

Abstract
Coronary artery anomalies are common congenital disorders with serious consequences in adult life. Coronary circulation begins when the coronary stems form connections between the aorta and the developing vascular plexus. We recently identified the WNT signaling modulator R-spondin 3 (Rspo3), as a crucial regulator of coronary stem proliferation. Using expression analysis and tissue-specific deletion we now demonstrate that Rspo3 is primarily produced by cardiomyocytes. Moreover, we have employed CRISPR/Cas9 technology to generate novel Lgr4-null alleles that showed a significant decrease in coronary stem proliferation and thus phenocopied the coronary artery defects seen in Rspo3 mutants. Interestingly, Lgr4 mutants displayed slightly hypomorphic right ventricles, an observation also made after myocardial specific deletion of Rspo3. These results shed new light on the role of Rspo3 in heart development and demonstrate that LGR4 is the principal R-spondin 3 receptor in the heart.
Keywords: Coronary artery, Secondary heart field, endothelial proliferation, R-spondin3 (Rspo3), Wnt signaling, Leucine Rich Repeat G Protein Coupled Receptor (Lgr4)
INTRODUCTION
Coronary arteries are essential for supplying blood to the heart and coronary artery anomalies can lead to serious consequences such as sudden cardiac death (Riley and Smart, 2011). Furthermore, coronary heart disease, resulting from obstruction of the coronary arteries, is one of the leading causes of death worldwide (Aisagbhoni et al., 2014). Hence, understanding the molecular mechanisms guiding coronary artery formation is crucial for developing novel treatments for heart disease.
The coronary arteries are derived from the primitive vascular plexus of the heart in a complex remodeling process that begins at embryonic day (E) 11.5 (Red Horse et al., 2010) and continues well into the post-natal period (Tian et al., 2014). Coronary circulation begins when coronary artery stems form connections between the aorta and the vascular plexus at highly stereotyped locations (Tian et al., 2013, Chen et al., 2014). Proper development of the coronary stems is essential for supporting the coronary arteries and defects in stem formation can lead to improper development of the arterial tree (Chen et al., 2014). Cardiomyocytes are one of many cell types essential for coronary vessel development. By secreting soluble growth factors, they promote formation of the coronary arteries as well as the coronary stems (Wu et al., 2012, Lavine et al., 2006).
R-spondins (RSPO1–4) are secreted activators of both canonical (β-catenin dependent) and non-canonical Wnt signaling pathways with highly pleiotropic roles during embryonic development and adult homeostasis (de Lau et al., 2012; Scholz et al., 2016). By binding to leucine-rich-repeat-containing G protein-coupled receptors (LGRs) they potently increase the availability of WNT receptors thus enhancing WNT signalling (de Lau et al., 2014). Of particular interest is R-spondin3 (Rspo3), which has been shown to play an important role in placental vasculature remodeling and the zonation of various organs (Aoki et al., 2007; Kazanskaya et al., 2008; Vidal et al., 2016; Rocha et al., 2015). In the heart, tissue-specific ablation of Rspo3 with the Islet1Cre line led to decreased expansion of the secondary heart field (SHF), resulting in outflow tract abnormalities, hypomorphic right ventricles and early embryonic lethality (Cambier et al., 2014).
More recently, we have shown that Rspo3 also plays a critical role in coronary artery formation. Rspo3 is highly expressed around the developing coronary stems and temporal ablation of Rspo3 with the ubiquitously expressed CAGGCreER™ line (CAGR3 mutants) (Hayashi and McMahon, 2002) leads to improper formation of the arterial tree. We have also shown that RSPO3 acts through the Wnt/β-catenin signaling pathway to promote arterial specific proliferation of the coronary stems and ensure their proper development (Da Silva et al., 2017). The cell types expressing Rspo3 and the receptors that transduce the Rspo3 signal remain, however, unknown.
R-spondins can bind to three different LGR receptors, LGR4, LGR5 and LGR6 (Carmon et al., 2011; Glinka et al., 2011; de Lau et al., 2011). LGR4 and LGR5 mutants die postnatally from various complications in the intestines, kidneys and lungs, while LGR6 mutants are viable (Mazerbourg et al., 2004; Morita et al., 2004; Snippert et al., 2010). To date, neither coronary nor progenitor defects have been reported in either LGR4, LGR5 or LGR6-null embryos, which may suggest that RSPO3 acts through different receptors in the heart. However, a previous study has revealed that approximately 40% of LGR4 mutants die during gestation (Mazerbourg et al., 2004; Kinzel et al., 2014). Although the causes are unclear, the premature lethality occurring in LGR4 null embryos may be indicative of cardiac defects.
Here we show that Rspo3 is specifically expressed in the cardiomyocytes surrounding the coronary stems and that myocardial-specific deletion of Rspo3 phenocopies the coronary artery defects seen in CAGR3 mutants. In addition, by using CRISPR-Cas9 technology we have generated novel Lgr4 mutants, that, strikingly, display cardiac abnormalities resembling the phenotype in Rspo3 mutants
MATERIALS AND METHODS
Mice
All animal work was conducted according to national and international guidelines and was approved by the local ethics committee (PEA-NCE/2013/88). The Rspo3flox, mTmG, CAGGCre-ER™, Myh6MerCreMer, WT1CreERT2, and Pax3Cre lines have been described previously (Rocha et al., 2015; Muzumdar et al., 2007; Hayashi and McMahon, 2002; Yan et al., 2015; Zhou et al., 2008; Engleka et al., 2005). Cre activation was obtained by a single administration (gavage) of 150 mg/kg tamoxifen (Sigma-Aldrich) dissolved in corn oil (Sigma-Aldrich) to pregnant females aged around 8 weeks carrying E8.5 embryos. For proliferation assays BrdU (Sigma-Aldrich) dissolved in 0.9% NaCl was administered to pregnant dams one hour before sacrificing via intraperitoneal (IP) injection at a dose of 50mg/kg. Embryos were analyzed at various time-points (E13.5, E16.5 and E17.5) and gender was not taken into consideration.
