injury, in the presence or absence of reperfusion. Cardiovasc Res. 2004;62:154–166. doi: 10.1016/j.cardiores.2004.01.009. [DOI] [PubMed] [Google Scholar]
- 32.Liu MH, Li GH, Peng LJ, Qu SL, Zhang Y, Peng J, Luo XY, Hu HJ, Ren Z, Liu Y, et al. PI3K/Akt/FoxO3a signaling mediates cardioprotection of FGF-2 against hydrogen peroxide-induced apoptosis in H9c2 cells. Mol Cell Biochem. 2016;414:57–66. doi: 10.1007/s11010-016-2658-5. [DOI] [PubMed] [Google Scholar]
- 33.Singla DK, Singla RD, Abdelli LS, Glass C. Fibroblast growth factor-9 enhances M2 macrophage differentiation and attenuates adverse cardiac remodeling in the infarcted diabetic heart. PLoS One. 2015;10:e0120739. doi: 10.1371/journal.pone.0120739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Liu SQ, Roberts D, Kharitonenkov A, Zhang B, Hanson SM, Li YC, Zhang LQ, Wu YH. Endocrine protection of ischemic myocardium by FGF21 from the liver and adipose tissue. Sci Rep. 2013;3:2767. doi: 10.1038/srep02767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Joki Y, Ohashi K, Yuasa D, Shibata R, Ito M, Matsuo K, Kambara T, Uemura Y, Hayakawa S, Hiramatsu-Ito M, et al. FGF21 attenuates pathological myocardial remodeling following myocardial infarction through the adiponectin-dependent mechanism. Biochem Biophys Res Commun. 2015;459:124–130. doi: 10.1016/j.bbrc.2015.02.081. [DOI] [PubMed] [Google Scholar]
- 36.Patel V, Adya R, Chen J, Ramanjaneya M, Bari MF, Bhudia SK, Hillhouse EW, Tan BK, Randeva HS. Novel insights into the cardio-protective effects of FGF21 in lean and obese rat hearts. PLoS One. 2014;9:e87102. doi: 10.1371/journal.pone.0087102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chanoit G, Lee S, Xi J, Zhu M, McIntosh RA, Mueller RA, Norfleet EA, Xu Z. Exogenous zinc protects cardiac cells from reperfusion injury by targeting mitochondrial permeability transition pore through inactivation of glycogen synthase kinase-3beta. Am J Physiol Heart Circ Physiol. 2008;295:H1227–H1233. doi: 10.1152/ajpheart.00610.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cong WT, Ling J, Tian HS, Ling R, Wang Y, Huang BB, Zhao T, Duan YM, Jin LT, Li XK. Proteomic study on the protective mechanism of fibroblast growth factor 21 to ischemia-reperfusion injury. Can J Physiol Pharmacol. 2013;91:973–984. doi: 10.1139/cjpp-2012-0441. [DOI] [PubMed] [Google Scholar]
- 39.Han MM, Wang WF, Liu MY, Li DS, Zhou B, Yu YH, Ren GP. FGF-21 protects H9c2 cardiomyoblasts against hydrogen peroxide-induced oxidative stress injury. Yao Xue Xue Bao. 2014;49:470–475. In Chinese. [PubMed] [Google Scholar]
- 40.Aita VM, Liang XH, Murty VV, Pincus DL, Yu W, Cayanis E, Kalachikov S, Gilliam TC, Levine B. Cloning and genomic organization of beclin 1, a candidate tumor suppressor gene on chromosome 17q21. Genomics. 1999;59:59–65. doi: 10.1006/geno.1999.5851. [DOI] [PubMed] [Google Scholar]
- 41.Yang S, Guo Y, Zhang W, Zhang J, Zhang Y, Xu P. Effect of FGF-21 on implant bone defects through hepatocyte growth factor (HGF)-mediated PI3K/AKT signaling pathway. Biomed Pharmacother. 2019;109:1259–1267. doi: 10.1016/j.biopha.2018.10.150. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The datasets used or analyzed during the current study are available from the corresponding author on reasonable request.