

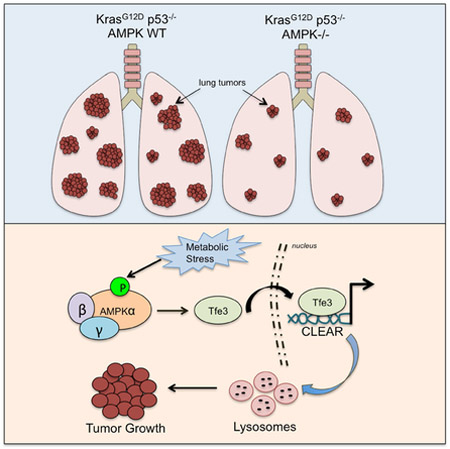
SUMMARY
AMPK, a conserved sensor of low cellular energy. can either repress or promote tumor growth depending on the context. However, no studies have examined AMPK function in autochthonous genetic mouse models of epithelial cancer. Here, we examine the role of AMPK in murine KrasG12D mediated non-small cell lung cancer (NSCLC), a cancer type in humans that harbors frequent inactivating mutations in the LKB1 tumor suppressor - the predominant upstream activating kinase of AMPK and twelve related kinases. Unlike LKB1 deletion, AMPK deletion in KrasG12D lung tumors did not accelerate lung tumor growth. Moreover, deletion of AMPK in KrasG12D p53f/f tumors reduced lung tumor burden. We identified a critical role for AMPK in regulating lysosomal gene expression through the Tfe3 transcription factor, which was required to support NSCLC growth. Thus, AMPK supports the growth of KrasG12D dependent lung cancer through the induction of lysosomes, highlighting an unrecognized liability of NSCLC.
Keywords: Cancer, lung, metabolism, Kras, AMPK, lysosomes, Tfeb, Tfe3, LKB1, tumor
Graphical Abstract
eTOC
AMPK can either inhibit or promote tumor growth depending on tumor type and context. XXX et al show that AMPK, unlike LKB1, has a pro-tumorigenic role in a Kras-dependent non-small cell lung cancer model through the regulation of Tfe3, a master regulator of lysosomes, which supports lung tumor growth.
INTRODUCTION
A fundamental property of cancer cells is to couple nutrient consumption to macromolecular biosynthesis and energy production in order to facilitate tumor growth and survival (Cairns et al., 2011). Thus, the elucidation of genes and details of signaling pathways that control the biosynthetic and bioenergetic demands of a growing tumor is critical for understanding the molecular basis of how metabolism is rewired in different forms of cancer. In 2003, one of the early links between a bona fide cancer gene and a metabolism gene was discovered when the bona fide tumor suppressor, LKB1 (Liver Kinase B1; also known as Serine Threonine Kinase 11 (STK11)), was discovered to be the upstream kinase responsible for activation of the central metabolic regulator AMP-activated Protein Kinase (AMPK) under low energy conditions (Hardie and Alessi, 2013). LKB1 was originally identified as the causal tumor suppressor gene on human chromosome 19p3 mutated in the inherited autosomal dominant cancer disorder Peutz Jeghers Syndrome (Hemminki et al., 1998). Subsequent studies identified LKB1 mutations in sporadic cancers, particularly in non-small cell lung cancers (NSCLC), where LKB1 mutations are the third most frequent genetic alteration (~20%) and are often concurrent with Kras mutations (Ding et al., 2008; Sanchez-Cespedes et al., 2002). Soon after, AMPK was shown to inhibit mTORC1 signaling via its phosphorylation and activation of the TSC2 tumor suppressor, solidifying the concept of AMPK as the biochemical step between two well-documented human tumor suppressors (LKB1, TSC2). This presented the hypothesis that AMPK would itself be a critical tumor suppressor (Shackelford and Shaw, 2009) (Hardie and Alessi, 2013).
However, soon after LKB1 was also found to directly phosphorylate and activate a family of 12 additional kinases in the AMPK superfamily (Lizcano et al., 2004), hence loss of LKB1 in tumors is accompanied by loss of the activity of AMPK as well as the other 12 AMPK-related kinases. Subsequently, CAMKK2 was demonstrated to serve as an alternative upstream kinase for AMPK activation, but unlike low ATP conditions, CAMKK2 activates AMPK in response to cellular calcium flux. Though genetic studies in mice found that LKB1 deletion triggered loss of most AMPK activity, CAMKK2 was found to provide some basal level of AMPK activation in the absence of LKB1, meaning such tumors were not abolished of AMPK activity (Garcia and Shaw, 2017).
Given that AMPK inhibits major biosynthetic pathways like mTORC1, fatty acid and sterol synthesis, it was assumed that AMPK largely functions to suppress tumorigenesis via inhibition of pro-growth pathways needed for cell proliferation (Shackelford and Shaw, 2009). Supporting this, direct activation of AMPK by small molecules has been shown to suppress tumor growth in several experimental tumor models (Huang et al., 2008; Lee et al., 2011; Zadra et al., 2014). Furthermore, suppression of AMPK activity in tumors outside of LKB1 mutation has been suggested to promote tumor growth, including via inhibitory phosphorylation of AMPKα1 at Ser487 by AKT (Hawley et al., 2014) or degradation of AMPK by E3 ligases such as TRIM28 or UBE20 (Pineda et al., 2015; Vila et al., 2017). Consistent with the idea that AMPK itself may behave as a tumor suppressor, one of the first genetic studies of AMPK function in mouse models of cancer revealed that whole-body AMPKα1 knockout mice increase tumor burden in the Eu-Myc model of Burkitt’s lymphoma (Faubert et al., 2013). More recently, the whole-body AMPKβ1 knockout mice were crossed to p53-null mice which spontaneously develop T cell lymphomas, and in this tumor context, systemic AMPKβ1-deficiency accelerated lymphoma tumor burden (Houde et al., 2017).
However, other recent studies examining AMPK in cancer models have demonstrated that AMPK can also support tumor growth by strongly promoting tumor cell survival, revealing that the role of AMPK in cancer is multifaceted and much more complex than originally hypothesized. AMPK in orthotopically transplanted mammary tumors was demonstrated to be required to prolong cell survival by regulating the redox state of the tumor cells (Jeon et al., 2012). Furthermore, genetic deletion of AMPK in three distinct mouse models of leukemia (AML, pre-B ALL, T-ALL) resulted in tumor cell death and dramatically improved mouse survival from leukemia (Chan et al., 2017)(Kishton et al., 2016)(Saito et al., 2015). In one model, deletion of AMPKα1 and α2 rendered the leukemic cells sensitive to cell death induced by dietary restriction through redox imbalance (Saito et al., 2015). Thus, it is increasingly evident that there is no single unifying role of AMPK as either a tumor suppressor or tumor promoter in cancer.
Notably, all of the aforementioned studies of AMPK deletion have been exclusively performed in blood borne cancer models, and while AMPK function in xenograft tumor models has been studied via shRNA, the conditional genetic deletion of AMPK in genetically engineered autochthonous solid tumor models has not been described to date. Here we report the phenotype of genetic AMPK loss in KrasG12D dependent mouse models of NSCLC, the tumor type where its upstream kinase LKB1 is the 3rd most frequent genetic alteration.
RESULTS
AMPK loss does not phenocopy LKB1 loss in KrasG12D lung tumors
The catalytic subunits of AMPK are encoded by the AMPKα1 and AMPKα2 genes (Prkaa1 and Prkaa2), which have been shown to be functionally redundant in cell types where both are expressed. We examined the expression of AMPKα1 and AMPKα2 in human NSCLC cell lines and in a murine KrasG12D model of NSCLC and found that proteins for each appear well-expressed (Figure S1A), suggesting that genetic depletion of both Prkaa1 and Prkaa2 would be needed to functionally remove all AMPK activity, as has been reported previously (Jorgensen et al., 2005; Laderoute et al., 2006).
One of the best-established measures of AMPK activity in vivo is phosphorylation of its first-reported and most documented substrate, Acetyl-CoA Carboxylase (ACC). In tissue-specific KO studies, depletion of both AMPKα1 and AMPKα2 results in complete loss of phosphorylated-ACC at serine 79 (P-ACCS79) (Howell et al., 2017; Laderoute et al., 2006). We validated immunohistochemistry using the P-ACCS79 antibody as a readout of AMPK-dependent phosphorylation in mouse liver and lung. AMPK deletion lead to complete loss of P-ACCS79 detection in AMPKα1/α2 null livers (Figure S1B). We determined the specificity for the P-ACCS79 immunostain in KrasG12D p53−/− lung tumors that had been treated with the ACC inhibitor ND-646, which leads to complete dephosphorylation of its AMPK phosphorylation site (Svensson et al., 2016) (Figure S1B). We confirmed that loss of both AMPKα1 and AMPKα2 was required in KrasG12D lung tumors to fully genetically deplete AMPK activity (Figure S1C).
To assess genetic AMPK function in KrasG12D mediated NSCLC, we generated mice harboring the following alleles (1) KrasLSLG12D for conditional activation of oncogenic Kras, (2) homozygous floxed alleles of catalytic AMPKα1 and α2 subunits (Prkaa1L/L and Prkaa2L/L), and (3) ROSA26LSLluciferase for conditional activation of firefly luciferase. In these mice, activation of KrasG12D in the lung epithelium via intranasal instillation of adenovirus expressing Cre-recombinase (Ad-Cre) is coupled to simultaneous deletion of Prkaa1 and Prkaa2 and activation of firefly luciferase to allow for non-invasive bioluminescence imaging (BLI) of NSCLC development. We designated mice from this model wild-type for AMPK as “K,” mice with loss of AMPKα1 and AMPKα2 activity as “KA,” and mice with loss of AMPK α 1 or AMPK α 2 alone as “KA1” or “KA2,” respectively. We performed a number of experiments by nasally adminstering adenoviral-Cre into the various Kras AMPK allelic combinations, and found no overall differences in lung tumor burden. Using a combination of western blotting on tumors (Figure S1D), laser-capture microdissection (Figure S1E), and P-ACCS79 immunohistochemistry (Figure S1J-K), it became clear that we were getting very inefficient deletion of AMPKα1 and AMPKα2 in the tumors. Incomplete deletion of conditional alleles has previously been reported in the KrasG12D model of NSCLC, where essential proliferation genes such as Dicer (Kumar et al., 2009) and Rac1 (Kissil et al., 2007) had escaped recombination and retained expression in lung tumors. Our data demonstrate that a number of lung tumors in KA mice escape Ad-Cre mediated recombination, suggesting that more efficient AMPK deletion is required to determine the effect of genetic AMPK loss in KA lung tumors.
We reasoned that lenti-viral mediated Cre recombinase expression (Lenti-Cre) would enable more efficient AMPK deletion due to the ability of Lenti-Cre to integrate into the genome of KA lung tumors and produce stable Cre expression. We directly compared the efficiency of AMPK deletion in Ad-Cre versus Lenti-Cre initiated lung tumorigenesis in K, KA1, KA2, and KA mice and also in KrasG12D mice harboring floxed alleles of the LKB1 (STK11) tumor suppressor (“KL”), to compare the phenotype of complete AMPK loss to LKB1 loss (Figure 1A, S1F). While no difference was observed between Kras alone and Kras combined with any combination of AMPK alleles in the Adeno-Cre treated animals (Figure S1G-I), when we monitored lung tumor growth in K and KA tumors initiated with Lenti-Cre we found that KA mice displayed reduced lung tumor development compared to K mice (Figures 1B-C, S1I). KL tumors initiated by Lenti-Cre displayed enhanced growth in a manner identical to that observed in Ad-Cre cohorts (Figure 1B, S1H). Notably, KA1 and KA2 mice did not display altered lung tumor growth compared to K mice using either Ad-Cre or Lenti-Cre induction (Figure 1B, S1H), and still retained P-ACC immunostaining (Figure S1C), so we focused the majority of our subsequent analysis on KA mice. Quantitation of tumor growth as a function of fold change in bioluminescence (BLI) during the entire imaging period revealed a reduced increase in BLI when tracking KA lung tumor development compared to K lung tumors when utilizing Lenti-Cre (Figure 1C), although the differences were not statistically significant. In contrast, KL mice had significantly increased fold change in BLI compared to K and KA mice (Figure 1C, S1G). Given that KA lung tumor growth does not phenocopy LKB1 loss using either system, these data demonstrate that AMPK deficiency is not sufficient to mimic the tumor suppressor effects of LKB1 in KrasG12D NSCLC.