In situ hybridization
Tissues were fixed overnight in 4% paraformaldehyde, progressively dehydrated and embedded in paraffin. 7 micrometer thick sections were cut then rehydrated and hybridization was performed as described in Wilkinson, 1992. Hybridized DIG-RNA probes were detected with alkaline phosphatase-coupled anti-digoxygenin antibody (1:4000, Roche). After washing, the chromogenic reaction was performed with NBT-BCIP substrate (Promega) for several days at room temperature. For the RNAscope analysis the Lgr4, Lgr5 and Rspo3 probes were purchased from the manufacturer (Advanced Cell Diagnostics) and the protocol was performed according to the manufacturer’s instructions using the chromogenic Fast Red dye that can be visualized using light or fluorescence microscopy.
Immunofluorescence and histological analysis
For immunofluorescence experiments, tissues were fixed overnight in 4% paraformaldehyde, progressively dehydrated and embedded in paraffin. 5 micrometer thick sections were rehydrated, boiled in a pressure cooker for 2 minutes with Antigen Unmasking Solution (Vector laboratories) and blocked in PBS solution containing 10% normal donkey serum and 3% BSA. All antibodies were applied overnight at 4°C at the concentrations listed in the antibody table (see Table S1). Secondary antibodies were diluted 1:400 and applied at room temperature for 1 hour. For histological analysis 5 micrometer thick sections were stained with haematoxylin and eosin according to standard procedures.
Wholemount Immunohistochemistry
Hearts were fixed in 4% PFA for 1 hour, washed in PBS, incubated in methanol/hydrogen peroxide (8:1) for 1 hour and then blocked in PBSST (5% Skim milk powder and 0.5%Triton X-100 dissolved in PBS) for five hours. The primary antibody used was Rabbit anti-mouse Connexin 40 (Alpha Diagnostic International, 1:200). Donkey anti-Rabbit IgG (Santa Cruz, 1:100) was used as a secondary antibody and visualization was performed by incubating with DAB substrate (Sigma Aldrich). All antibodies were diluted in PBSST and incubations were carried out at 4°C overnight. Following each overnight incubation, tissues were washed three time 1 hour each at 4°C with PBSST
Isolation and treatment of primary endothelial cells from embryonic hearts
Isolation of endothelial cells from E15.5 hearts was performed as described elsewhere (Da Silva et al., 2017). Briefly, hearts were dissected, minced, pooled together and digested for 30 minutes at 37°C. The cell suspension was then washed and incubated for 15 minutes at 4°C with magnetic CD31 microbeads (Miltenyi) in PBS supplemented with 2% FBS and 2mM EDTA Cells were then passed through two MS columns (Miltenyi) attached to a magnet, applying several washes and then eluting according to the manufacturer’s protocol. The eluted cells were then resuspended in endothelial cell growth medium (DMEM + 20%FBS + 100μg/ml ECGC (Sigma Aldrich)) and grown until confluence. The non-bound flow-through cells were kept and grown in the same media as the isolated endothelial cells. RNA was extracted using the RNAeasy microkit (QIAGEN) according to the manufacturer’s protocol and gene expression levels analyzed by RT-qPCR as described in the supplemental materials and methods.
Generation of CRISPR/Cas9 Lgr4 mutant embryos
The publically available Zhanglab CRISPR software (addgene) was used to design the guide RNA template targeting exon 3 of the Lgr4 gene. Potential off-targets were also identified using this software. Primers encoding the guide RNA sequence were cloned into the pX330 plasmid (addgene) and a T7 promoter sequence was added on by PCR to the final construct for transcription. Cytoplasmic microinjection of the guide RNA along with Cas9 RNA into fertilized B6D2 zygotes was then performed. F1 heterozygotes containing a 5bp deletion immediately before the cut site were identified by sequencing the PCR product and then backcrossed for three generations before being analyzed.
Statistical Analyses
Statistical Analyses were performed according to the two tailed unpaired Student’s t-test, *p<0.05 **p<0.01, ***p<0.001. Error estimates are expressed as standard error of mean (SEM). Details of the statistical analyses and the programs used for quantification can be found in the figure legends. The letter “n” refers to the number of individual samples/hearts (embryonic dissections) or the number of wells (in vitro experiments).
RESULTS
R-spondin3 is expressed by cardiomyocytes around the developing coronary stems
Our previous work demonstrated that Rspo3 was highly expressed around the developing coronary stems during critical time-points in their development. Genetic deletion was performed using the CAGGCreER™ line that induces deletion in a ubiquitous manner. To determine whether the Rspo3-expressing cells were derived from the cardiac neural crest (CNC) or the proepicardial organ (PEO) we crossed an Rspo3 floxed allele (Rspo3fl; Rocha et al., 2015 ) with the Pax3Cre (CNC-specific) (Engleka et al., 2005) or WT1CreERT2 (PEO-specific) (Zhou et al., 2008) lines (Fig. S1A, schematic), respectively. The presence of the mTmG reporter allele, which expresses GFP after Cre-induced recombination (Muzumdar et al., 2007), allowed detection of cells in which recombination had occurred. In situ hybridization analysis on sections of the heart near the developing coronary stems revealed persistent expression of Rspo3 when deletion was induced with the Pax3Cre or Wt1CreERT2 lines (Fig. S1A). Furthermore, neither the Pax3Cre nor the Wt1CreERT2 deletion of Rspo3 led to coronary defects (Fig. S1B), demonstrating that Rspo3 is not expressed by cardiac neural crest or epicardial-derived cells.
