

Abstract
Background
Niemann-Pick disease type C1 (NPC1) is a rare, neurodegenerative cholesterol storage disorder. Diagnostic delay of more than 5 years is common due to the rarity of the disease and non-specific early symptoms. To improve diagnosis and facilitate early intervention, we previously developed a newborn screening assay based on newly identified plasma bile acid biomarkers. Because the newborn screen had been validated using dried blood spots (DBS) from already diagnosed NPC1 patients, an unanswered question was whether the screen would be able to detect individuals with NPC1 at birth.
Methods
To address this critical question, we obtained the newborn DBS for already diagnosed NPC1 subjects (n=15) and carriers (n=3) residing in California, New York, and Michigan, states that archive residual DBS in biorepositories. For each of the DBS, we obtained two neighbor controls – DBS from patients born on the same day and in the same hospital as the NPC1 patients and carriers. 3β,5α,6β-trihydroxycholanic acid (bile acid A) and trihydroxycholanic acid glycine conjugate (bile acid B) were measured in the DBS using a liquid chromatography-tandem mass spectrometry (LC-MS/MS) assay.
Results
Bile acid B, the more specific biomarker for which the fully validated DBS assay was developed, was detected in 8/15 NPC1 patients, and elevated above the cut-off in 2/15 patients (the two samples with the shortest storage time). Bile acid B was detected in 2/2, 6/10, and 0/7 NPC1 samples that have been stored for < 10.5 years, 13–20 years, and >20 years, respectively, indicating that the glycine conjugate is detectable in DBS but may have reduced long term stability compared with bile acid A, the precursor trihydroxycholanic acid, which was elevated in 15/15 NPC1 subjects, but not in carriers and controls.
Conclusions
These results demonstrate that newborn screening for NPC1 disease is feasible using bile acid biomarkers.
Keywords: Niemann-Pick C disease, biomarker, bile acid, newborn screening
1. Introduction
Niemann–Pick C (NPC) disease is a rare, neurodegenerative, lysosomal cholesterol storage disorder caused by mutations in NPC1 (95%) and NPC2 (5%).[1] The incidence of classical juvenile NPC disease has been estimated to be on the order of 1/100,000, though more common variants associated with late onset disease are predicted based on allele frequencies in the general population.[2, 3] The disease often presents in early childhood with ataxia and progressive impairment of motor and intellectual function, and affected individuals usually die in adolescence.[2] Increasingly, NPC disease is being recognized among adults with cognitive defects and movement disorders.[4] Interventions, such as substrate reduction therapy (e.g., miglustat) or alleviation of cholesterol storage (e.g., cyclodextrin), can reduce morbidities and slow disease progression.[5–7] Diagnosis of NPC is challenging due its rare disease status, disease heterogeneity, and non-specific early visceral and neurological symptoms.[8] As a result, the disease frequently goes unrecognized, leading to diagnostic delays of >5 years,[9, 10] during which time the neurological symptoms progress and opportunities for intervention are lost.
To enable early diagnosis of NPC, we recently developed a newborn screening test based on quantification of a newly discovered bile acid biomarker, the glycine conjugate of trihydroxycholanic acid, in dried blood spots (DBS).[11] Because the newborn screen had been validated using DBS from already diagnosed NPC1 patients (ages 4 months to 58 years), an unanswered question was whether the screening test would be able to detect NPC1 patients at birth. Here, we report further validation of the screen by analysis of bile acids in archived DBS collected at birth from NPC1 patients and carriers paired with neighbor controls.
2. Materials and Methods
2.1. DBS Samples
The study was approved by the Washington University School of Medicine Human Research Protection Office. All participants or guardians provided written informed consent to allow retrospective analysis of the archived newborn DBS. The residual de-identified DBS from newborn NPC1 patients, newborn NPC1 carriers, and normal newborns were obtained from biorepositories in California, New York, and Michigan with the approval of the California Department of Public Health, New York State Department of Health, and the Michigan Department of Health and Human Services, respectively. The DBS collected in California, New York, and Michigan were stored at −20 °C, 4 °C, uncontrolled temperature, respectively. For each NPC1 and carrier DBS, two de-identified DBS collected from normal neonates on the same day at the same hospital and stored at the same location served as “neighbor” controls. De-identified DBS from older NPC1 patients (age > 1 year) were provided by NIH and Rush University and stored at −20 °C.