Figure 1. AMPK loss does not phenocopy LKB1 loss in KrasG12D dependent NSCLC.

(A) Schematic of experimental design in Kras (K), Kras-AMPKα1 (KA1), Kras-AMPKα2 (KA2), Kras-AMPKα1/α2 (KA) and Kras-LKB1 (KL) floxed mouse models administered Lenti-Cre.
(B) Longitudinal BLI data from Lenti-Cre induced tumors. Average BLI is plotted.
(C) Fold change in BLI during the imaging period.
(D) Representative H&E stained sections at endpoint.
(E) Representative images of P-ACC IHC on lung sections at endpoint. E, escaper tumor; D, AMPK deleted tumor.
(F) Quantitation of tumors from (D) cohorts that have escaped AMPK deletion vs. deleted AMPK, as assessed by P-ACC IHC.
(G-I) Post hoc tumor burden analyses from H&E sections of Lenti-Cre induced tumors. KA tumors that escaped deletion of AMPK were identified by P-ACC IHC analysis of serial sections to those stained with H&E and excluded from the data shown. (G) Tumor area as a percentage of total lung area per mouse. (H) Average tumor number per mouse. (I) Tumor size. Each dot represents a single tumor.
(J) Western blots on single primary K, KL and KA tumor lysates.
Scale bars, 2000μm. Data are represented as mean ± SEM. * P<0.05 ** P<0.01 *** P<0.001 relative to K mice. NS = non-significant compared to K mice determined by students t test at the 95% confidence level.
We found that lung tumor burden measured by BLI was consistent with histopathology of hematoxylin and eosin (H&E) stained lung sections of K and KA mice. We noted similar disease burden in K and KA lung tumors initiated by Ad-Cre but reduced lung tumor burden in KA mice compared to K mice using Lenti-Cre (Figure 1D). These data suggest that the discrepancy between KA phenotypes in Ad-Cre versus Lenti-Cre cohorts may be a function of the deletion efficiency of AMPK. We performed IHC for P-ACCS79 expression on Ad-Cre and Lenti-Cre initiated KA lung sections to quantitate and compare AMPK deletion efficiency in these two systems. We found that P-ACCS79 expression was detected in some tumors and completely absent in others suggesting that AMPK was deleted in some tumors and retained in others (Figures 1E-F, S1K). We confirmed that the P-AMPKT172 staining pattern matched that of the P-ACCS79 IHC (Figure S1J), consistent with P-ACCS79 functioning as a robust in-vivo read-out of AMPK activity and deletion status in primary tumors. Quantitation of deleted versus escaped tumors via P-ACCS79 analysis revealed markedly less escaped tumors using Lenti-Cre (40% escaped tumors in Ad-Cre versus 16% in Lenti-Cre) (Figure 1F). Thus, the phenotype of KA NSCLC growth is masked in Ad-Cre initiated tumors due to the large number of tumors that escape full deletion of AMPKα1 and AMPKα2.
To specifically assess the impact of AMPK deletion on KrasG12D mediated NSCLC growth we quantitated and compared lung tumor burden between K and KA mice. For analysis, we excluded tumors that had escaped AMPK deletion in KA mice. We found that when we removed the escaper tumors, AMPK deletion showed a trend of reduced lung tumor burden compared to K mice in Lenti-Cre induced tumors, but was not statistically significant (Figures 1G). When we calculated tumor number we found a reduction in the number of tumors in KA mice compared to K mice (Figure 1H), however, we did not find any differences in the average size of the tumors (Figure 1I) suggesting that the reduction in total lung tumor burden that occurs when AMPK is deleted is mostly due to reduction in the number of tumors and not tumor size. No differences in these parameters were observed between K and KA mice in the Ad-Cre induced tumors (Figure S1L-N). Finally, to corroborate our BLI results from KA1 and KA2 mice we performed post-hoc analyses and detected no significant changes in tumor burden or tumor number upon Lenti-Cre induced single deletion of either AMPKα1 or AMPKα2, consistent with the redundancy observed between both catalytic subunits of AMPK (Figure S1O, P). Taken together, these data demonstrate that AMPK loss does not phenocopy LKB1 loss in murine KrasG12D-dependent NSCLC.
Given that LKB1 is the major activating kinase of AMPK and its deletion should result in near complete loss of AMPK activity (which spurred the hypothesis that AMPK loss should mimic LKB1 loss), we directly compared AMPK signaling in lysates from individual tumors isolated from K, KA and KL mice to determine whether KL tumors had in fact completely lost AMPK activity comparable to KA (Figure 1J). We found that while AMPK activity was lower in KL tumors compared to K, there was still significant retention of AMPK activation in KL tumors compared to KA tumors, suggesting that additional upstream kinases such as CAMKK2 must be promoting AMPK activity upon LKB1 loss. P-ACCS79 analysis by IHC revealed that expression was lower in KL tumors compared to K but still detectable (Figure S1Q). Thus KL tumors still retain residual AMPK activity compared to KA tumors, which are completely nullified for AMPK and thus helps explains why LKB1 deletion would not have the same inability to restore energetic balance in the face of metabolic stress as AMPK deletion does.
AMPK deletion is detrimental to the growth of KrasG12D p53−/− non-small cell lung cancer
Given that AMPK is required for optimal growth of KrasG12D NSCLC, we reasoned that AMPK deletion may impose a metabolic liability during NSCLC development. To explore this further we examined the impact of AMPK deletion in KrasG12D lung tumors that were also conditionally deleted for the tumor suppressor Trp53 (“KP” and “KPA” mice). Tumors from the KP model proliferate far more rapidly and hence likely encounter more metabolic stress demands. We initiated lung tumors using Lenti-Cre in KP and KPA mice and compared also to KP mice harboring floxed alleles of LKB1 (“KPL”) (Figure 2A). BLI demonstrated that growth of KPA tumors was markedly impaired compared to KP tumors (Figure 2B-C). Interestingly, KPL mice displayed significantly enhanced rates of lung tumorigenesis, succumbing to lung cancer at a time when KP mice displayed only moderate disease burden. KPL mice also developed a large number of local and distant metastases (Figure S2A), demonstrating that simultaneous deletion of p53 and LKB1 leads to enhanced lung tumor growth and more aggressive metastatic NSCLC compared to single deletion of either tumor suppressor alone. Quantitation of tumorigenesis as a function of fold change in BLI between days 56-96 post Cre revealed that KP tumors grew 47-fold compared to only 9-fold growth of KPA tumors, demonstrating that AMPK deletion suppresses lung tumorigenesis (Figure 2D). Analysis of H&E stained lung sections from KP, KPA and KPL mice revealed striking differences in lung tumor burden between the three genotypes (Figure 2E). To determine whether any KPA lung tumors had escaped AMPK recombination, we isolated gDNA from single lung tumors by LCM in FFPE sections and analyzed recombination of AMPKα1 and α2. We found that several lung tumors in KPA mice had escaped recombination, despite the use of Lenti-Cre (Figure S2B). P-ACCS79 immunostain quantitation revealed that ~16% of tumors were classified as escapers (Figures 2F and S2C-D).
Figure 2. AMPK deletion is detrimental to the growth of KrasG12D p53−/− (KP) NSCLC.

(A) Schematic of experimental design in KP, KP-AMPK floxed (KPA) and KP-LKB1 floxed (KPL) mouse models.
(B) Representative BLI images. Scale bar represents photons/sec/cm2/sr.
(C) Average longitudinal BLI data.
(D) Fold change in BLI during the imaging period (Day 56 to Day 96).
(E) Representative H&E stained sections at endpoint. Scale bar, 2000μm.
(F) Representative image of P-ACC IHC in KPA tumors. Inset: quantitation of escapers across entire cohort. Scale bar, 2000μm.
(G-K) Post hoc analyses from H&E sections. KPA tumors that escaped AMPK deletion were determined by P-ACC IHC analysis of sections serial to H&E sections and are included or excluded as indicated.
(G) Tumor area as a percentage of total lung area per mouse.
(H) Average tumor number per mouse.
(I-J) Individual tumor size. (I) Each dot represents a single tumor. KPA tumors that escaped AMPK deletion are indicated in red. (J) Average tumor size. (K) Tumors from (I) binned by size.
(L-M) BrdU IHC. (L) Representative BrdU IHC images. (M) Percent BrdU positivity normalized to tumor cell number excluding KPA tumors that escaped AMPK deletion. Scale bar, 50μm.
(N) Representative histopathology images.
Data are represented as mean ± SEM. * P<0.05 ** P<0.01 *** P<0.001 relative to KP determined by students t test.
Next, we performed extensive post-hoc analyses of H&E stained KP and KPA lung sections to determine the effect of AMPK deletion at the histological level. Quantitation of total lung tumor burden in KP and KPA mice revealed that AMPK deletion resulted in a significant decrease in lung tumor burden (Figures 2G and S2E), and when escaper tumors were excluded, AMPK deletion reduced lung tumor burden by 85% compared to KP mice. AMPK deletion also led to a 40% decrease in total tumor number (Figures 2H and S2F). Next, we measured the size of each individual lung tumor in KP and KPA mice and found that KPA lung tumors were significantly smaller. Interestingly, when we correlated each individual KPA tumor to AMPK deletion status determined by P-ACCS79 staining of serial sections, we found that the largest tumors observed in KPA mice had escaped AMPK deletion (Figure 2I). When we excluded the escaped tumors from the analysis, we found that KPA lung tumors were significantly reduced in size by 75% compared to KP tumors (Figure 2J). We binned each tumor from KP and KPA mice into three separate sizes ranging from small to large, and found that only 2% of KPA tumors were categorized as large, compared to 24% of tumors in KP mice (Figure 2K). These data demonstrate that AMPK deletion in KrasG12D p53−/− NSCLC leads to significant reduction in lung tumor burden that is due to reduced tumor number and a dramatic reduction in tumor size in the tumors present, suggesting that AMPK is required for the optimal growth of KP lung tumors. Consistent with this, BrdU positivity was significantly reduced in KPA tumors (Figures 2L-M and S2G). Next, we performed IHC for cleaved Caspase 3 to determine whether the decreased tumor burden that occurs upon AMPK deletion might also be explained by increased cell death, however no differences were observed by genotype at the timepoint examined (Figures S2H-I). KP and KPA tumors were then evaluated and classified in histopathology according to established criteria (Nikitin et al., 2004) (Figure 2N). Both KP and KPA mice had a continuum of lesions ranging from alveolar hyperplasia to adenomas and adenocarcinomas of varying sizes. Tumors in KPA mice were mostly classified as small mixed adenomas to medium sized mixed adenocarcinomas with some cellular atypia, compared to tumors in KP mice that were larger, more pleomorphic and mostly classified as papillary to mixed adenomas to very large papillary adenocarcinomas. Several KP tumors also showed airway and vascular invasion, inflammation and necrosis as well as cellular and nuclear atypia. Taken together, KPA tumors were overtly less aggressive at the histological level compared to KP.
AMPK promotes lysosomal gene expression (CLEAR network) during glucose starvation in NSCLC cells
To further dissect how AMPK activation may be impacting control of growth and metabolism in Kras mutant lung tumors, we performed CRISPR/Cas9 deletion of AMPKα1 and α2 in murine KP lung adenocarcinoma cells in vitro. We transfected guide RNAs (gRNAs) designed to delete AMPKα1 and α2 into two luciferase-expressing mouse NSCLC cell lines derived from KP lung tumors, 634T (Liu et al., 2013) and 368T1 (Winslow et al., 2011), and screened clones for AMPKα1 and α2 deletion (designated as double knockout, DKO) by immunoblotting. We isolated 3 DKO clones from 634T cells and 1 DKO clone from 368T1 cells and created isogenic cell lines that stably expressed either vector control (Vec), wild-type AMPKα1 (α1) or a kinase dead mutant of AMPKα1 (α1-KD). AMPK deletion led to a complete loss in AMPK signaling that was fully restored with stable re-expression of AMPKα1 but not kinase dead AMPKα1 (Figures 3A and S3A). We found that proliferation of AMPK DKO 634T and 368T1 cells was identical to AMPKα1 restored cells under high glucose media conditions (Figure 3B), however the proliferation of all DKO clones was significantly slower than isogenic AMPKα1 expressing cells under conditions of complete glucose deprivation or low glucose conditions (Figures 3C and S3B).