To determine if Rspo3 was expressed by cardiomyocytes, we performed immunostaining with the myocyte-specific myosin heavy chain 1E (MF20) antibody on sections of the heart near the coronary stems previously stained with an Rspo3 in situ hybridization probe. Indeed, close inspection of the staining pattern revealed co-expression of Rspo3 mRNA and MF20 (Fig. 1A). We then performed RNAScope analysis with a fluorescent probe specific to Rspo3 mRNA followed by immunostaining with the endothelial-specific Platelet endothelial cell adhesion molecule (PECAM) antibody. Upon close observation, Rspo3 was not detected in the endothelial cells of the coronary stems (Fig, 1A).
Fig. 1. R-spondin3 is expressed by cardiomyocytes around the developing coronary stems.
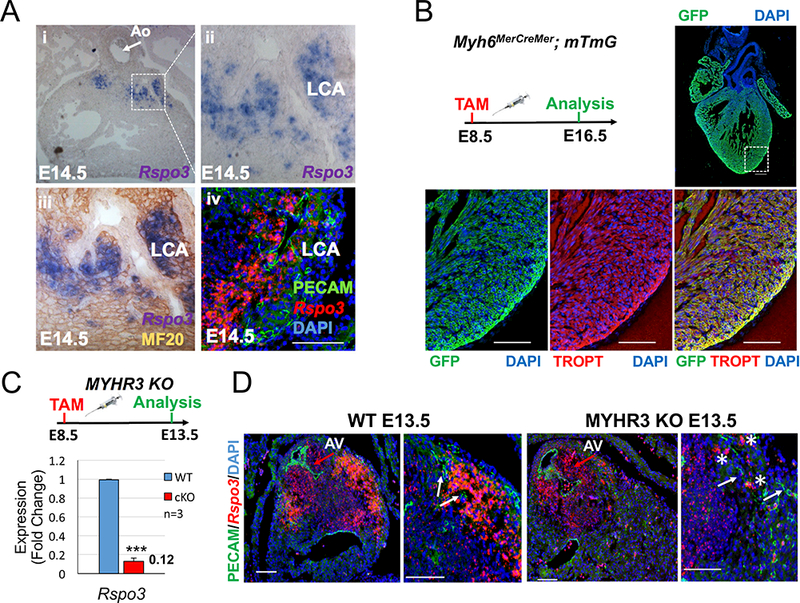
(A) Rspo3 in situ hybridization analysis (i-iii) overlayed with immunostaining for the myocyte-specific MF20 protein (iii) demonstrates Rspo3 is expressed by cardiomyocytes around the developing left coronary artery stem. (iv) RNAScope analysis reveals Rspo3 is not expressed by endothelial cells (PECAM-positive) of the developing left coronary stem (Scale bar 200 μM). (B) The mTmG reporter allele was crossed with the Myh6MerCreMer line to evaluate Cre-specific activity in the myocardium. GFP and Troponin T (TROPT) co-immunostaining shows high recombination efficiency in the myocardium of the atria and ventricles when tamoxifen was administered at E8.5 and analyzed at E16.5. (C) Analysis of Rspo3 expression levels by qPCR reveals an 88% reduction in Myh6MerCreMer ; Rspo3fl/fl (MYHR3-mutant) hearts. Data are expressed as fold change vs. controls and columns are means ± SEM. (D) RNAScope analysis of Rspo3 expression in MYHR3 mutant hearts reveals a specific reduction around the developing left coronary artery vessels (white arrows). Expression in the aortic valves (red arrows) is not affected in MYHR3 mutants. Some Rspo3 expression is still detected around the coronary stems in MYHR3 mutants (asterisks). For all statistical analyses the paired student t-test was used assuming unequal variances, ***p<0.001. LCA=left coronary artery, AV=aortic valve.
Myocardial-specific ablation of Rspo3 leads to coronary artery defects and hypomorphic right ventricles
To test whether cardiomyocyte-specific deletion of Rspo3 would reproduce the coronary defects observed in our CAGR3 deletion, we employed the inducible Myh6MerCreMer line (Myh6 MerCreMer ; Rspo3fl/fl from now on called MYHR3 mutants) (Yan et al., 2015). We first attempted to delete Rspo3 at E11.5, a critical time-point in coronary artery development. However, deletion at this time-point resulted in a mere 60% reduction (p=0.001) in Rspo3 expression (Fig. S2A, B), which was drastically less efficient than the CAGR3 deletion (97%, p=0.00028) (Fig. S2C, D). To examine why the deletion efficiency was so low we analyzed Myh6MerCreMer hearts crossed with the mTmG line. In accordance with Yan et al., 2015, and consistent with the Myh6 expression pattern (Ng et al., 1991), the recombination rate with the E11.5/12.5 pulse was very low in the ventricular myocytes of the heart (where Rspo3 is expressed) in comparison to atrial myocytes (Fig. S2A). To test if we could improve the deletion efficiency of Rspo3, we performed deletion at E8.5 (Fig 1B, schematic). With this early deletion we observed very efficient recombination in all compartments of the heart as testified by activation of the GFP protein (Fig. 1B). More importantly, qPCR analysis revealed an 88% decrease (p=0.0007) in Rspo3 expression levels when compared to controls (Fig. 1C). RNAScope analysis confirmed a strong decrease in Rspo3 expression around the coronary stems (PECAM-positive), whereas its expression in the interstitial cells of the aortic valves was maintained (Fig. 1D). In some MYHR3 mutants, however, persistent expression of Rspo3 was detected around the coronary stems after deletion indicating variable recombination rates with the Myh6MerCreMer line (Fig. S2E).