2.2. Sample analysis
The DBS bile acid assays for N-(3β,5α,6β-trihydroxy-cholan-24-oyl)glycine (bile acid B) and its related precursor 3β,5α,6β-trihydroxycholanic acid (bile acid A) were performed as previously described.[11] Twenty-five DBS samples from older NPC patients that were analyzed two years earlier and stored at −20 °C were included as positive NPC controls in the same LC-MS/MS run. Samples consisting of 8-point calibration standards in duplicate, a blank, a blank with internal standard, three levels of quality control (QC) samples (low QC, middle QC and high QC), and unknown clinical samples were analyzed. The LC-MS/MS run met the acceptance criteria as indicated in Food and Drug Administration (FDA) recommendations.[12]
3. Results
Fifteen newborn NPC1 subjects and three newborn NPC1 carriers were recruited to the study (Table 1). The storage times of the archived newborn DBS ranged from 6.0 – 28.2 years and 3.8 – 17.5 for NPC1 subjects and NPC1 carriers, respectively. Clinical data was available for eight of these NPC1 subjects. At birth, hepatosplenomegaly was present in 5/8 subjects, and prolonged jaundice reported in 4/8 subjects. Onset of neurological symptoms ranged from 0.5 – 6 years. Time to diagnosis from initial presentation of symptoms varied widely from 0.3 – 14 years.
Table 1:
Demographic characteristics of affected patients and controls.
| Newborn controls | Newborn NPC1 carriers |
Newborn NPC1 | NPC1 (> 1 year) | |
|---|---|---|---|---|
| Number of subjects, n |
36 | 3 | 15 | 25 |
| Gender (M/F), n | 12/24 | 0/3 | 6/9 | 18/7 |
| Age of sample collection |
0 | 0 | 0 | 1.7 – 26.1 |
| Age of diagnosis | - | - | 0.3 – 14 | - |
| Hepatosplenomegaly (n/m)* |
- | - | 5/8 | - |
| Prolonged jaundice (n/m)* |
- | - | 4/8 | - |
| Age of neurological symptom onset (year) |
- | - | 0.5 – 6 | - |
| Age of DBS (year) | 3.8 – 28.2** | 3.8 – 17.5** | 6.0 – 28.2** | 8.7 – 2.6*** |
n: number of patients presented with symptom; m: number of patients with clinical information available.
: stored at −20 °C, or 4 °C, or uncontrolled temperature.
: stored at −20 °C; -: not available.
The results of the bile acid biomarker analyses are shown in Figure 1. The bile acid B concentrations in the two NPC1 specimens that had been stored for 10.5 years or less were above the cutoff of 13.5 ng/mL.[11] The remaining NPC1 specimens stored for more than 13 years were below the cutoff, though six DBS showed detectable bile acid B ranging from 2.4 – 7.3 ng/mL. Thus, among the archived DBS from newborn NPC1 subjects, there was an inverse correlation between the amount of time that the DBS had been archived and levels of bile acid B (r2=0.44, p = 0.007) (Figure 1B). Bile acid B was undetectable in all neighbor newborn controls and in the three newborn NPC1 carriers, including one that has been stored for 3.8 years (Figure 1A). Among the positive control DBS from NPC1 patients, all values were above the cutoff (Figure 1A). The difference between initial and repeat determination of bile acid B was within 20% for the vast majority (96%) of the samples, suggesting our method is highly reproducible and bile acid B is stable when stored at −20 °C for 2 years.
Figure 1. Detection of bile acid biomarkers in newborn dried blood spots.