Figure 3. AMPK promotes lysosomal gene expression upon glucose starvation in NSCLC cells.

(A) Western blots for AMPK signaling in lysates from 368T1 and 634T mouse NSCLC cell lines deleted for AMPK by CRISPR/Cas9 and reconstituted stably with vector control (Vec), AMPKα1 (α1) or kinase-dead AMPKα1 (α1-KD) in the presence (+) or absence (−) of glucose (Gluc) for 18hrs.
(B) Proliferation rates of 634T and 368T1 isogenic cell lines in high glucose after 48hrs. Data are represented as percent proliferation of Vector.
(C) Proliferation rates of 634T and 368T1 isogenic cell lines in no glucose after 48hrs. Data are represented as percent proliferation of cells in high glucose.
(D-J) RNA-seq analysis of AMPKα1α2 KO 368T1 cells with vector control (DKO) or AMPKα1 (A1) stably reconstituted and grown in high or no glucose conditions for 12 or 18 hours.
(D) Cluster analysis of no glucose RNA-seq data. 548 genes cluster among the No Glucose samples, 210 of which (red box) are up regulated in AMPKα1 expressing cells in an AMPK-dependent manner.
(E) David Analysis of the 210 genes identified in (D). All terms with Benjamini <E−10 are shown.
(F) Fold expression from the RNA-seq data for each gene within the Lysosome KEGG Pathway and GOTerm enriched in (E).
(G) David Analysis of statistically significant (p<0.05) genes up regulated in AMPKα1 expressing cells (A1) compared to DKO after 12 hours of glucose deprivation (>0.5 fold change) across both RNA-seq replicates. All terms with Benjamini <E−5 are shown.
(H) Fold expression change from the RNA-seq data for each gene within the Lysosome KEGG Pathway and GOTerm enriched in (G).
(I) Heatmap depicting relative mRNA expression of each lysosomal gene from (H) across all RNA-seq conditions.
(J) Gene set enrichment analysis (GSEA) plot from RNA-seq data sorted by p-value, considering both replicates. The KEGG lysosome gene set is the only enriched gene set with an FDR <0.1. (NES) Normalized enrichment score; (FDR) false discovery rate.
(K) GSEA plot for the KEGG-Lysosome gene set analyzed from Saito et al 2015 transcriptional profiling data describing AMPK deletion (DKO) in leukemia-initiating cells. Data are represented as mean ± SEM. * P<0.05 ** P<0.01 *** P<0.001 relative to a1 in B and C as determined by students t test.
To determine if the residual AMPK activity left in KP tumor cells lacking LKB1 was sufficient to mediate growth and survival, we used CRISPR/Cas9 to delete AMPKα1 and α2 in two mouse lung adenocarcinoma cell lines derived from KPL lung tumors harboring LKB1 inactivation, 821T4 and 807LN (Liu et al., 2013) (Figure S3C). Similarly to KP cells, AMPK deletion did not affect proliferation rates of KPL cells in high glucose, but AMPK deletion did blunt proliferation of both KPL cell lines compared to WT cells when cultured in low glucose or no glucose (Figure S3D). Moreover, when KPL cells with or without AMPK deletion were placed into soft agar, tumor cells retaining AMPK showed greater survival and growth (Figure S3E), indicating that even the low level of AMPK activity left in LKB1-deficient KP lung tumor cells is sufficient to support survival.
Since AMPK activity is highest in the presence of LKB1, we continued our in vitro studies in KP cell lines. We further profiled the cell growth properties of the 368T AMPK DKO clones and found that these cells were sensitized to the apoptotic effects of glucose deprivation and phenformin treatment (Figure S3F). Under glucose deprivation, these cells also progressed more slowly through the cell cycle (Figure S3G), and exhibited a blunted ability to form colonies in a clonogenic assay (Figure S3H). Taking glucose deprivation as a metabolic stress condition that growing tumors are likely to face, to examine how AMPK might impart selective advantage we performed RNA sequencing (RNA-seq) analysis of one of our isogenic cell lines (368T1.DKO#1:Vec and 368T1.DKO#1:AMPKα1) subjected to high glucose or complete glucose deprivation for 12hrs or 18hrs. Unsupervised hierarchical clustering of the datasets revealed that samples clustered by glucose treatment, demonstrating that glucose status dominated the transcriptional response (Figure S3I). However, when we considered only the samples that had undergone glucose deprivation, cluster analysis revealed a set of 210 genes that were induced in an AMPK dependent manner (genes enriched in AMPKα1 versus DKO cells) (Red box, Figure 3D). The most AMPK dependent of these genes also clustered together when high and no glucose conditions were analyzed simultaneously (Red box, Figure S3I). Gene ontology (GOTerm) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis revealed that this gene set associated uniquely with Lysosome and related terms with high statistical significance (Figure 3E). This lysosomal gene set (CLEAR network) was induced by AMPKα1 expressing cells only under conditions of glucose deprivation, as we found no difference in expression of these genes in the isogenic cell lines under high glucose conditions (Figure 3F). In an independent analysis, we next considered all genes significantly upregulated in AMPKα1 expressing cells compared to DKO after 12 hours of glucose deprivation across two independent biological RNA-seq replicates. The most significant KEGG and GOTerms were again Lysosome (Figure 3G), which accurately reflected an AMPK-dependent induction of a core set of lysosomal genes upon glucose deprivation (Figure 3H-I). Gene Set Enrichment Analysis (GSEA) considering both biological replicates revealed that KEGG Lysosome was the only significant term associated with the RNA-seq dataset after 12 hours of glucose deprivation, and that the KEGG Lysosome gene set positively correlates with genes upregulated in AMPKα1 expressing cells compared to DKO (Figure 3J). Consistently, when we applied GSEA to a previously published RNA-seq dataset from primary leukemia cells (Saito et al., 2015), the same KEGG Lysosome gene set was significantly enriched in an AMPK-dependent manner (Figure 3K). Taken together, this data suggests that AMPK promotes lysosomal gene expression in cancer cells in response to glucose deprivation. Finally, to confirm our findings in an additional cell line and to analyze the transcriptome of AMPK DKO cells compared to WT parental cells i.e. not in isogenic AMPK add-back cells, we performed RNA-seq analysis of 634T cells that were deleted for AMPKα1/α2 in polyclonal pools and compared them to the WT polyclonal controls under conditions of glucose deprivation (Figure S3J). In this new cell line also, AMPK deletion led to significant down-regulation of the lysosomal gene network (Figure S3K-M) and AMPK DKO cells were unable to dephosphorylate Tfe3 during low glucose or treatment with 991 (Figure S3N).
AMPK activation promotes lysosomal gene expression through a Tfe3 dependent mechanism.
We recently uncovered a dynamic role for AMPK in the control of lysosomal gene expression in embryonic stem cells, where AMPK activity was required for the transcriptional activity of Tfeb to promote lysosomal biogenesis and lineage specification (Young et al., 2016). The TFE family of transcription factors, including Tfeb and Tfe3, are master transcription factors that control lysosomal gene expression and biogenesis and are tightly regulated by nutrient status. Under high nutrient conditions, Tfeb and Tfe3 are hyper-phosphorylated, predominantly by mTOR, and retained in the cytoplasm at the lysosome where they bind to 14-3-3; and during nutrient deprivation they become de-phosphorylated and translocate to the nucleus where they induce the expression of genes in the CLEAR network (Raben and Puertollano, 2016). We hypothesized that TFE transcription factors could be responsible for the AMPK-dependent induction of lysosomal target genes observed in the lung cancer cells. To examine the dynamic AMPK-dependent regulation of lysosomal gene expression at the biochemical level, we cultured 368T1-DKO#1 Vec/AMPKα1/AMPKα1-KD isogenic cells in high glucose or glucose-starved conditions for 6hrs and 12hrs and immunoblotted for Tfeb and Tfe3. Given that the activity of Tfeb and Tfe3 is controlled by phosphorylation status, the phosphorylated versus de-phosphorylated state of each can be visualized by pronounced electrophoretic mobility shifting. We found that under high glucose conditions, Tfeb and Tfe3 were hyper-phosphorylated in each isogenic cell line (Figure 4A and S4B). However, glucose starvation induced a dramatic downshift in the molecular weight of Tfeb and Tfe3 only in 368T1 cells with functional AMPKα1, demonstrating that AMPK activation promotes de-phosphorylation of Tfeb and Tfe3 in NSCLC cells during glucose starvation (Figure 4A). Of note, Tfe3 is alternatively spliced to generate two major isoforms (long and short) that are expressed in mouse NSCLC cells and each isoform undergoes a dramatic molecular weight shift upon glucose starvation in 368T1 cells expressing AMPKα1. This mobility shift in Tfeb and Tfe3 during glucose starvation was equal to that induced by pharmacologic mTOR inhibition (Figure S4A), demonstrating that Tfeb and Tfe3 are maximally activated by AMPK during glucose deprivation. Interestingly, glucose starvation of 634T cells induced a more pronounced AMPK dependent down-shift of Tfe3 than Tfeb, suggesting that Tfe3 may be more dynamically regulated by AMPK than Tfeb in some contexts (Figure S4B). We found that AMPK activation using the direct synthetic AMPK activator 991 induced Tfeb and Tfe3 de-phosphorylation only in 634T and 368T1 cells that expressed functional AMPKα1 (Figures 4B and S4C). Cells lacking AMPK expression or cells expressing kinase dead AMPKα1 only expressed hyper-phosphorylated Tfeb and Tfe3 after treatment with 991. Additionally, treatment of 634T-DKO isogenic cells with a panel of AMPK activators led to pronounced dephosphorylation of Tfeb and Tfe3 only in cells with functional AMPKα1 (Figure S4D). Taken together, these data demonstrate that physiological and pharmacological activation of AMPK promotes acute dephosphorylation of Tfeb and Tfe3, two master transcriptional regulators of lysosomal gene expression. Given that de-phosphorylation of Tfeb and Tfe3 is associated with their nuclear translocation, we examined this and found that glucose starvation or 991 treatment led to enrichment of Tfeb and Tfe3 in the nucleus only in cells expressing functional AMPKα1, demonstrating that AMPK activation promotes nuclear translocation of these two transcription factors (Figure 4C). Next we investigated whether this nuclear translocation promotes the transcriptional activity of Tfeb and Tfe3. We isolated RNA from isogenic 368T1 cells that were grown in high or no glucose for 12hrs and 18hrs and performed RT-qPCR for two canonical CLEAR targets, hexosaminidase subunit alpha (Hexa) and Cd63. Under high glucose conditions, Hexa and Cd63 were expressed equally between AMPK null and proficient cells (Figures 4D and S4E). However, during glucose deprivation the expression of Hexa and Cd63 was significantly induced only in AMPKα1 expressing cells. Notably, after 18hrs of glucose starvation we noted a modest induction of Hexa and Cd63 in AMPK deficient cells, however the magnitude of induction in AMPK proficient cells was significantly increased compared to that in AMPK deleted cells (Figures 4D and S4E). Direct AMPK activation by 991 treatment produced the same result; Hexa expression was only induced in AMPK expressing cells but not in AMPK deficient cells (Figure 4E). These data demonstrate the requirement for AMPK in promoting maximal transcriptional activity of Tfeb and Tfe3 during conditions of AMPK activation including glucose deprivation. Given that Tfeb and Tfe3 are known to promote the expression of many of the same genes and also have overlapping function, we examined their relative contribution in promoting the expression of lysosomal genes during AMPK activation. Our RNA-seq analysis revealed that Tfe3 is the predominant MITF/TFE family member expressed in both mouse and human NSCLC cells (Figures S4F-G). We performed siRNA knockdown of Tfeb and Tfe3 in 368T1-DKO and AMPKα1 restored cells under high glucose or no glucose culture conditions and measured the expression levels and induction of Hexa, Cd63 and Neu1 by RT-qPCR. We found that the induction of Hexa, Cd63 and Neu1 during AMPK activation under no glucose was minimally impacted by Tfeb knockdown, despite efficient Tfeb depletion (Figures 4F and S4H-K). However, Tfe3 knockdown significantly reduced the induction of these genes in AMPKα1 restored cells during glucose starvation, down to basal expression levels measured under high glucose and equivalent to simultaneous knockdown of Tfeb and Tfe3 (Figures 4F and S4I-K). Importantly, Tfe3 knockdown had no effect on the expression of Hexa, Cd63 and Neu1 under high glucose culture conditions, consistent with its hyper-phosphorylation and cytoplasmic retention. These data demonstrate that AMPK is required for induction of Tfe3 transcriptional activity during periods of metabolic stress that promote AMPK activation, such as glucose deprivation.