Since deletion of Rspo3 with the Islet1Cre led to severely hypomorphic right ventricles and outflow tract deformations (Cambier et al., 2014), we analyzed our MYHR3 mice for these abnormalities. Interestingly, 5 out of 13 embryos died before analysis at E16.5, and 4 out of 8 MYHR3 mutant hearts displayed hypomorphic right ventricles (Fig. 2A-B). However, no outflow tract defects were detected, which is most likely due to the myocyte specific expression of the Myh6 gene, in comparison to Islet1 which is more broadly expressed in the SHF (Sun et al., 2007). Since a smaller right ventricle is commonly associated with defects in secondary heart field expansion we decided to analyze the expression pattern of Rspo3 at earlier time-points. RNAScope analysis on E9.5 hearts revealed that Rspo3 was highly and specifically expressed in regions with secondary heart field progenitors such as the outflow tract and atrio-ventricular canal (Fig. S1C).
Fig. 2. Myocardial-specific deletion of Rspo3 leads to hypomorphic right ventricles and coronary artery defects.
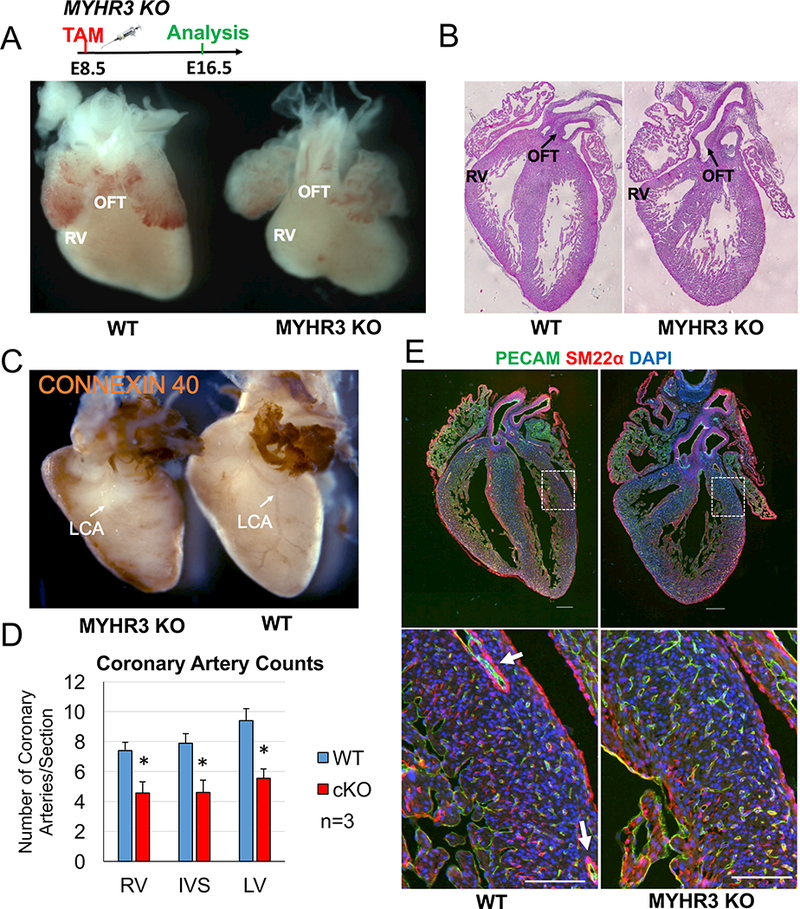
(A) Administration of tamoxifen at E8.5 followed by analysis at E16.5 reveals MYHR3 mutant hearts have hypomorphic right ventricles when compared to controls. (B) H & E staining on sections reveals MYHR3 hearts have smaller right ventricles (RV) when compared to controls. No outflow tract (OFT) abnormalities were detected in MYHR3 mutant hearts (n=8, 4 KO hearts had smaller right ventricles). (C) Whole-mount immunostaining with the arterial-specific Connexin 40 antibody reveals gross morphological differences in the arterial tree of MYHR3 mutants. Lateral view of hearts shows the left coronary artery is completely absent in MYHR3 mutants (white arrows). The atria and outflow tracts of WT and MYHR3 mutant hearts were trimmed to better visualize the coronary artery staining. (D) Quantification of coronary vessel number reveals MYHR3 mutant hearts have less coronary arteries in all compartments of the heart. (n=3, 2 litters). Columns are means ± SEM. (E) Co-immunostaining with PECAM and SM22ɑ antibodies reveals an absence of large coronary arteries (white arrows) in MYHR3 hearts (n=8, 5 KO hearts had coronary defects). All scale bars 200 μM. For all statistical analyses the paired student t-test was used assuming unequal variances, *p<0.05.
To analyze coronary artery development in our MYHR3 mutants we performed wholemount immunostaining with the arterial-specific Connexin 40 antibody. Analysis of MYHR3 hearts revealed a drastically underdeveloped arterial tree in 5 out of 8 mutant hearts analyzed. Indeed, in mutants with coronary vessel defects the main left coronary artery branch was almost completely absent (Fig. 2C). Quantification of coronary arterial vessel numbers in MYHR3 mutants revealed a significant decrease in all compartments of the heart when compared to controls (Fig. 2D). Co-immunostaining of PECAM with the smooth muscle marker smooth muscle actin 22 alpha (SM22ɑ) confirmed the absence of larger arteries (white arrows) (Fig. 2E). However, contrary to the CAGR3 mutants (Da Silva et al., 2017), many smaller arteries were still detected in MYHR3 hearts (Fig. 2E). To confirm that the phenotype was related to defective coronary stem proliferation we quantified endothelial proliferation rates at E13.5 with BrdU and PECAM co-staining. As expected MYHR3 mutants exhibited a significant decrease (−34%, p=0.0486) in proliferation of the coronary stems when compared to controls (Fig. S2F), which is slightly less, but consistent with that observed in CAGR3 mutants (−35–50%).