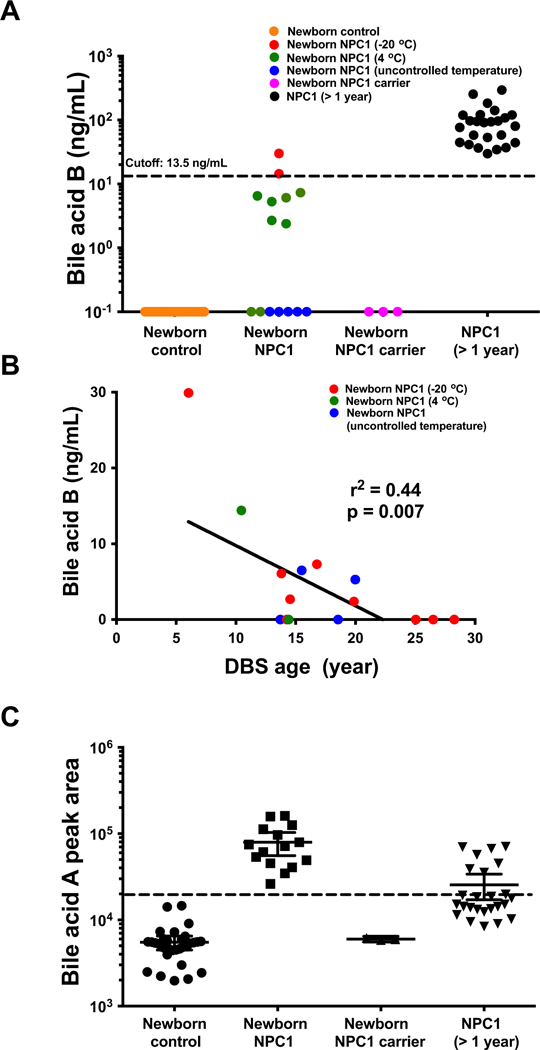
(A) Bile acid B in newborn NPC1 (n = 15), newborn control (n = 30), newborn NPC1 carrier (n = 3) and NPC1 at other age (> 1 year; n = 29) dried blood spot samples. DBS stored 10.5 years or less are shown in red symbols, DBS stored for > 10.5 years but < 20 years are shown in green symbols, and DBS stored for >20 years are shown in blue symbols. Individual data points are means ± 95% CI concentration. P < 0.0001 for NPC1 at other age versus newborn controls. (B) Bile acid B levels detected in newborn NPC1 subjects as a function of storage time of the archived DBS. DBS stored at −20 °C, 4 °C and uncontrolled temperature are shown in red, green, and blue symbols, respectively. (C) Bile acid A in newborn NPC1 (n = 15), newborn control (n = 30), newborn NPC1 carrier (n = 3) and NPC1 at other age (> 1 year; n = 25) dried blood spot samples. Individual data points are means ± 95% CI peak area. P < 0.0001 for newborn NPC1 versus newborn controls, and NPC1 at other age versus newborn controls.
Since a validated assay for bile acid A had not been developed, relative quantification was used to compare newborn NPC1 subjects, NPC1 carriers, and controls. In contrast to bile acid B, bile acid A clearly differentiated all the newborn NPC1 patients from controls and NPC1 carriers. However, bile acid A values for three newborn controls overlapped with positive NPC1 controls (Figure 1C).
There is significant correlation between bile acid A and bile acid B levels in the same sample (r2 = 0.40, p= 0.00067) in archived newborn NPC samples. However, there is no significant correlation in positive control NPC patients with age >1 year (r2 = 0.069, p = 0.096). It should be noted that the correlation analysis may be confounded by inaccuracy in bile acid A as the method is not fully validated. Bile acid A and B levels in patients manifesting visceral phenotype are higher than those without manifestations. However, the differences are not significant due to large variation in patients with manifestations and small number in each group.
4. Discussion
Diagnosis of NPC1 disease is frequently delayed due to the limited awareness of this rare disease in general clinical practice, the non-specificity of early symptoms, and the heterogeneity of symptoms.[2, 8, 10, 13–15] In retrospect, onset of neurological disease often presents as a delay in motor or speech milestones, clumsiness or difficulty in school. However, the clinical presentation of NPC1 may vary widely depending on the disease stage, and can even differ between individuals with similar genotypes. Implementation of diagnostic assays based on sensitive and specific blood-based biomarkers, including cholestane-3β,5α,6β-triol,[16–23] its bile acid metabolites,[11] and lysoSM509[24–26] have contributed to more timely diagnosis of NPC1, but have not eliminated the diagnostic delay. The general consensus among NPC experts if that screening of the general population is needed to identify neurologically pre-symptomatic subjects, those most likely to benefit from early intervention based on preclinical studies.[27] Miglustat, an inhibitor of glycosphingolipid synthesis, has shown limited efficacy and is approved for treatment of neurologically symptomatic disease in 45 countries.[6, 7] More promising is 2-hydoxypropyl-β-cyclodextrin (HPβCD), which has shown significant neuroprotection and survival benefit in preclinical models[27–29], and was shown to be safe and effective in an early stage clinical trial.[5] HPβCD (VTS-270) is currently being evaluated in a phase 3 trial. Neither of these treatments, however, are able to reverse the neurological impairment, underscoring the need for early diagnosis.