Figure 4. AMPK activation promotes de-phosphorylation, nuclear translocation and transcriptional activity of Tfeb and Tfe3.

(A) Western bot analysis of isogenic 368T1 AMPK DKO clones stably expressing either vector control (Vec), AMPKα1 (α1) or kinase-dead AMPKα1 (α1-KD) in the presence (+) or absence (−) of glucose (Gluc). Arrows highlight molecular weight of de-phosphorylated TFEB and TFE3.
(B) Western bot analysis of isogenic AMPK DKO 634T clones left untreated (−) or treated (+) with 991 for 3hrs. (C) Western blot analysis in nuclear and cytoplasmic fractions of 368T1 AMPK DKO clones in the presence (+) or absence (−) of glucose for 6hrs or left untreated or treated with 991 for 1hr.
(D) qPCR for Hexa expression in 368T1 isogenic clones cultured in the presence or absence of glucose for 12hrs or 18hrs. (E) qPCR for Hexa expression in 368T1 isogenic clones left untreated or treated with 991 for 12hrs or 18hrs.
(F) qPCR for Hexa expression in 368T1 isogenic clones with siRNA knockdown of either control, Tfeb, Tfe3, Tfeb+Tfe3 and cultured in the presence or absence of glucose for 18hrs.
Data are represented as mean ± SEM. * P<0.05 ** P<0.01 *** P<0.001 determined by students t test or ANOVA with Tukeys method for multiple comparison.
Requirement for Tfe3 in lung tumorigenesis
Oncogenic Tfeb and Tfe3 gene fusions have been described in renal cell carcinoma (Kauffman et al., 2014) and Tfeb and Tfe3 were shown to play a crucial role in autophagy-lysosome function to promote the growth of pancreatic ductal adenocarcinoma (PDAC) (Perera et al., 2015). Thus, a critical role for Tfeb, Tfe3 and the lysosome is recognized in oncology, however whether these factors contribute to NSCLC is unknown. We analyzed published RNA sequencing datasets (Ambrogio et al., 2016; Sweet-Cordero et al., 2005) to determine whether Tfeb, Tfe3 and the CLEAR network were differentially expressed in lung tumors compared to normal lung tissue. We found that Tfeb and Tfe3 mRNA levels were significantly up regulated in Kras mutant (Krasmut) mouse lung tumors (Figure S5A), and GSEA revealed significant induction of a Lysosome/Autophagy gene set (Figures 5A-B and S5B) and the KEGG_Lysosome gene set (Figure S5C) in Krasmut tumors compared to normal lung tissue. To dissect the function of Tfeb and Tfe3 in lung tumorigenesis, we performed CRISPR/Cas9 deletion of each in polyclonal pools of luciferase expressing 634T cells using two different gRNAs (designated as KO#1 and KO#2) (Figure 5C). Deletion of Tfeb and Tfe3 led to significant loss of protein expression in polyclonal pools of cells (Figure 5D) and, consistent with Figure 4F, Tfe3 deletion but not Tfeb deletion led to a loss of Hexa induction upon 991 treatment (Figure S4D). Neither Tfeb nor Tfe3 inactivation resulted in basal proliferation rate changes (Figure S5E), but both were required for maximal protection against glucose deprivation (Figure S5F), mirroring our observations in AMPK KO lung cancer cells (Figure 3B-C). Next, we injected our panel of cells intravenously into SCID/Beige mice and monitored their ability to form lung tumors over a period of 24 days (Figure 5C). BLI revealed no significant differences in tumor growth between Tfeb KO and control cells. However, Tfe3 deletion led to a significant reduction in lung tumor growth in both KO cell lines (Figures 5E-G). This was further supported by quantitative analysis of lung tumor burden from H&E stained sections where Tfe3 deletion led to significant reduction in lung tumor burden (Figure 5H-I), attributable to reduced tumor number (Figure 5J) but not tumor size (Figure S5G). Notably, Tfeb deletion led to modest reduction in lung tumor burden that did not reach statistical significance. Taken together these data reveal a critical role for Tfe3 in promoting tumorigenesis of mouse NSCLC.
Figure 5. Tfe3 deletion impairs the growth of mouse NSCLC cells in vivo.

(A) GSEA plot for the lysosome-autophagy gene set from mouse Krasmut lung tumors versus normal tissue using a geneset from (Ambrogio et al., 2016).
(B) Heat map depicting relative mRNA expression of the “lysosomal-autophagy” genes from (A)
(C) Schematic of experimental design to asses the role of Tfeb and Tfe3 in lung tumorigenesis.
(D) Western blot analysis of Tfeb and Tfe3 deletion by CRISPR/Cas9 in polyclonal lysates in KP 634T.Luc cells.
(E) Bioluminescence overlay images after 24 days of lung tumor growth. Scale bar represents photons/sec/cm2/sr.
(F) Longitudinal BLI quantitation. Average photon flux for each cell line is plotted for each imaging time point.
(G) Fold change in photon flux during the tumor growth period (Day 0 to Day 24).
(H) Representative H&E stained lung sections from each group.
(I) Tumor area was calculated as a percentage of total lung area.
(J) Tumor number per mouse.
Data are represented as mean ± SEM. * P<0.05 ** P<0.01 *** P<0.001 determined by ANOVA with Tukeys method for multiple comparison.
AMPK deletion impacts the CLEAR network in primary NSCLC tumors
To further delineate the role of AMPK and to determine whether the CLEAR network is also deregulated in KPA lung tumors we analyzed the impact of AMPK deletion in vivo using several methodologies. Given that AMPK deletion led to a dramatic reduction in KP tumor size (Figure 2) we wondered if AMPK activity might be higher in small tumors to support their outgrowth. To examine this hypothesis, we performed IHC for activated AMPK (P-AMPKT172) in KP lung tumors and found that small tumors displayed stronger staining intensity for P-AMPK compared to large tumors (Figures 6A-B). Additionally, we noted that in smaller tumors (<3,000 cells) P-AMPK was predominantly found in the center of the tumor and not at the periphery close to surrounding normal cells, whereas in larger tumors P-AMPK was lower and now only found at the peripheral edge of the tumor (Figure 6A). These distinct cell populations bearing the highest P-AMPK was mirrored by P-ACC staining (Figure S6A). To assess the impact of AMPK loss on AMPK signaling components we plucked individual lung tumors from KP and KPA and immunoblotted for AMPK signaling. As expected, AMPK deletion led to loss of phosphorylation of the well-documented AMPK sites in the direct AMPK substrates ACC, Raptor and ULK1 (Figure S6B). We also detected an increase in P-S6 signal in KPA tumors consistent with the role of AMPK as a negative regulator of mTOR (Shaw, 2009). IHC for P-S6235/236 in KP and KPA lung tumors revealed that P-S6 expression was highest in small KPA tumors (Figure S6C-D). Also consistent with earlier studies of AMPK-deficiency in acute myeloid leukemia models (Saito et al., 2015), GLUT1 levels were lower in the AMPK-deficient lung tumors, as seen by GLUT1 IHC in small tumors that were either KP or KPA (Figures S6E-F). Loss of GLUT1 was shown to contribute to elevated ROS and cell death from metabolic dysfunction in those models (Saito et al., 2015). Notably, the greatest difference between KP and KPA tumors in P-S6 and GLUT1 signal was observed in small tumors, which correlates with the observation that AMPK activity is highest in small tumors, and suggests a functional AMPK-dependent event in vivo.
Figure 6. AMPK deletion impacts the lysosomal pathway in KP lung tumors in vivo.

(A) Representative P-AMPK IHC images in KP tumors. Tumors were binned by size and representative staining for P-AMPK is shown for each size bin. Scale bar, 100μm.
(B) Quantitation of P-AMPK IHC signal intensity binned by tumor size.
(C) Representative images from Lamp2 IHC in KP and KPA tumors. Left scale bar, 100um. Right scale bar,20um.
(D) Quantitation of Lamp2 staining intensity.
(E) Plot of tumor size (cell number per tumor) for KP tumors with High Lamp2 IHC signal intensity. Pearson correlation coefficient is shown.
(F) Lamp2 IHC signal intensity binned by tumor size (cell number per tumor).
(G) Representative images illustrating correlation between Lamp2 and P-ACC IHC signal intensity in KP tumors from serial sections. Left scale bar, 2000μm. Right scale bar, 20μm.
(H) Representative images from Neu1 IHC. (I) Quantitation of staining intensity. (J) Tumors from (I) with High signal intensity plotted according to tumor size (cell number per tumor). Scale bar, 20μm.
(K) qPCR for lysosomal gene expression in primary tumors.
(L) Representative images from LC3A/B IHC. Scale bar, 20μm.
(M) Western blots on single primary KP and KPA tumor lysates.
IHC quantitation: number of tumors analyzed indicated above each column. Data are represented as mean ± SEM. * P<0.05 ** P<0.01 *** P<0.001 determined by students t test.
Next, to determine if the lysosome is affected by AMPK in vivo, we investigated the impact of AMPK deletion in KPA tumors on the expression of lysosomal associated membrane protein-2, LAMP2, a well-established lysosomal marker, by IHC. LAMP2 staining intensity was significantly reduced in KPA tumors compared to KP (Figure 6C-D), consistent with LAMP2 down-regulation in AMPK KO tumor cells (Figure 3). Interestingly, LAMP2 staining intensity correlated with KP tumor size, with the highest staining intensity observed in the smallest tumors (Figures 6E-F). Furthermore, LAMP2 staining intensity matched the staining intensity of P-ACC (Figure 6G), suggesting that LAMP2 protein expression levels parallel AMPK activity. To determine whether other CLEAR network members were similarly de-regulated in KPA tumors we assessed protein and RNA levels of other CLEAR network genes. Using IHC, we found that Neu1 and Slc17A5 staining intensity was significantly lower in KPA tumors compared to KP (Figures 6H-J and SG-I). Using RT-qPCR, we found that Sult1c2 and Atp6v0b mRNA transcripts levels were also significantly down-regulated in KPA tumors (Figure 6K). Together, these data support a role for AMPK in the regulation of the CLEAR network in KP tumors in-vivo. Finally, given that AMPK deletion impairs autophagy we analyzed the localization and expression of LC3A/B in KPA tumors by IHC and immunoblotting. LC3A/B positive puncta were infrequently observed in KP tumors, but KPA tumors exhibited a consistent accumulation of LC3A/B puncta that was independent of expression level (Figure 6L). This corresponded with western blot results from individual primary lung tumors which revealed an accumulation of the lipidated form of LC3A/B (LC3-II), which associates with autophagic vesicles, in KPA tumors compared to KP (Figure 6M). This suggests that unprocessed autophagosomes accumulate in KPA tumors. Since lysosomes are responsible for the final step of autophagy wherein autophagic cargo is degraded after autophagosome-lysosome fusion, this data is consistent with maintained autophagic flux in the presence of reduced lysosomal capacity in KPA tumors.