Lgr4 is the main R-spondin receptor expressed in the heart
We next attempted to determine the Lgr family member(s) that may act as R-spondin receptors in the developing heart. RT-PCR analysis with intron spanning primers on cDNA from E13.5 hearts revealed strong bands for Lgr4, while Lgr5 and Lgr6 showed very little amplification (Fig. 3A). This was confirmed with RNAscope analysis, which demonstrated high Lgr4 and very little Lgr5 expression in the heart (Fig. 3B). Limited Lgr5 expression was detected in the heart valves and endocardial cushions, which are areas of high WNT signaling (data not shown) (Alfieri et al., 2010). Interestingly, upon closer inspection we noticed Lgr4 was specifically expressed in the myocardium of the heart and was absent from the endocardial cushions and valves (Fig. 3C, Fig. S3A). qPCR analysis of RNA extracted from whole hearts revealed Lgr4 expression levels remained similar from E10.5 to E16.5 (Fig. 3D). To determine, if Lgr4 was expressed in the endothelium of the heart we isolated primary endothelial cells from E15.5 hearts using magnetic beads coupled to a PECAM antibody (Fig 3E, schematic). Relative expression analysis by qPCR and RT-PCR indicated Lgr4 mRNA levels were high in both the flow through as well as the isolated endothelial cells (Fig. 3F). This sharply contrasts expression of Rspo3, which is not detectable in the endothelium of the heart (Da Silva et al., 2017). Expression levels for Lgr5 and Lgr6 were dramatically lower when compared to Lgr4 expression (Fig. 3G-H). Interestingly, we also detected Lgr4 expression in adult hearts with RNAScope analysis. Immunostaining with an anti-PECAM antibody further revealed that Lgr4 is expressed in the endothelium of adult hearts (Fig. S3B).
Fig. 3. Lgr4 is the main R-spondin receptor expressed in the heart.
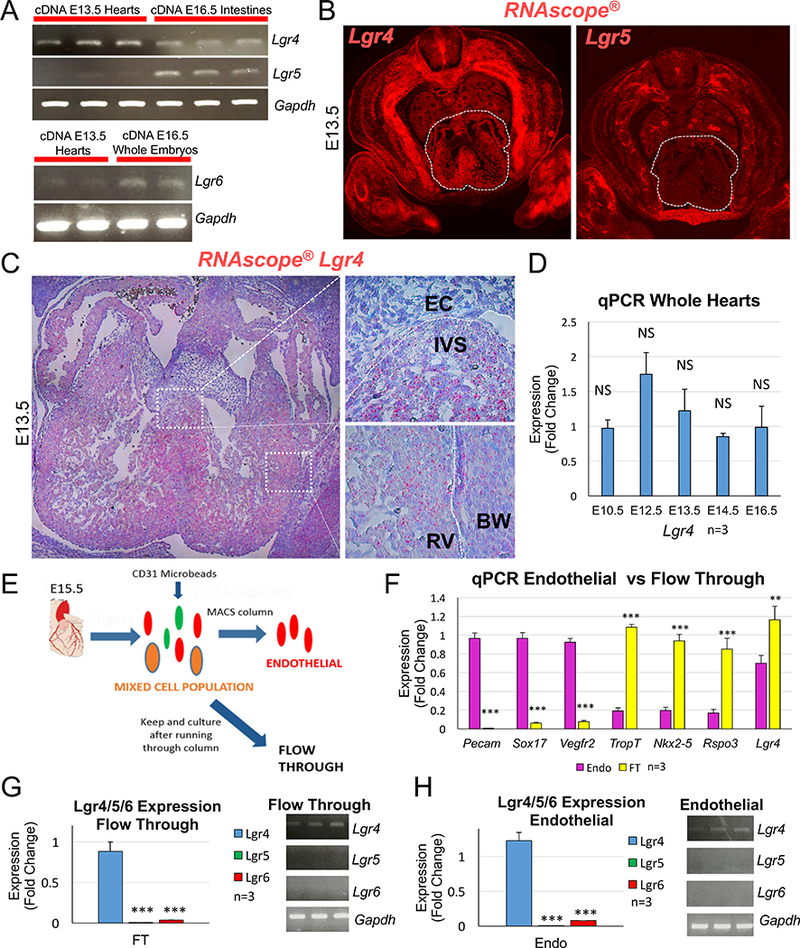
(A) RT-PCR analysis on cDNA from E13.5 hearts with intron-spanning primers demonstrates Lgr4 is expressed in the heart. Very little amplification of Lgr5 is detected. Amplification of both Lgr4 and Lgr5 was detected in control cDNA from intestines (3 separate hearts and intestines were analyzed). Lgr6 amplification was also detected in E13.5 hearts, but at very low levels. cDNA extracted from whole embryos was used as a control for Lgr6 amplification (2 separate hearts and 2 whole embryos analyzed). (B) RNAscope® analysis with fluorescent probes for Lgr4 and Lgr5 reveals Lgr4 is highly expressed in the heart (white dashed outlines) at E13.5 while very little Lgr5 expression is detected. (C) Closer inspection of Lgr4 RNAScope® analysis with Fast Red dye staining reveals Lgr4 is expressed in the myocardium of the ventricles and interventricular septum, but not in the endocardial cushions. (D) qPCR analysis on RNA extracted from whole hearts shows Lgr4 is consistently expressed at similar levels from E10.5 to E16.5. (E) Schematic demonstrating the strategy used to isolate endothelial cells from E15.5 hearts with magnetic microbeads coupled to a CD31 antibody. Non-bound flow through cells (FT) were kept to test the purity of the isolations. (F) qPCR analysis of RNA extracted from isolated cells demonstrates high enrichment of endothelial-specific genes (Pecam1, Sox17 and Vegfr2) in the endothelial cells while FT cells express high levels of myocyte-specific markers (TropT and Nkx2–5). Lgr4 expression is greater in the FT but is still highly expressed in the endothelial cells. Rspo3 was used as a control gene that is not expressed in the endothelium of the heart. (G-H) Analysis of Lgr5 and Lgr6 expression by qPCR and RT-PCR reveals both are expressed at relatively low levels compared with Lgr4 in the FT (G) and endothelial cells (H). All columns are means ± SEM. For all statistical analyses the paired student t-test was used assuming unequal variances, **p<0.01, ***p<0.001. RV=right ventricle, BW=body wall, EC=endocardial cushion, IVS=interventricular septum.