Previously, we identified 3β,5α,6β-trihydroxycholanic acid (bile acid A) and its glycine conjugate (bile acid B) as NPC biomarkers.[11] Because bile acid B showed greater specificity, this biomarker was selected to develop a fully validated DBS assay. In a validation set of 4992 newborn controls, 134 NPC1 carriers (> 1 year old), and 44 NPC1 subjects (> 3 months old), the assay showed 100% sensitivity and specificity. A limitation of the earlier study was that previously diagnosed NPC1 subjects that were beyond the newborn stage were used to develop the DBS test. Therefore, it remained unknown whether the marker was sufficiently elevated in newborns such that the test could recognize NPC1 disease at birth. Here, we show that one of the bile acid markers, bile acid A, was significantly elevated in all newborn NPC subjects examined. Similar to previous observation,[11] the bile acid A showed overlap between three newborn controls and NPC1 subjects at other ages, likely due to unresolved chromatographic interferences or to elevation of bile acid A in these controls. Further studies including development of an optimal method for quantification of bile acid A are needed to determine the cause(s). Elevation of the bile acid A in newborn NPC1 patients is consistent with previous case reports demonstrating significant elevation of the bile acid precursor cholestane-3β,5α,6β-triol in NPC1 and NPC2 neonates.[21]
Surprisingly, bile acid B was above the cut off in only two NPC1 subjects. There are several possible reasons for the failure to identify the remaining NPC1 subjects with the bile acid B assay. First, there may have been chemical degradation of bile acid B during long-term storage (up to 28 years) of DBS. Although we found that bile acid B in DBS was stable in DBS stored at −20 °C for two years, it may have degraded under storage conditions in the California, New York, and Michigan biobanks (−20 °C, 4 °C and uncontrolled temperature, respectively) in samples greater than 13 years of age. Second, we experienced low recovery of bile acid B from older DBS samples. We found that old DBS (>13 years) could not be dissolved in 1% sodium dodecyl sulfate (SDS) and 50 mM trisodium citrate solution at pH 12, which was used to disrupt the interaction of bile acid B the iron of hemoglobin.[11] The recovery of bile acid B from these DBS was low because the extraction of bile acid B bound to the iron of insoluble denatured hemoglobin was inefficient, and in turn, the low recovery led to lower determined bile acid B values. Third, we cannot rule out the possibility that conversion of bile acid A to its glycine conjugate (bile acid B) is impaired in the newborn NPC1 subjects.
As the bile acid A but not bile acid B can distinguish all the newborn NPC1 subjects from controls and carriers, there is a concern whether the elevation of bile acid A was caused by degradation of bile acid B or generation from its precursor cholestane-3β ,5α ,6β-triol in DBS. We have not evaluated stability of bile acid A in dried blood spots. Evaluation of stability of bile acid A over more than 10 years is beyond the scope of this study. Conversion of bile acid B to bile acid A is a hydrolysis reaction of amide, which is extremely slow under neutral condition and very unlikely in the medium of dried blood spot that lacks the water reactant. As far as we know, there are no known non-enzymatic routes to produce bile acid A. The biosynthetic route for conversion of cholestane-3β,5α,6β-triol to bile acid A occurs via the alternative bile acid pathway requires the enzymatic activity of Cyp27A1.[30] For this reason, even if cholestane-3β,5α,6β-triol was generated under oxidative conditions, the reaction would not progress to generation of the bile acids. Therefore, all bile acid A and B in the samples were generated in vivo.
In population screen for NPC disease, false positives would occur and will need to be adjudicated with second tier testing such as genetic analyses. Other disorders of sterol metabolism, such as acid sphingomyelinase deficiency and Wollman disease, are expected to generate rare positives during routine screening. We fully expect that an occasional NPC1 or NPC2 carrier may generate false positives and will need to be adjudicated with a plasma sample. Based on our published assay validation, the expectation is that the positive predictive value of the assay will be comparable to currently adopted newborn screens. However, the goal of this study was not to test the predictive value; rather, it was to address the question of whether these highly unusual bile acid biomarkers could be detected in newborn DBS. We believe the data supports the conclusion that these biomarkers are indeed present in NPC1 newborns.