Elevated lysosomal gene expression correlates with accelerated disease recurrence in human lung adenocarcinoma patients
To query the role of lysosomal function in human lung cancer, we turned to the largest Lung Adenocarcinoma dataset (522 samples), Lung Adenocarcinoma (TCGA, Provisional), within The Cancer Genome Atlas (TCGA) (Cancer Genome Atlas Research, 2014). In Figure 3I we defined a core set of AMPK-dependent lysosome genes in lung cancer cells, so we applied the 16 most AMPK dependent of these genes as a lysosomal lung cancer gene signature. We queried the expression of this gene signature according to RNA-seq mRNA expression data, separating all 230 complete tumors within this dataset into two groups; 43% of the samples exhibited high expression of at least one of the lysosomal genes (Altered) and 57% of samples had low expression of all of the lysosomal genes (Unaltered) (Figure 7A). The majority of samples in the Altered group had elevated levels of multiple lysosomal gene signature genes, suggestive of broad lysosomal gene upregulation. Correspondingly, many lysosomal genes that were not part of the 16-gene lysosomal gene signature were also significantly upregulated in the Altered group compared to the Unaltered group (Figure 7B-C). This confirms that our 16-gene signature can identify tumors with elevated lysosomal gene expression, and suggests that these Altered tumors may have heightened lysosomal function. Importantly, disease recurred significantly faster in patients with tumors in the Altered category compared to patients with Unaltered tumors (Figure 7E). Patients with Altered tumors also exhibited a trend toward poor survival (Figure 7D). Together, this suggests that elevated lysosomal gene expression in lung cancer patients correlates with poor patient outcome. To query the generalizability of this observation, we screened all 23 datasets from other cancer types within the TCGA with greater than 100 patient samples by mRNA expression of our 16-gene signature. Four other cancer types, Brain lower grade glioma, Kidney renal clear cell carcinoma, Sarcoma and Skin cutaneous melanoma, also exhibited statistically significant disease outcome patterns in survival and/or disease free status, all of which correlated with worse outcome for the Altered cohort (Figure S7).
Figure 7. Elevated lysosomal gene expression correlates with accelerated disease recurrence in human lung adenocarcinoma patients.

(A) The 230 complete tumors within the Lung Adenocarcinoma TCGA dataset (TCGA, Provisional, 522 samples) were queried for alterations in mRNA levels of any of the 16 AMPK-dependent lysosomal lung cancer gene signature genes defined in Figure 3I. The RNA-seq z-score cut-off was set at +/− 2. Patient samples were divided into two groups; those with modulation of mRNA levels of lysosomal gene signature genes, termed “altered,” or those without major mRNA alterations in any of these genes, termed “unaltered.” Heatmap illustrates mRNA expression of each gene across all patient samples.
(B) All 9 V-ATPase genes that were not part of the lysosomal gene signature genes but are significantly misregulated (q-value < 0.05) between the “altered” and “unaltered” tumors are upregulated in the “altered” gene set. 3 examples are shown.
(C) Two cathepsin genes that were not part of the lysosomal gene signature genes are significantly upregulated (q-value < 0.05) in the “altered” gene set. One example is shown.
(D) Overall patient survival after stratification of the data according to alterations in the lysosomal gene signature (A).
(E) Disease free survival after stratification of the data according to alterations in the lysosomal gene signature (A).
(F) The Lung Adenocarcinoma TCGA dataset was queried according to mutation status in STK11, segregating the data into tumors with mutations in STK11, “Altered,” and those without mutations in STK11, “Unaltered”. Plot of AMPK phosphorylation at T172 (PRKAA1_PT172) within the associated proteomics data (left). Total PRKAA1 protein levels (right).
(G) mRNA levels of lysosomal gene signature genes.
(H) mRNA levels of a V-ATPase gene.
(I) mRNA levels of a cathepsin gene.
We then queried the Lung Adenocarcinoma dataset for LKB1 mutant tumors, breaking the dataset into an Altered group with mutation in LKB1 (STK11) or an Unaltered group with functional LKB1. As expected, the LKB1-dependent phosphorylation of AMPK at T172 was significantly reduced in LKB1 mutant tumors, while total AMPK protein level was up regulated (Figure 7F). Moreover, mRNA levels of lysosomal genes were significantly reduced in Altered tumors (Figure 7G-I), suggesting that low AMPK activity also correlates with low lysosomal gene expression in human lung tumors.
DISCUSSION
This study represents the first genetic analysis of AMPK function in any genetically engineered mouse model of solid tumor growth described to date. Importantly, we report a pro-tumorigenic role for AMPK in autochthonous KrasG12D-dependent NSCLC and we conclude that AMPK is either not the key target of LKB1 in suppressing NSCLC, or AMPK deficiency alone is not sufficient to mimic LKB1 loss, suggesting that there are likely unrecognized tumor suppressor roles for the other 12 AMPK related kinases in this context.
Our evolving understanding of AMPK in cancer suggests that AMPK plays a multifaceted role, as emerging evidence suggests that it can either inhibit or promote tumor growth depending on the tumor type and context (Zadra et al., 2015). Despite the discovery of LKB1 as the kinase that mediates phosphorylation and activation of AMPK, whether AMPK is responsible for mediating LKB1 tumor suppressor function in NSCLC had not been elucidated. Indeed some genetic studies, crossing germline whole-body knockouts of AMPKα1 or AMPKβ1 to the Eμ-Myc B-cell lymphoma model or the p53-null T-cell lymphoma model each revealed an acceleration of tumor burden, suggesting AMPK may be functioning as a tumor suppressor in some regard (Faubert et al., 2013; Houde et al., 2017).
In contrast, our discovery that deletion of both AMPKα1 and AMPKα2 suppresses rather than promotes growth of KrasG12D and KrasG12D p53−/− lung tumors adds to the growing list of evidence of a requirement for AMPK activation to support tumorigenesis. Interestingly, most studies describing a protumor growth role of AMPK demonstrate that the requirement for AMPK to support tumor growth is most essential during periods of metabolic catastrophe, consistent with the highly conserved role of AMPK as essential for cell and organismal survival under energy-depleted conditions (Hardie, 2011). In orthotopically transplanted MCF7 mammary tumor cells, shRNA targeting AMPKα1 reduced tumor burden (Jeon et al., 2012). In AML, genetic deletion of both AMPKα1 and AMPKα2 suppressed growth and survival of AML cells exclusively in metabolically stressed environments such as the bone marrow, which is glucose starved and hypoxic (Saito et al., 2015). In T cell acute lymphoblastic leukemia (T-ALL) oncogene-induced metabolic stress activates AMPK, which is essential to promote mitochondrial pathways that mitigate metabolic stress and apoptosis (Kishton et al., 2016). In pre-B-cell ALL, B-lymphoid transcription factors enforce a state of chronic energy deprivation which results in constitutive activation of AMPK. AMPK deletion or pharmacologic inhibition in B-ALL led to leukemic cell death and improved survival from ALL (Chan et al., 2017). However, in contrast, AMPK loss stimulated proliferation in cell populations that were characterized as energy replete, such as myeloid cells and mature B cell lymphoma, thus reflecting a dependence on AMPK for survival only in cancer cells that are metabolically compromised and energetically stressed.
The finding that AMPK loss does not mimic LKB1 loss might be counterintuitive to some, given that LKB1 null tumors should lose AMPK activity. However, it is important to note that we show in Figure S1 that LKB1 null lung tumors actually maintain residual AMPK signaling. Thus, the reduction in AMPK signaling from LKB1 loss is not analogous to full genetic depletion of AMPK. Given that AMPK is still active in LKB1 null tumors it is interesting to speculate that maintaining some level of AMPK activity, through activation of other kinases such as CAMKK2 that are known to phosphorylate AMPK but not the 12 AMPK related kinases (Fogarty et al., 2010; Garcia and Shaw, 2017), might be required in some capacity to support the growth of LKB1 null tumors, given that complete loss of AMPK via genetic depletion inhibits tumorigenesis. Consistent with this, we show that CRISPR mediated deletion of AMPK in LKB1 null tumor cell lines reduces proliferation under glucose deprivation conditions and is essential for growth in soft agar (Figure S3D-E). Interestingly, AMPK was also recently shown to protect cells from genotoxic stress and cell death induced by etoposide treatment in LKB1-null cancer cell lines, through calcium dependent activation of CAMKK2, suggesting that AMPK activation can hinder the anti-cancer effects of genotoxic based therapies (Vara-Ciruelos et al., 2017). Whether short-term pharmacological inhibition of AMPK may have a role as a therapeutic avenue, and in what genetic or combination therapy context remain open questions for future studies.
Limitations of Study
We do not suggest a single unifying function of AMPK in cancer. Whether AMPK is required to support or inhibit tumorigenesis is context dependent and most likely relies on the temporal energetic demands of the tumor. Tumors that exist in metabolically compromised environments or those that are under sustained energy deprivation during their evolution are likely to be dependent on AMPK activity to maintain ATP homeostasis and cell survival. In this setting acute AMPK inhibition could be considered as a therapeutic strategy to suppress tumor growth. In contrast, tumors that are energy replete and not dependent on AMPK activity would not respond to AMPK inhibition and perhaps a better strategy to suppress the growth of these tumors would be to activate AMPK to suppress the major biosynthetic pathways including mTORC1 and lipogenesis that AMPK negatively regulates. Thus, the identification of specific tumor types and biomarkers that facilitate our understanding and rationale to either inhibit or activate AMPK as a therapeutic strategy in cancer is of great interest.
It is important to note that we are not implying that TFEB or TFE3 are the most important targets downstream of AMPK in its tumor cell survival function; we can only state that TFE3 function is required. One might expect additional downstream targets may also be required in this tumor type if they are examined genetically in the future in some of these NSCLC models. It is also not clear that AMPK plays no part in tumor suppression in lung. It may well be that a severe but not complete attenuation of AMPK signaling, as occurs with LKB1 loss, still retain enough AMPK activity to provide the survival signal, yet lowered enough to elevate mTORC1 and lipogenesis. Such highly reduced - but not abolished - AMPK function might then synergize with pro-tumorigenic pathways which are induced upon loss of function of AMPK-related kinases.
Taken together, our study describing the requirement for AMPK to promote the growth of NSCLC warrants further investigation into AMPK function in different cancers to enhance our understanding of the complex and dynamic relationship between AMPK, metabolism, and cancer. In addition, rigorous dissection of the roles of the AMPK related kinases in lung cancer is now needed and will likely reveal novel mechanistic insights into how LKB1 functions to suppress lung tumorigenesis.
STAR METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Reuben Shaw (shaw@salk.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell culture and cell lines
All cell lines were incubated at 37 °C and were maintained in an atmosphere containing 5% CO2. 634T, 821T4 and 807LN cells were a kind gift from Kwok Wong and 368T1 cells were a kind from Monte Winslow. Luciferase expressing 634T.Luc cells were generated by stable infection of retrovirus expressing luciferase cDNA and hygromycin resistance (pBabe-Luc-hygro). Cells were tested for Mycoplasma (Lonza) using manufacturer’s conditions and were deemed negative. Cells were grown in Dulbecco’s modified Eagles medium (DMEM) plus 10% fetal bovine serum (Hyclone) and were continuously maintained under antibiotic selection for stable cell lines. For glucose deprivation assays, dialyzed FBS was used. Proliferation assays were performed using Cyquant (Life Technologies #C35011) in 96 well culture plates under manufacturers conditions. For quantitation under high glucose (25mM) conditions, values from experimental cells were divided by values from vector control cells and expressed as percent proliferation of vector control. For quantitation under glucose starved (no glucose) or low glucose (2.5mM) conditions, values from these samples were divided by their respective values under high glucose conditions and expressed as a percentage of growth compared to high glucose media at the indicated time points. Doses used for AMPK activating drugs were as follows, 991 (50μM), Phenformin (2.5mM), Aicar (2.5mM), Rotenone (1μM), all for 1hr.
Mouse studies
All procedures using animals were approved by the Salk Institute Institutional Animal Care and Use Committee (IACUC).
Genetically engineered mouse model studies:
10 week old FVB/n KrasLSLG12D/+ R26LSL:luc/luc (Kluc), KrasLSLG12D/+; p53fl/fl R26LSL:luc/luc (KPluc) and KrasLSLG12D/+; LKB1fl/fl R26LSL:luc/luc (KLluc) mice have been described previously (Shackelford et al., 2013). AMPKα1fl/fl and AMPKα2fl/fl mice were a kind gift from Benoit Violet and were backcrossed into FVB/n mice for 10 generations before the generation of KrasLSLG12D/+; AMPKα1fl/fl, KrasLSLG12D/+; AMPKα2fl/fl, KrasLSLG12D/+; AMPKα1fl/fl AMPKα2fl/fl or KrasLSLG12D/+; p53fl/fl AMPKα1fl/fl AMPKα2fl/fl mice. All experiments utilized a mixture of female and male mice. For studies using adenovirus, Ad5-Cre (University of Iowa) was instilled intranasally into mice at a viral titer of 1*10^6 PFU per mouse according to the protocol by Du Page et al 2009. For studies using lenti virus, lenti-Cre (PGK-Cre, gift from Tyler Jacks) was intubated into the trachea of mice at a viral titer of 2* 10^6 PFU per mouse according to the protocol by (DuPage et al., 2009).