Lgr4-null embryos display hypomorphic right ventricles and coronary artery defects
Since Lgr4 was determined to be the only R-spondin receptor expressed at relatively high levels in the heart, we decided to search for putative cardiac defects in LGR4-null mice. Several mutants targeting exon 1 of Lgr4 have been previously generated, but results regarding the phenotypes have been conflicting (Mazerbourg et al., 2004; Medive et al., 2006; Kinzel et al., 2014; Mustata et al., 2011). Moreover, in silico analysis of the Lgr4 gene revealed a putative start codon in exon 2 (Ensembl, data not shown), which may lead to persistence of a truncated LGR4 protein in the existing knockout animals. We therefore decided to use CRISPR/Cas9 technology to target exon 3 of the Lgr4 gene (Fig. 4A, schematic). Sequencing analysis of genomic DNA from our LGR4 CRISPR mice revealed a 5 base pair deletion immediately before the predicted cut site of the guide RNA (Fig. 4B). This deletion is predicted to lead to a frameshift and a 90% truncation of the original LGR4 protein (CRLGR4 mutants) (data not shown). To exclude the possibility of off-target effects caused by unspecific binding of guide RNAs, we sequenced the top 9 possible off-targets. None of the top hits displayed mutations (data not shown).
Fig 4. Novel Lgr4 CRISPR/Cas9 mutants display cardiac defects.
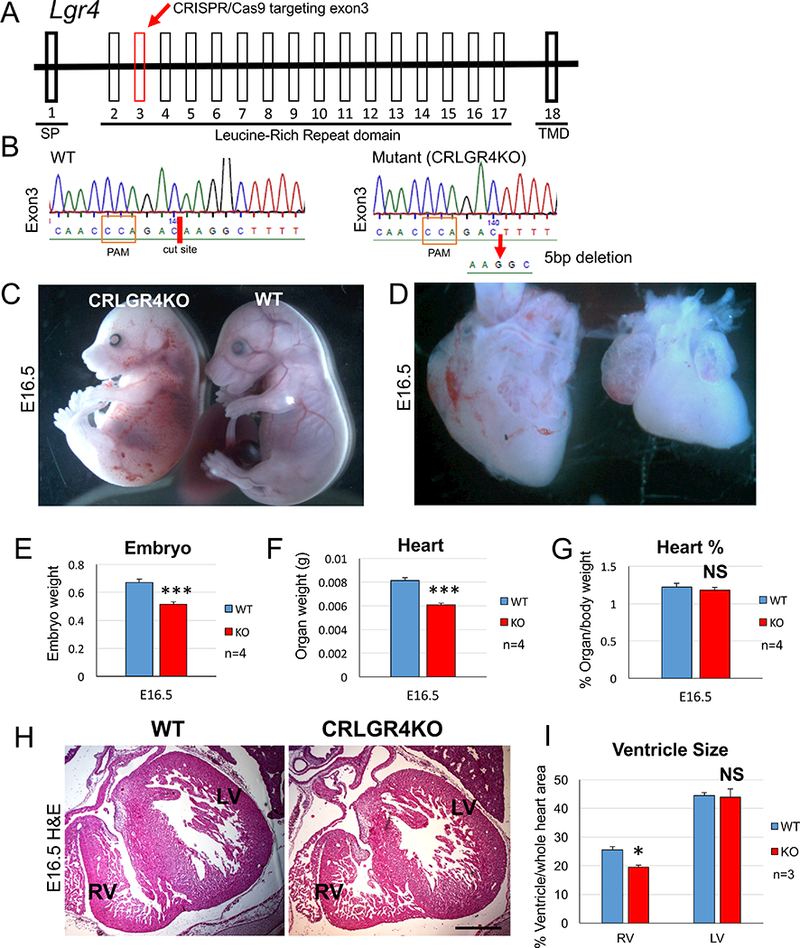
(A) Schematic demonstrating the targeting of exon 3 of the Lgr4 gene using CRISPR/Cas9 technology. (B) DNA sequencing of Lgr4 exon 3 from CRISPR/Cas9 Lgr4 mutants (CRLGR4) demonstrates a 5 base pair deletion was introduced into the cut site (red dash). Sequence shown is the reverse complement. (C) Analysis of CRLGR4 mutant embryos at E16.5 reveals they are smaller and occasionally display edema and haemorrhages when compared to wildtype (WT) controls. (D) CRLGR4 mutant hearts are smaller and display smaller right ventricles. The outflow tracts of CRLGR4 mutant hearts display normal morphology. (E) Quantification of embryo weight reveals CRLGR4 mutants are smaller when compared to controls. (F) CRLGR4 mutant hearts weigh significantly less compared to wildtype controls. (G) When taking embryo weight into account there is no significant difference in heart weight between CRLGR4 mutants and controls. (H) H&E staining reveals CRLGR4 mutant hearts have smaller right ventricles when compared to controls. Scale bar 200 μm. (I) Quantification of right ventricle size reveals a significant decrease in CRLGR4 mutants when compared to controls. Left ventricle size is not affected. All columns are means ± SEM. For all statistical analyses the paired student t-test was used assuming unequal variances, ***p<0.001. SP=signal peptide, TMD=Transmembrane domain, PAM=Protospacer Adjacent Motif, RV= right ventricle, OFT=outflow tract.