5. Conclusions
In summary, we demonstrate for the first time that bile acid biomarkers are elevated in NPC1 newborn subjects. Although bile acid B failed to identify all the newborn patients in this study due to the likely limitation of long term stored DBS, the finding that its precursor, bile acid A, was elevated in all samples tested, coupled with our previous validation of the newborn screening assay, suggests that bile acid B can be used effectively to screen for NPC1 disease in the general population. We are in the planning stage to pilot the NPC newborn screen based on bile acid B in two states, with a goal of screening 150,000 – 250,000 spots over the next several years. This will be a critical test of the biomarker and the assay.
Acknowledgments
We are grateful to the National Niemann-Pick Disease Foundation for their assistance in obtaining samples from NPC1 and NPC1 carrier subjects. The authors express their appreciation to the families and patients who participated in this study. This work was supported by grants from the National Niemann-Pick Disease Foundation (X.J.), Dana’s Angels Research Trust (D.S.O.), Support Of Accelerated Research for NPC Disease (D.S.O.), aby NIH Grant R01 NS081985 (D.S.O. and J.E.S.), and by the Intramural Research Program of NICHD, NIH (F.D.P. and N.Y.F). This work was performed in the Metabolomics Facility at Washington University (NIH P30 DK020579 and NIH P30 DK056341). The content of the article has not been influenced by the sponsors.
Abbreviations
- DBS
dried blood spots
- FDA
Food and Drug Administration
- HPβCD
2-hydoxypropyl-β-cyclodextrin
- LC-MS/MS
liquid chromatography-tandem mass spectrometry
- NPC
Niemann–Pick C disease
- SDS
sodium dodecyl sulfate
- QC
quality control
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of interest
D.S.O. and X.J. are named as co-inventors on a patent application pertaining to the use of bile acid biomarkers in NPC. Other authors report no conflicts of interest.
References
- [1].Runz H, Dolle D, Schlitter AM, Zschocke J, NPC-db, a Niemann-Pick type C disease gene variation database Hum Mutat 29 (2008) 345–350. [DOI] [PubMed] [Google Scholar]
- [2].Vanier MT, Niemann-Pick disease type C Orphanet J Rare Dis 5 (2010) 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Wassif CA, Cross JL, Iben J, Sanchez-Pulido L, Cougnoux A, Platt FM, Ory DS, Ponting CP, Bailey-Wilson JE, Biesecker LG, Porter FD, High incidence of unrecognized visceral/neurological late-onset Niemann-Pick disease, type C1, predicted by analysis of massively parallel sequencing data sets Genet Med 18 (2016) 41–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Bauer P, Balding DJ, Klunemann HH, Linden DE, Ory DS, Pineda M, Priller J, Sedel F, Muller A, Chadha-Boreham H, Welford RW, Strasser DS, Patterson MC, Genetic screening for Niemann-Pick disease type C in adults with neurological and psychiatric symptoms: findings from the ZOOM study Hum Mol Genet 22 (2013) 4349–4356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Ory DS, Ottinger EA, Farhat NY, King KA, Jiang X, Weissfeld L, Berry-Kravis E, Davidson CD, Bianconi S, Keener LA, Rao R, Soldatos A, Sidhu R, Walters KA, Xu X, Thurm A, Solomon B, Pavan WJ, Machielse BN, Kao M, Silber SA, McKew JC, Brewer CC, Vite CH, Walkley SU, Austin CP, Porter FD, Intrathecal 2-hydroxypropyl-beta-cyclodextrin decreases neurological disease progression in Niemann-Pick disease, type C1: a non-randomised, open-label, phase 1–2 trial Lancet 390 (2017) 1758–1768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Patterson MC, Mengel E, Vanier MT, Schwierin B, Muller A, Cornelisse P, Pineda M, investigators NPCR, Stable or improved neurological manifestations during miglustat therapy in patients from the international disease registry for Niemann-Pick disease type C: an observational cohort study Orphanet J. Rare Dis 10 (2015) 65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Patterson MC, Vecchio D, Prady H, Abel L, Wraith JE, Miglustat for treatment of Niemann-Pick C disease: a randomised controlled study Lancet Neurol 6 (2007) 765–772. [DOI] [PubMed] [Google Scholar]