Intravenous lung tumor studies:
8 week old female SCID/Beige mice (PrkdcscidLystbg-J/Crl:Fox Chase#250) were injected into the lateral tail vein with 5E4 luciferase expressing 634T cells and imaged via BLI 20 mins after i.v injection. BLI was performed once per week for a period of 24 days.
Bioluminescence Imaging:
All BLI was performed in an IVIS Spectrum (Caliper). Mice were injected with 150mg/kg D-Luciferin while being maintained on isoflurane and were imaged ventrally in an IVIS Spectrum 10 mins after injection. Mice were then imaged dorsally and a region of interest (ROI) was drawn around the thoracic cavity and the photon flux for ventral and dorsal images were calculated and combined to generate the total photon flux for each animal.
BrdU Injection:
BrdU (Sigma) was I.P. injected at 100mg/kg. Mice were sacrificed 4 hours post injection and lungs inflated for paraffin embedding and subsequent immunohistochemistry.
Post-Hoc analyses from tumor sections:
For GEMM studies, lungs were inflated with 10% neutral buffered formalin at the time of sacrifice, and subsequently paraffin embedded and sectioned. Tumor size of each individual tumor was measured from H&E stained sections using morphometric analysis in Panoramic viewer software (Perkin Elmer). IHC staining of a serial section with pACCS79 provided data about AMPK deletion (if a tumor deleted AMPK or escaped deletion) in KA and KPA sections. Each individual KA and KPA tumor measured from H&E sections was also analyzed for AMPK status. Average tumor number per mouse and tumor size was plotted from the data obtained during the tumor size analysis. To quantitate lung tumor area, individual tumor sizes were summed for each mouse and the percentage of tumor area normalized to total lung area was calculated per mouse. Total lung area was quantitated using Panoramic viewer software. For 634T cell lung allograft studies, tumor area was measured from H&E sections using Inform v2.1 image analysis software (Cambridge Research and Instrumentation). In brief, the Trainable Tissue Segmentation method was trained to identify tumor, normal lung, vessel and space. This program was then applied to all H&E images, and each of the resulting mapped images was then screened to verify that accurate tissue segmentation had occurred. The quantitation data from this analysis was then used to calculate the percentage of tumor area as normalized to total lung area (tumor area + normal lung area).
METHOD DETAILS
Immunohistochemistry and image analysis
Formalin fixed tumor-burdened lungs were transferred into 70% EtoH after 18-24hrs and paraffin embedded (FFPE) at the Salk Institute Histology Core. 5μm sections from FFPE tissues were prepared and stained with hematoxylin and eosin. For immunohistochemistry, slides were deparaffinized and rehydrated. Antigen retrieval was performed at high heat (~95°C) for 11 minutes for all antibodies except BrdU, for which antigen retrieval was performed for 30 minutes at high heat. Citrate buffer was used as the antigen retrieval buffer for all antibodies except Glut1, for which Tris-EDTA buffer was used. Endogenous peroxidase activity was quenched with hydrogen peroxide. Using the ImmPress HRP Ig (Peroxidase) Polymer Detection Kits (Vector Labs), slides were blocked, incubated overnight with primary antibody diluted in blocking buffer, and secondary antibody steps were carried out according to the manufacturer’s instructions. Staining was visualized using ImmPACT DAB Peroxidase Substrate (Vector Labs, SK-4105). Slides were counterstained with hematoxylin, dehydrated, and mounted with Cytoseal 60 (Thermo Scientific). Primary antibodies used are listed in the antibodies section. Immunostained slides were scanned using a Perkin Elmer Slide Scanner (Panoramic MIDI Digital Slide Scanner). Inform v2.1 image analysis software (Cambridge Research and Instrumentation) was used as a non-biased method to quantitate staining. Briefly, for each tumor analyzed, the image was prepared, the tissue was segmented, the cells were segmented and then the staining intensity was scored. Hematoxylin-stained nuclei were identified by the software and tallied to generate a total cell count per tumor. Nuclei or cytoplasm, depending on the target protein, were analyzed for diaminobenzidine (DAB) staining and were, correspondingly, binned by staining intensity score. A ratio between the number of DAB-positive cells per staining intensity bin and total cell number was used to calculate the % of cells staining positive. Quantitative analysis was performed on individually cropped tumors from all mice per experiment group, and all tumor images were analyzed by the same algorithm simultaneously in a batch analysis. The scoring of each image was visually verified to assure accuracy, and a blinded screen was performed to annotate and exclude any extreme outlier images from the final analysis. KPA tumors that escaped AMPK deletion were identified by pACCS79 staining of a serial section, and subsequent exclusion of the escaped tumors from analyses. Total cell numbers per tumor as determined by Inform analysis were also used to bin the data according to tumor size.
CRISPR/Cas9 studies
AMPK, Tfeb and Tfe3 knockouts were generated using the Cas9 strategy as described (Ran et al., 2013). gRNAs designed to knockout mouse AMPKα1 and AMPKα2 were described previously (Young et al., 2016). 634T and 368T1 cells were transfected with gRNAs and clones were screened for deletion of AMPKα1 and AMPKα2 by western blot. Isogenic clones were generated by stable re-expression of either vector control, WT-AMPKα1 or kinase dead (KD) AMPKα1. gRNAs were designed to target mouse Tfeb and Tfe3 genes using the optimized CRISPR design tool (http://crispr.mit.edu). Two separate gRNAs were designed for each gene. Oligonucleotides from IDT were phosphorylated, annealed and ligated into a BbsI-digested pSpCas9(BB)-2A-Puro (PX459) vector (Addgene, no. 48139). 634T cells were transfected with gRNAs for 24hrs and selected with 2ug/ml puromycin for 48hrs before being expanded and subjected to western blot to assess deletion of Tfeb and Tfe3 at the first passage. Polyclonal cells were injected into mice for lung tumor studies at the subsequent passage. Oligonucleotide sequences are listed in Supplemental Table S1.
Antibodies and reagents
For western blotting, antibodies from Cell Signaling Technologies (Denvers, MA USA) were diluted 1:1,000 and include P-ACCS79 (#3661), P-AMPKT172 (#2535), P-Raptor (#2083), ACC (#3662), AMPKα (#2532), for human cells AMPKα1 (#2795), for human cells AMPKα2 (#2757), LKB1 (#3047), Raptor (#2280), cleaved Caspase 3 (#9661), ULK (#8054), P-ULK1S555 (#5869), P-S6S235/236 (#2211), TXNIP (VDUP-1 #K0205-3), Tfe3 (#14779), 4EBP1 (#9452), P-S6K (#9205), S6 (#2217), and HDAC3 (#3949). Tfeb (Bethyl Laboratories #A303-673A) was used at a 1:2,000 dilution. For mouse cells, EMD Millipore (#07-350) was used to detect AMPKα1, and (#07-363) was used to detect AMPKα2. From Sigma-Aldrich, anti-actin (#A5441) was diluted 1:10,000 and anti-Tubulin (#T5168) was diluted 1:5,000. For IHC, the following antibodies were used: pACCS79 used at 1:400 (Cell Signaling #11818), pAMPKT172 used at 1:75 (Cell Signaling #2535), BrdU used at 1:200 (Abcam #6326), Cleaved Caspase 3 used at 1:200 (Cell Signaling #9661), Glut1 used at 5ug/mL (Alpha Diagnostics GT11-A), P-S6S235/236 used at 1:1,000 (Cell Signaling #4858), LAMP2 used at 1:100 (Abcam #13524), Neu1 used at 1:250 (GeneTex #GTX64524), Slc17a5 used at 1:500 (GeneTex #GTX115822), and LC3A/B used at 1:500 (Cell Signaling #12741). Antibody validation is provided on the manufacturers website for each antibody used.
Plasmids
A DONR vector containing full length wild-type human AMPKα1 was obtained from the human orfeome collection. Kinase dead (KD) AMPKα1 was generated from WT cDNA by site directed mutagenesis (K47R). cDNAs then underwent a LR clonase reaction into plenti-CMV-Destination vectors for stable cell line generation.
siRNA experiments
siRNAs targeting mouse Tfeb (#L-050607-02-0010) and mouse Tfe3 (#L-054750-00-0010) were purchased from Dharmacon. 368T1 cells were transfected with 20nM of each individual siRNA or a combination of both for 48hrs before being plated into high glucose or glucose starved conditions for a period of 12hrs. RNA was subsequently extracted from cells and qPCR performed to measure expression of CLEAR network genes.
Laser capture micro-dissection and PCR
Formalin fixed paraffin embedded (FFPE) lung sections were deparafinized and dehydrated before undergoing LCM. LCM was performed in a Zeiss Palm Microbeam IV microdisection system by capturing single tumors into adhesive caps (Zeiss) and subsequent genomic DNA was isolated by adding 30ul of Picopure DNA extraction solution (Arcturus) for 24hrs at 65°C. Samples were heat inactivated for 20 mins at 95°C and then subjected to PCR analysis for AMPK recombination. PCR conditions for recombined AMPK and floxed AMPK alleles were as follows. 94°C-4 min, 94°C-30 sec, 58°C-30 sec, 72°C-10 sec, 35 cycles, 72°C-2 min, 4°C-hold.
Western blots
For biochemical analysis of tumors, tumors were immediately snap frozen in liquid nitrogen and homogenized on ice in lysis buffer. Protein lysates were equilibrated for protein levels using a BCA protein assay kit (Pierce) and resolved on 8% SDS-PAGE gels depending on the experiment. For biochemical analysis of cells, cell lysates were prepared in lysis buffer, centrifuged and equilibrated for protein levels as above. Lysates were resolved on 8-12% SDS-PAGE gels depending on the experiment. Nuclear and cytoplasmic fractions were isolated using a NE-PER nuclear and cytoplasmic extraction kit (Thermofisher) under manufacturers conditions.
mRNA extraction and qPCR
RNA from snap-frozen cell pellets was isolated using the Quick-RNA MiniPrep kit (Zymo Research), including a DNase treatment. cDNA was synthesized from 1 μg of RNA using Superscript III (Life Technologies), and qPCR was carried out with diluted cDNA, appropriate primers, and SYBR Green PCR master mix (ThermoFisher Scientific) using a C1000 Thermal Cycler (BioRad). Relative mRNA levels were calculated using the 2−∆ ∆Ct method, with TBP serving as the internal control. All primers are listed in Supplemental Table S1.
mRNA-seq
RNA was isolated using the Quick-RNA MiniPrep kit (Zymo Research), including a DNase treatment. RNA integrity (RIN) numbers were determined using the Agilent TapeStation prior to library preparation. mRNA-seq libraries were prepared using the TruSeq RNA library preparation kit (version 2) according to the manufacturer's instructions (Illumina). Libraries were then quantified, pooled, and sequenced by single-end 50 base pairs using the Illumina HiSeq 2500 platform at the Salk Next-Generation Sequencing Core. Raw sequencing data were demultiplexed and converted into FASTQ files using CASAVA (version 1.8.2). Libraries were sequenced at an average depth of 80 million reads per sample.
mRNA-seq analysis
Sequenced reads were quality-tested using the online FASTQC tool (http://www.bioinformatics.babraham.ac.uk/projects/fastqc) and aligned to the mouse mm10 genome using the STAR aligner version 2.4.0k (Dobin et al., 2013). Raw gene expression was quantified across all annotated exons using HOMER (Heinz et al., 2010), and differential gene expression was carried out using the edgeR package version 3.6.8 using duplicates to compute within-group dispersion and correcting for batch effects (Robinson et al., 2010). Differentially expressed genes were defined as having a false discovery rate (FDR) <0.05 and a log2 fold change >1. GSEA (Mootha et al., 2003) was carried out with the GenePattern interface (https://genepattern.broadinstitute.org) using preranked lists generated from FDR values, setting gene set permutations to 1000 and using the c2 and c6 collections in MSigDB version5.0. Clustering was performed with Gene Cluster 3.0 (de Hoon et al., 2004) and visualized by heat maps using Java TreeView (version 1.1.6r4) (Saldanha, 2004). DAVID enrichment analysis was performed using the online DAVID tool (https://david.ncifcrf.gov; Huang et al. 2009).