Tests of several LGR4 antibodies on wildtype tissues were unsuccessful. Hence, to confirm that the CRISPR/Cas9 deletion led to the truncation of the LGR4 protein we cloned full length Lgr4 cDNA from WT and CRLGR4 hearts upstream of the enhanced green fluorescent protein (EGFP) into the pcDNA3.1 plasmid and then transfected the constructs into HEK293T cells. As a control we cloned wildtype Lgr4 cDNA with an artificial truncation at the predicted deletion site in exon 3 of CRLGR4 mutants. As predicted, the CRLGR4 mutant construct produced a truncated protein (45 kDa) with a similar size to the artificially truncated WT construct when analyzed by western blot with an anti-GFP antibody; thus, confirming that our CRLGR4 5 base pair deletion indeed leads to truncation of the LGR4 protein (Fig. S4).
To avoid counter-selection of severely affected embryos and better understand the full-spectrum of the phenotypes resulting from LGR4 deletion, we decided to analyze our mutant mice at E16.5 rather than at later time-points. In accordance with previously published data (Mazerbourg et al., 2004), CRLGR4-null mice were smaller and weighed less (−23%, n=4) than control littermates (Fig. 4C, E and Fig. S5A). Some mutant embryos displayed edema (Fig. 4C). CRLGR4 mutant hearts were also significantly smaller (−22%, n=4; Fig. 4D, F, and Fig. S5B), although this difference disappeared when taking into account body weight (Fig. 4G).
To ensure that the introduced CRISPR mutation recapitulated the previously published knockout strains, we analyzed other organs known to be affected by LGR4 deletion. LGR4 has been previously shown to be required for stem cell proliferation in the intestinal crypts (de Lau et al., 2011; Kinzel et al., 2014). As expected, CRLGR4 mutant mice displayed significantly decreased proliferation rates (−40%, p=0.03) in the intestinal crypts (Fig. S5C, D). In the kidneys, LGR4 ablation leads to dilated tubules and cyst formation (Kinzel et al., 2014) and a similar phenotype was detected in CRLGR4 mice (Fig. S6).
Next we decided to analyze various CRLGR4 mutant hearts to see if we could detect SHF defects and/or coronary artery anomalies. Interestingly, in 5 out of 18 mutant embryos we observed slightly hypomorphic right ventricles. Three of these mutants also had noticeable edema (Fig. 4C). qPCR analysis at E10.5 revealed decreased expression of Islet1 (p=0.07) which although was not significant, may be indicative of decreased canonical Wnt signaling since Islet1 is a direct target of β-catenin (Fig. 5A). In previous works deletion of β-catenin (Lin et al. 2007) and Rspo3 (Cambier et al. 2014) in the secondary heart field with the Islet1 Cre both led to decreased proliferation rates in the heart. To determine if our CRLGR4 mutants also displayed decreased proliferation rates we performed BrdU staining at E10.5. Interestingly, CRLGR4 mutants displayed a slight (−15%) but significant decrease (p=0.043) in proliferation. Quantification of the number of coronary arteries revealed that 6 out of 18 CRLGR4 mutant embryos analyzed had less coronary arteries in all compartments of the heart when compared to controls (Fig. 5C, D). Whole-mount immunostaining with a Connexin 40 antibody showed that the major branches of the left and right coronary arteries were malformed in CRLGR4 mutants (Fig. 5E). However, many smaller coronary arteries were still detected, altogether suggesting the phenotype to be less severe than in CAGR3 mutants (Da Silva et al., 2017).
Fig. 5. Lgr4 mutants exhibit decreased proliferation and have defects in coronary artery formation.
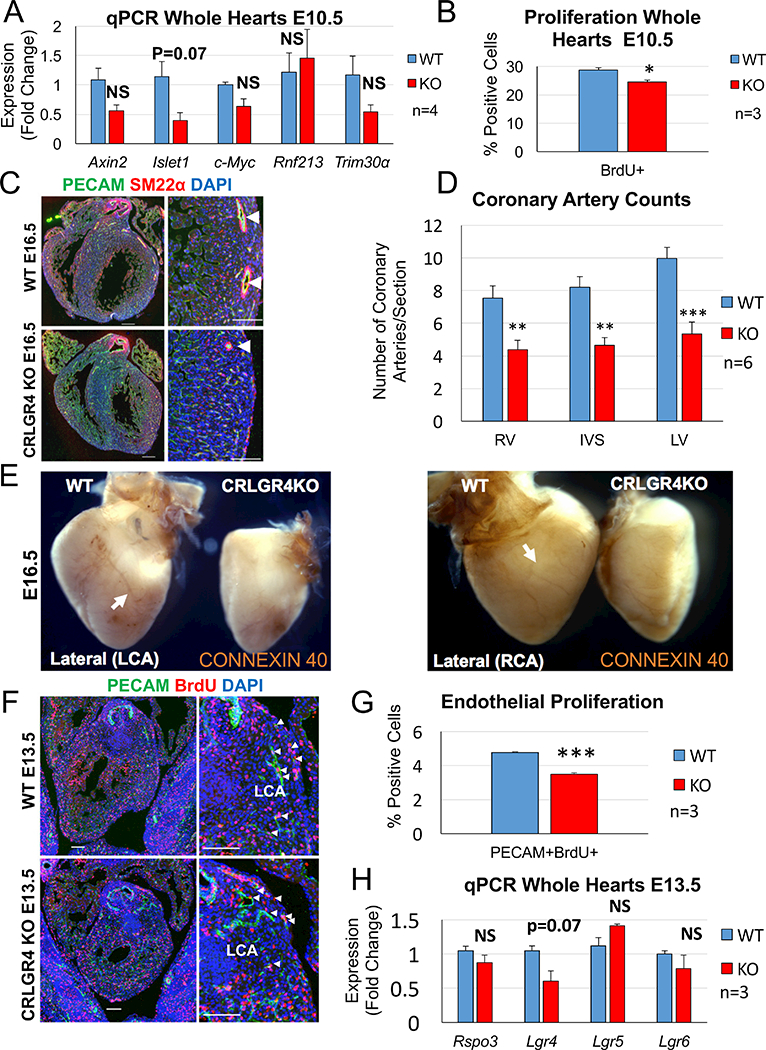
(A) qPCR analysis of CRLGR4 KO hearts at E10.5 reveals a decrease in the secondary heart field marker and β-catenin target Islet1 when compared to littermate controls although this did not reach statistical significance (n=4, 3 litters) (B) Quantification of proliferation in CRLGR4 KO hearts at E10.5 reveals a significant decrease when compared to controls (n=3, 2 litters) (C) Co-Immunostaining with PECAM and SM22ɑ antibodies demonstrates a reduction in coronary arteries (white arrowheads) in CRLGR4 mutants. Scale bars: mosaics 200 μM, close ups 100 μM. (D) Quantification of coronary arteries demonstrates a significant reduction in all compartments of the heart in CRLGR4 mutants when compared to controls. (E) Whole-mount