- [8].Vanier MT, Gissen P, Bauer P, Coll MJ, Burlina A, Hendriksz CJ, Latour P, Goizet C, Welford RW, Marquardt T, Kolb SA, Diagnostic tests for Niemann-Pick disease type C (NP-C): A critical review Mol Genet Metab 118 (2016) 244–254. [DOI] [PubMed] [Google Scholar]
- [9].Sevin M, Lesca G, Baumann N, Millat G, Lyon-Caen O, Vanier MT, Sedel F, The adult form of Niemann-Pick disease type C Brain 130 (2007) 120–133. [DOI] [PubMed] [Google Scholar]
- [10].Yanjanin NM, Velez JI, Gropman A, King K, Bianconi SE, Conley SK, Brewer CC, Solomon B, Pavan WJ, Arcos-Burgos M, Patterson MC, Porter FD, Linear clinical progression, independent of age of onset, in Niemann-Pick disease, type C Am J Med Genet B Genet 153B (2010) 132–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Jiang X, Sidhu R, Mydock-McGrane L, Hsu FF, Covey DF, Scherrer DE, Earley B, Gale SE, Farhat NY, Porter FD, Dietzen DJ, Orsini JJ, Berry-Kravis E, Zhang X, Reunert J, Marquardt T, Runz H, Giugliani R, Schaffer JE, Ory DS, Development of a bile acid-based newborn screen for Niemann-Pick disease type C Sci Transl Med 8 (2016) 337ra363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].US Department of Health and Human Services, Food and Drug Administration, Center for Drug Evaluation and Research and Center for Veterinary Medicine (2001) Guidance for Industry: Bioanalytical Method Validations. http://www.fda.gov/downloads/Drugs/Guidances/ucm070107.pdf. Accessed 7 June 2017.
- [13].Crocker AC, Farber S, Niemann-Pick disease: a review of eighteen patients Medicine (Baltimore) 37 (1958) 1–95. [DOI] [PubMed] [Google Scholar]
- [14].Imrie J, Dasgupta S, Besley GT, Harris C, Heptinstall L, Knight S, Vanier MT, Fensom AH, Ward C, Jacklin E, Whitehouse C, Wraith JE, The natural history of Niemann-Pick disease type C in the UK J Inherit Metab Dis 30 (2007) 51–59. [DOI] [PubMed] [Google Scholar]
- [15].Stampfer M, Theiss S, Amraoui Y, Jiang X, Keller S, Ory DS, Mengel E, Fischer C, Runz H, Niemann-Pick disease type C clinical database: cognitive and coordination deficits are early disease indicators Orphanet J Rare Dis 8 (2013) 35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Porter FD, Scherrer DE, Lanier MH, Langmade SJ, Molugu V, Gale SE, Olzeski D, Sidhu R, Dietzen DJ, Fu R, Wassif CA, Yanjanin NM, Marso SP, House J, Vite C, Schaffer JE, Ory DS, Cholesterol oxidation products are sensitive and specific blood-based biomarkers for Niemann-Pick C1 disease Sci Transl Med 2 (2010) 56ra81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Jiang X, Sidhu R, Porter FD, Yanjanin NM, Speak AO, te Vruchte DT, Platt FM, Fujiwara H, Scherrer DE, Zhang J, Dietzen DJ, Schaffer JE, Ory DS, A sensitive and specific LC-MS/MS method for rapid diagnosis of Niemann-Pick C1 disease from human plasma J Lipid Res 52 (2011) 1435–1445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Boenzi S, Deodato F, Taurisano R, Martinelli D, Verrigni D, Carrozzo R, Bertini E, Pastore A, Dionisi-Vici C, Johnson DW, A new simple and rapid LC-ESI-MS/MS method for quantification of plasma oxysterols as dimethylaminobutyrate esters. Its successful use for the diagnosis of Niemann-Pick type C disease Clinica chimica acta; international journal of clinical chemistry 437 (2014) 93–100. [DOI] [PubMed] [Google Scholar]