Apoptosis Analysis by FACS
0.75 million cells were plated on P60 dishes. The next day, cells were washed once with PBS and incubated in either low glucose (2.5mM) or high glucose (25mM) containing media (DMEM containing 10% dialyzed FBS) for 36-48 hours, or no glucose (0mM) or high glucose (25mM) for 24 hours. Cells were collected at the indicated time point, washed once in PBS, trypsinized, and pelleted. Cells were then washed in 1X AnnexinV buffer and stained with AnnexinV and 7-amino-actinomycin D (7-AAD) according to the manufacturer’s protocol (BD Pharmingen, San Diego, CA). Briefly, cells were re-suspended in AnnexinV buffer to a concentration of 1×106/mL. Cells were then stained with 5 μL of phycoerythrin (PE)-conjugated AnnexinV antibody (Cat #556422 BD Pharmingen) and 5 μL of 7AAD (Cat# 51-2359KC BD Pharmingen) and incubated at room temperature for 15 minutes. 400 μL of AnnexinV buffer was then added to each sample with gentle mixing. Stained cells were analyzed using a FACScan flow cytometer (Becton Dickinson, San Jose, CA) and flow cytometry data was analyzed using FlowJo v. 10.1 software (Tree Star Inc., Ashland, OR).
BrdU and cell cycle analysis
0.75 million cells were plated on P60 dishes in full media. The next day, cells were washed once with PBS and incubated in either low glucose (2.5mM) or high glucose (25mM) containing media (DMEM with 10% dialyzed FBS) including 10μM BrdU (Cat# 51-2420KC, BD Pharmingen). Cells were collected 12 hours later, then fixed and stained using the FITC BrdU Flow Kit (Cat # 559619 BD Pharmingen) and 7-AAD according to the manufacturer’s instructions. Briefly, cells were washed once with PBS, trypsinized, pelleted, and resuspended in CytoFix/Cytoperm buffer for 15 minutess at room temperature. Cells were then permeabilized in 100μL CytoPerm Plus buffer for 10 minutes on ice, followed by a 5 minute re-fixation in CytoFix/Cytoperm buffer. To expose incorporated BrdU, cells were incubated with 30μg DNase in FACS staining buffer (PBS containing 5% FBS) for 1hr at 37 degrees. Lastly, cells were stained with 1μL FITC conjugated anti-BrdU antibodies for 20 minutes at room temperature in FACS staining buffer, washed, and resuspended in 20uL 7AAD followed by 1mL of staining buffer. Between each step of the protocol, cells were washed with 500μL of 1 × Wash Buffer and pelleted. The analysis was performed using a Becton-Dickinson LSR II flow cytometer (BD Biosciences) and FlowJo v. 10.1 software.
Colony Formation Assay
Cells were plated into 6-well plates, and media was changed to high or low glucose the next day. After 4 days in high or low glucose, cells were washed in PBS, fixed in 4% PFA diluted in PBS and stained with 0.1% crystal violet, washed with PBS and then imaged. The plate area covered by cells was quantitated using ImageJ.
TCGA analysis
The results shown are in whole based upon data generated by the TCGA Research Network: http://cancergenome.nih.gov/. TCGA datasets were queried using cBioPortal (www.cbioportal.org) (Cerami et al., 2012; Gao et al., 2013). Methods for data generation, normalization and bioinformatics analyses were previously described in the TCGA LUAD publication (Cancer Genome Atlas Research, 2014). mRNA data used for this analysis was RNA Seq V2 RSEM. All z-score thresholds used were 2.0.
Analysis of TCGA data from cancers beyond Lung Adenocarcinoma was performed on all TCGA datasets containing >100 patient samples, which at the time of the analysis were the following: Bladder Urothelial Carcinoma (TCGA, Provisional), Brain Lower Grade Glioma (TCGA, Provisional), Breast Cancer (Pereira et al. Nat Commun 2016), Breast Invasive Carcinoma (TCGA, Provisional), Cervical Squamous Cell Carcinoma and Endocervical Adenocarcinoma (TCGA, Provisional), Colorectal Adenocarcinoma (TCGA, Provisional), Esophageal Carcinoma (TCGA, Provisional), Glioblastoma Multiforme (TCGA, Provisional), Head and Neck Squamous Cell Carcinoma (TCGA, Provisional), Kidney Renal Clear Cell Carcinoma (TCGA, Provisional), Kidney Renal Papillary Cell Carcinoma (TCGA, Provisional), Liver Hepatocellular Carcinoma (TCGA, Provisional), Lung Squamous Cell Carcinoma (TCGA, Provisional), Ovarian Serous Cystadenocarcinoma (TCGA, Provisional), Pancreatic Adenocarcinoma (TCGA, Provisional), Prostate Adenocarcinoma (TCGA, Provisional), Sarcoma (TCGA, Provisional), Skin Cutaneous Melanoma (TCGA, Provisional), Stomach Adenocarcinoma (TCGA, Nature 2014), Testicular Germ Cell Cancer (TCGA, Provisional), Thymoma (TCGA, Provisional), Thyroid Carcinoma (TCGA, Provisional), Uterine Corpus Endometrial Carcinoma (TCGA, Provisional).
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical analyses
Statistical parameters including the exact value of n, dispersion and precision measures (mean ± SEM) and statistical significance are reported in the Figures and Figure Legends. Data is judged to be statistically significant when p < 0.05 by two-tailed Student’s t test. In figures, asterisks denote statistical significance as calculated by Student’s t test (*, p < 0.05; **, p < 0.01; ***, p < 0.001; ****, p < 0.0001). Statistical analyses were performed using Graph Pad Prism 7.
DATA AND SOFTWARE AVAILABILITY
Raw RNA sequencing data files have been deposited at Mendeley (data.mendeley.com).
DOI for 368T1 cell data, “RNA-seq profiling of 368T1 murine KP NSCLC cells deleted for AMPK with AMPKal add-back subjected to high or no glucose conditions”: 10.17632/gj763hyrny.1.
DOI for 634T cell data, “RNA-seq profiling of 634T murine KP NSCLC cells deleted for AMPK subjected to no glucose conditions”: 10.17632/3cy28w7v9k.1.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit polyclonal anti-Phospho-ACC S79 | Cell Signaling Technology | Cat# 3661 |
| Rabbit (40H9) anti-Phospho-AMPKα T172 | Cell Signaling Technology | Cat# 2535 |
| Rabbit polyclonal anti-Phospho-Raptor S792 | Cell Signaling Technology | Cat# 2083 |
| Rabbit polyclonal anti-ACC | Cell Signaling Technology | Cat# 3662 |
| Rabbit polyclonal anti-AMPKα | Cell Signaling Technology | Cat# 2532 |
| Rabbit polyclonal anti-AMPKα1 | Cell Signaling Technology | Cat# 2795 |
| Rabbit polyclonal anti-AMPKα2 | Cell Signaling Technology | Cat# 2757 |
| Rabbit (D60C5) anti-LKB1 | Cell Signaling Technology | Cat# 3047 |
| Rabbit (24C12) anti-Raptor | Cell Signaling Technology | Cat# 2280 |
| Rabbit polyclonal anti-Cleaved Caspase 3 | Cell Signaling Technology | Cat# 9661 |
| Rabbit (D8H5) anti-Ulk1 | Cell Signaling Technology | Cat# 8054 |
| Rabbit (D1H4) anti-Phospho-Ulk1 S555 | Cell Signaling Technology | Cat# 5869 |
| Rabbit polyclonal anti-Phospho-S6 S235/236 | Cell Signaling Technology | Cat# 2211 |
| Mouse (JY2) anti-TXNIP | MBL | Cat# K0205-3 |
| Rabbit anti-Tfe3 | Cell Signaling Technology | Cat# 14779 |
| Rabbit polyclonal anti-4EBP1 | Cell Signaling Technology | Cat# 9452 |
| Rabbit polyclonal anti-Phospho-S6K (T389) | Cell Signaling Technology | Cat# 9205 |
| Rabbit (5G10) anti-S6 | Cell Signaling Technology | Cat# 2217 |
| Mouse (7G6C5) anti-Hdac3 | Cell Signaling Technology | Cat# 3949 |
| Rabbit polyclonal anti-Tfeb | Bethyl Laboratories | Cat# A303-673A |
| Rabbit polyclonal anti-AMPKα1 | EMD Millipore | Cat# 07-350 |
| Rabbit polyclonal anti-AMPKα2 | EMD Millipore | Cat# 07-363 |
| Mouse (AC-15) anti-Actin | Sigma-Aldrich | Cat# A5441 |
| Mouse (B-5-1-2) anti-α-Tubulin | Sigma-Aldrich | Cat# T5168 |
| Rabbit (D7D11) anti-Phospho-ACC S79 | Cell Signaling Technology | Cat# 11818 |
| Rat (BU1/75 (ICR1)) anti-BrdU | Abcam | Cat# 6326 |
| Rabbit polyclonal anti-Glut1 | Alpha Diagnostics | Cat# GT11-A |
| Rabbit (D57.2.2E) anti-Phospho-S6 S235/236 | Cell Signaling Technology | Cat# 4858 |
| Rat (GL2A7) anti-Lamp2 | Abcam | Cat# 13524 |
| Rabbit polyclonal anti-Neu1 | GeneTex | Cat# GTX64524 |
| Rabbit polyclonal anti-Slc17a5 | GeneTex | Cat# GTX115822 |
| Rabbit (D3U4C) anti-LC3A/B | Cell Signaling Technology | Cat# 12741 |
| Bacterial and Virus Strains | ||
| Biological Samples | ||
| Chemicals, Peptides, and Recombinant Proteins | ||
| 991 | Glixx Laboratories | Cat# GLXC-09267 |
| Phenformin hydrochloride | Sigma-Aldrich | Cat# P7045 |
| Aicar | Toronto Research Chemicals | Cat# A611700 |
| Rotenone | Sigma-Aldrich | Cat# R8875 |
| D-Luciferin potassium salt | PerkinElmer | Cat# 122796 |
| BrdU | Sigma | B5002 |
| Critical Commercial Assays | ||
| Cyquant | LifeTechnologies | Cat# C35011 |
| Annexin V-PE Apoptosis Detection | BD Pharmingen | Cat# 559763 |
| FITC BrdU Flow Kit | BD Pharmingen | Cat# 559619 |
| Deposited Data | ||
| Raw data files from RNA seq on 368T1 KP NSCLC cells | Mendeley (data.mendeley.com) | DOI 10.17632/gj763hyrny.1 |
| Raw data files from RNA seq on 634T KP NSCLC cells | Mendeley (data.mendeley.com) | DOI 10.17632/3cy28w7v9k.1 |
| TCGA Research Network | (Cancer Genome Atlas Research, 2014) | http://cancergenome.nih.gov/. |
| Experimental Models: Cell Lines | ||
| 368T1 mouse NSCLC cells | Dr. Monte Winslow | (Winslow et al., 2011) |
| 634T mouse NSCLC cells | Dr. Kwok Wong | (Liu et al., 2013) |
| 821T4 mouse NSCLC cells | Dr. Kwok Wong | (Liu et al., 2013) |
| 807LN mouse NSCLC cells | Dr. Kwok Wong | (Liu et al., 2013) |
| Experimental Models: Organisms/Strains | ||
| KrasLSLG12D/+ mice (K Luc) on FVB | (Shackelford et al., 2013) | N/A |
| KrasLSLG12D/+; p53fl/fl; R26LSLLuc/Luc mice (KP Luc) on FVB | (Shackelford et al., 2013) | N/A |
| KrasLSLG12D/+; LKB1fl/fl; R26LSLLuc/Luc mice (KL Luc) on FVB | (Shackelford et al., 2013) | N/A |
| KrasLSLG12D/+; p53fl/fl; LKB1fl/fl; R26LSLLuc/Luc mice (KPL Luc) on FVB | This paper | N/A |
| AMPKα1fl/fl mice | Dr. Benoit Violet | N/A |
| AMPKα2fl/fl mice | Dr. Benoit Violet | N/A |
| KrasLSLG12D/+; R26LSLLuc/Luc; AMPKα1fl/fl mice on FVB | This paper | N/A |
| KrasLSLG12D/+; R26LSLLuc/Luc; AMPKα2fl/fl mice on FVB | This paper | N/A |
| KrasLSLG12D/+; R26LSLLuc/Luc; AMPKα1fl/fl; AMPKα2fl/fl mice on FVB | This paper | N/A |
| KrasLSLG12D/+; p53fl/fl; R26LSLLuc/Luc; AMPKα1fl/fl; AMPKα2fl/fl mice on FVB | This paper | N/A |
| SCID/Beige mice: PrkdcscidLystbg-J/Crl:Fox Chase | Charles River | Cat# 250 |
| Oligonucleotides | ||
| LCM_PCR Rec_AMPKα1_Fwd: CCCATGAGCTCCAGAAGAAG | This paper | N/A |
|
LCM_PCR Rec_AMPKα1_Rev: GCCAGACACAGGTGAAGACA |
This paper | N/A |
|
LCM_PCR Rec_AMPKα2_Fwd: GATCTGTCTGCTTCTGCGTTC |
This paper | N/A |
|
LCM_PCR Rec_AMPKα2_Rev: CGATGGCCGCTCTAGATAAC |
This paper | N/A |
|
LCM_PCR Lox_AMPKα1_Fwd: AATAGCCCATGAGCTCCAGA |
This paper | N/A |
|
LCM_PCR Lox_AMPKα1_Rev: TGCAGCCCTACACTGAAATG |
This paper | N/A |
|
LCM_PCR Lox_AMPKα2_Fwd: ATGGCTTGGCGCTGTCTA |
This paper | N/A |
|
LCM_PCR Lox_AMPKα2_Rev: ACTTTCTGGCCAGGCAGATG |
This paper | N/A |
| See Table S1 for additional oligonucleotides. | ||
| Recombinant DNA | ||
| pSpCas9(BB)-2A-Puro (PX459) | Addgene | Cat# 48139 |
| Lenti-Pgk-Cre | Dr. Tyler Jacks | (DuPage et al., 2009) |
| human AMPKα1 cDNA in DONR vector | Human Orfeome collection | |
| Software and Algorithms | ||
| Inform v2.1 | Cambridge Research and Instrumentation | |
| STAR aligner v2.4.0k | (Dobin et al., 2013) | |
| HOMER | (Heinz et al., 2010) | |
| EdgeR package v3.6.8 | (Robinson et al., 2010) | |
| GSEA | (Mootha et al., 2003) | |
| Gene Cluster 3.0 | (deHoon et al., 2004) | |
| Java TreeView v1.1.6r4 | (Saldanha et al., 2004) | |
| DAVID tool | (Huang et al., 2009) | http://david.ncifcrf.gov |
| cBioPortal | (Cerami et al., 2012) (Gao et al., 2013) | www.cbioportal.org |
| Other | ||
HIGHLIGHTS.