- [19].Pajares S, Arias A, Garcia-Villoria J, Macias-Vidal J, Ros E, de Las Heras J, Giros M, Coll MJ, Ribes A, Cholestane-3beta,5alpha,6beta-triol in lipidosis: High levels in Niemann Pick type C, Cerebrotendinous Xanthomatosis and Lysosomal Acid Lipase deficiency J Lipid Res (2015). [DOI] [PMC free article] [PubMed]
- [20].Polo G, Burlina A, Furlan F, Kolamunnage T, Cananzi M, Giordano L, Zaninotto M, Plebani M, Burlina A, High level of oxysterols in neonatal cholestasis: a pitfall in analysis of biochemical markers for Niemann-Pick type C disease Clin Chem Lab Med (2015). [DOI] [PubMed]
- [21].Reunert J, Fobker M, Kannenberg F, Du Chesne I, Plate M, Wellhausen J, Rust S, Marquardt T, Rapid Diagnosis of 83 Patients with Niemann Pick Type C Disease and Related Cholesterol Transport Disorders by Cholestantriol Screening EBioMedicine 10.1016/j.ebiom.2015.12.018 (2015). [DOI] [PMC free article] [PubMed]
- [22].Klinke G, Rohrbach M, Giugliani R, Burda P, Baumgartner MR, Tran C, Gautschi M, Mathis D, Hersberger M, LC-MS/MS based assay and reference intervals in children and adolescents for oxysterols elevated in Niemann-Pick diseases Clinical biochemistry 48 (2015) 596–602. [DOI] [PubMed] [Google Scholar]
- [23].Reunert J, Lotz-Havla AS, Polo G, Kannenberg F, Fobker M, Griese M, Mengel E, Muntau AC, Schnabel P, Sommerburg O, Borggraefe I, Dardis A, Burlina AP, Mall MA, Ciana G, Bembi B, Burlina AB, Marquardt T, Niemann-Pick Type C-2 Disease: Identification by Analysis of Plasma Cholestane-3beta,5alpha,6beta-Triol and Further Insight into the Clinical Phenotype JIMD Rep 23 (2015) 17–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Giese AK, Mascher H, Grittner U, Eichler S, Kramp G, Lukas J, te Vruchte D, Al Eisa N, Cortina-Borja M, Porter FD, Platt FM, Rolfs A, A novel, highly sensitive and specific biomarker for Niemann-Pick type C1 disease Orphanet J Rare Dis 10 (2015) 78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Kuchar L, Sikora J, Gulinello ME, Poupetova H, Lugowska A, Malinova V, Jahnova H, Asfaw B, Ledvinova J, Quantitation of plasmatic lysosphingomyelin and lysosphingomyelin-509 for differential screening of Niemann-Pick A/B and C diseases Anal Biochem 525 (2017) 73–77. [DOI] [PubMed] [Google Scholar]
- [26].Polo G, Burlina AP, Kolamunnage TB, Zampieri M, Dionisi-Vici C, Strisciuglio P, Zaninotto M, Plebani M, Burlina AB, Diagnosis of sphingolipidoses: a new simultaneous measurement of lysosphingolipids by LC-MS/MS Clin Chem Lab Med 55 (2017) 403–414. [DOI] [PubMed] [Google Scholar]
- [27].Vite CH, Bagel JH, Swain GP, Prociuk M, Sikora TU, Stein VM, O’Donnell P, Ruane T, Ward S, Crooks A, Li S, Mauldin E, Stellar S, De Meulder M, Kao ML, Ory DS, Davidson C, Vanier MT, Walkley SU, Intracisternal cyclodextrin prevents cerebellar dysfunction and Purkinje cell death in feline Niemann-Pick type C1 disease Sci Transl Med 7 (2015) 276ra226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Davidson CD, Ali NF, Micsenyi MC, Stephney G, Renault S, Dobrenis K, Ory DS, Vanier MT, Walkley SU, Chronic cyclodextrin treatment of murine Niemann-Pick C disease ameliorates neuronal cholesterol and glycosphingolipid storage and disease progression PLoS One 4 (2009) e6951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Liu B, Li H, Repa JJ, Turley SD, Dietschy JM, Genetic variations and treatments that affect the lifespan of the NPC1 mouse J Lipid Res 49 (2008) 663–669. [DOI] [PubMed] [Google Scholar]
- [30].Russell DW, The enzymes, regulation, and genetics of bile acid synthesis Annu Rev Biochem 72 (2003) 137–174. [DOI] [PubMed] [Google Scholar]