AMPK does not phenocopy the lung tumor suppressor role of its activator LKB1
AMPK deletion reduces tumor burden in Krasg12d and Kras p53−/− lung cancer models
AMPK is required during glucose deprivation to induce Tfeb and Tfe3 activation
Tfe3 activity is required for growth of mouse lung tumors
ACKNOWLEDGEMENTS
This study was supported by grants to R.J.S. from the National Institutes of Health (R35CA220538, P01CA120964), the Samuel Waxman Cancer Research Foundation, and The Leona M. and Harry B. Helmsley Charitable Trust grant #2012-PG-MED002. R.U.S. was supported by a postdoctoral fellowship from the American Cancer Society (ACS#124183-PF-13-023-01-CSM). L.J.E. is supported by a postdoctoral fellowship from the American Cancer Society (PF-15-037-01-DMC). S.N.B was supported by training grant 5T32CA009370 to the Salk Institute Cancer Center and 5F32CA206400. The Salk CCSG P30 CA014195 and Helmsley Charitable Trust supported the Functional Genomics Core and the Bioinformatics Core, as well as partial support for K. McIntyre and the histology core of the Salk Institute.
Footnotes
COMPETING FINANCIAL INTERESTS
None
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Cairns RA, Harris IS, and Mak TW (2011). Regulation of cancer cell metabolism. Nat Rev Cancer 11, 85–95.h [DOI] [PubMed] [Google Scholar]
- Chan LN, Chen Z, Braas D, Lee JW, Xiao G, Geng H, Cosgun KN, Hurtz C, Shojaee S, Cazzaniga V, et al. (2017). Metabolic gatekeeper function of B-lymphoid transcription factors. Nature 542, 479–483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding L, Getz G, Wheeler DA, Mardis ER, McLellan MD, Cibulskis K, Sougnez C, Greulich H, Muzny DM, Morgan MB, et al. (2008). Somatic mutations affect key pathways in lung adenocarcinoma. Nature 455, 1069–1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faubert B, Boily G, Izreig S, Griss T, Samborska B, Dong Z, Dupuy F, Chambers C, Fuerth BJ, Viollet B, et al. (2013). AMPK is a negative regulator of the Warburg effect and suppresses tumor growth in vivo. Cell Metab 17, 113–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fogarty S, Hawley SA, Green KA, Saner N, Mustard KJ, and Hardie DG (2010). Calmodulin-dependent protein kinase kinase-beta activates AMPK without forming a stable complex: synergistic effects of Ca2+ and AMP. Biochem J 426, 109–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia D, and Shaw RJ (2017). AMPK: Mechanisms of Cellular Energy Sensing and Restoration of Metabolic Balance. Mol Cell 66, 789–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardie DG, and Alessi DR (2013). LKB1 and AMPK and the cancer-metabolism link - ten years after. BMC biology 11, 36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawley SA, Ross FA, Gowans GJ, Tibarewal P, Leslie NR, and Hardie DG (2014). Phosphorylation by Akt within the ST loop of AMPK-alpha1 down-regulates its activation in tumour cells. Biochem J 459, 275–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houde VP, Donzelli S, Sacconi A, Galic S, Hammill JA, Bramson JL, Foster RA, Tsakiridis T, Kemp BE, Grasso G, et al. (2017). AMPK beta1 reduces tumor progression and improves survival in p53 null mice. Molecular oncology 11, 1143–1155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howell JJ, Hellberg K, Turner M, Talbott G, Kolar MJ, Ross DS, Hoxhaj G, Saghatelian A, Shaw RJ, and Manning BD (2017). Metformin Inhibits Hepatic mTORC1 Signaling via Dose-Dependent Mechanisms Involving AMPK and the TSC Complex. Cell Metab 25, 463–471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeon SM, Chandel NS, and Hay N (2012). AMPK regulates NADPH homeostasis to promote tumour cell survival during energy stress. Nature 485, 661–665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jorgensen SB, Wojtaszewski JF, Viollet B, Andreelli F, Birk JB, Hellsten Y, Schjerling P, Vaulont S, Neufer PD, Richter EA, et al. (2005). Effects of alpha-AMPK knockout on exercise-induced gene activation in mouse skeletal muscle. FASEB J 19, 1146–1148. [DOI] [PubMed] [Google Scholar]
- Kauffman EC, Ricketts CJ, Rais-Bahrami S, Yang Y, Merino MJ, Bottaro DP, Srinivasan R, and Linehan WM (2014). Molecular genetics and cellular features of TFE3 and TFEB fusion kidney cancers. Nat Rev Urol 11, 465–475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kishton RJ, Barnes CE, Nichols AG, Cohen S, Gerriets VA, Siska PJ, Macintyre AN, Goraksha-Hicks P, de Cubas AA, Liu T, et al. (2016). AMPK Is Essential to Balance Glycolysis and Mitochondrial Metabolism to Control T-ALL Cell Stress and Survival. Cell Metab 23, 649–662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kissil JL, Walmsley MJ, Hanlon L, Haigis KM, Bender Kim CF, Sweet-Cordero A, Eckman MS, Tuveson DA, Capobianco AJ, Tybulewicz VL, et al. (2007). Requirement for Rac1 in a Kras induced lung cancer in the mouse. Cancer Res 67, 8089–8094. [DOI] [PubMed] [Google Scholar]
- Kumar MS, Pester RE, Chen CY, Lane K, Chin C, Lu J, Kirsch DG, Golub TR, and Jacks T (2009). Dicer1 functions as a haploinsufficient tumor suppressor. Genes Dev 23, 2700–2704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laderoute KR, Amin K, Calaoagan JM, Knapp M, Le T, Orduna J, Foretz M, and Viollet B (2006). 5′-AMP-Activated Protein Kinase (AMPK) Is Induced by Low-Oxygen and Glucose Deprivation Conditions Found in Solid-Tumor Microenvironments. Mol Cell Biol 26, 5336–5347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Marks K, Cowley GS, Carretero J, Liu Q, Nieland TJ, Xu C, Cohoon TJ, Gao P, Zhang Y et al. (2013). Metabolic and functional genomic studies identify deoxythymidylate kinase as a target in LKB1-mutant lung cancer. Cancer Discov 3, 870–879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lizcano JM, Goransson O, Toth R, Deak M, Morrice NA, Boudeau J, Hawley SA, Udd L, Makela TP, Hardie DG, et al. (2004). LKB1 is a master kinase that activates 13 kinases of the AMPK subfamily, including MARK/PAR-1. Embo J 23, 833–843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perera RM, Stoykova S, Nicolay BN, Ross KN, Fitamant J, Boukhali M, Lengrand J, Deshpande V, Selig MK, Ferrone CR, et al. (2015). Transcriptional control of autophagy-lysosome function drives pancreatic cancer metabolism. Nature 524, 361–365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pineda CT, Ramanathan S, Fon Tacer K, Weon JL, Potts MB, Ou YH, White MA, and Potts PR (2015). Degradation of AMPK by a cancer-specific ubiquitin ligase. Cell 160, 715–728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raben N, and Puertollano R (2016). TFEB and TFE3: Linking Lysosomes to Cellular Adaptation to Stress. Annu Rev Cell Dev Biol 32, 255–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saito Y, Chapple RH, Lin A, Kitano A, and Nakada D (2015). AMPK Protects Leukemia-Initiating Cells in Myeloid Leukemias from Metabolic Stress in the Bone Marrow. Cell Stem Cell 17, 585–596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shackelford DB, and Shaw RJ (2009). The LKB1-AMPK pathway: metabolism and growth control in tumour suppression. Nat Rev Cancer 9, 563–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vara-Ciruelos D, Dandapani M, Gray A, Egbani EO, Evans AM, and Hardie DG (2017). Genotoxic Damage Activates the AMPK-alpha1 Isoform in the Nucleus via Ca(2+)/CaMKK2 Signaling to Enhance Tumor Cell Survival. Mol Cancer Res. [DOI] [PubMed] [Google Scholar]
- Young NP, Kamireddy A, Van Nostrand JL, Eichner LJ, Shokhirev MN, Dayn Y, and Shaw RJ (2016). AMPK governs lineage specification through Tfeb-dependent regulation of lysosomes. Genes Dev 30, 535–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zadra G, Photopoulos C, Tyekucheva S, Heidari P, Weng QP, Fedele G, Liu H, Scaglia N, Priolo C, Sicinska E, et al. (2014). A novel direct activator of AMPK inhibits prostate cancer growth by blocking lipogenesis. EMBO Mol Med 6, 519–538. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Raw RNA sequencing data files have been deposited at Mendeley (data.mendeley.com).
DOI for 368T1 cell data, “RNA-seq profiling of 368T1 murine KP NSCLC cells deleted for AMPK with AMPKal add-back subjected to high or no glucose conditions”: 10.17632/gj763hyrny.1.
DOI for 634T cell data, “RNA-seq profiling of 634T murine KP NSCLC cells deleted for AMPK subjected to no glucose conditions”: 10.17632/3cy28w7v9k.1.