immunostaining with the arterial-specific connexin 40 antibody reveals the major branches of the left and right coronary arteries (white arrows) are malformed in CRLGR4 mutants. The atria and outflow tracts of WT and CRLGR4 mutant hearts were trimmed to better visualize the coronary artery staining. (F) Co-immunostaining with PECAM and BrdU antibodies on sections of the heart near the developing left coronary artery reveals CRLGR4 mutants have decreased rates of endothelial proliferation. Scale bars mosaics 100 μM, close ups 100 μM. (G) Quantification of endothelial proliferation around the developing left coronary artery demonstrates a significant reduction in CRLGR4 mutants when compared to controls. Columns are means ± SEM. (H) qPCR analysis of RNA extracted from the upper halves of CRLGR4 mutant hearts does not demonstrate any significant change in the expression of Rspo3 or the Lgr4,5,6 receptors (n=3, 2 litters). Note that Lgr4 message carries the introduced deletion mutation, which may account for the slightly reduced expression levels. Data are expressed as fold change vs. controls and columns are means ± SEM. For all statistical analyses the paired student t-test was used assuming unequal variances, *p<0.05, ***p<0.001. RV= right ventricle, IVS=interventricular septum, LV=left ventricle, CA=coronary artery, LCA=left coronary artery, RCA= right coronary artery.
Finally, to see if the coronary artery defects observed in our CRLGR4 mutants was due to decreased proliferation of the coronary stems (as was previously observed in CAGR3 mutants) we performed co-immunostaining with anti-BrdU and PECAM antibodies on sections of the heart near the developing left coronary (a region with high Rspo3 expression) (Fig. 5F). Quantification of PECAM/BrdU-positive cells revealed a highly significant 25% reduction (p=0.0005, n=3) in CRLGR4 mutant hearts (Fig. 5G). However, the reduction in proliferation was less severe than that observed in CAGR3 mutants (35% to 50%) (Da Silva et al., 2017), possibly accounting for the milder phenotype in our CRLGR4 mutants. Quantification showed a small reduction of Lgr4 RNA (p=0.07), which may reflect reduced stability of the mutated RNA. However, Lgr5 and Lgr6 expression persisted in the CRLGR4 mutants, which may have permitted low levels of RSPO3 signaling (Fig. 5H).
DISCUSSION
Coronary arteries are essential to support the heart with oxygen and coronary artery abnormalities can lead to serious consequences in adult life (Riley and Smart, 2011). We have previously shown that localized expression of the WNT signaling modulator Rspo3 promotes proliferation of the coronary stems through the β-catenin signaling pathway (Da Silva et al., 2017). Here we demonstrated that the major cell-types responsible for expressing Rspo3 around the coronaries are cardiomyocytes and, myocardial-specific deletion of Rspo3 phenocopied the coronary artery defects observed after ubiquitous deletion of Rspo3. These results validate the critical role of Rspo3 in coronary stem proliferation and they further elucidate the importance of cardiomyocytes in promoting coronary vessel development. Particularly striking is the highly restricted regional expression pattern of Rspo3 around the coronaries, and it will be important to identify the factors involved in activating Rspo3 expression.
It is worth noting that although myocardial-specific deletion of Rspo3 led to an overall decrease in coronary arteries, the phenotype was less severe than in CAGR3 mutants. This can be explained by incomplete deletion resulting from the Cre line used. Indeed, previous work has shown that the Myh6 gene shows variable expression in the ventricles during embryonic development (Ng et al., 1991). Alternatively, Rspo3 may not exclusively be expressed by cardiomyocytes and indeed we were able to detect sparse expression of Rspo3 around the coronaries in all MYHR3 mutants analyzed (Fig. 1D, asterisks). It is also possible that expression of Rspo3 in the aortic valves contributes to the development of the coronary orifices and/or anastomosis of the coronary arteries with the aorta.
In addition to the analysis of tissue-specific deletions for Rspo3, we also report here a novel mutant allele for Lgr4 that carries a 5 base pair deletion in exon 3. Homozygous mutants recapitulated the intestinal and kidney phenotypes reported for previous Lgr4 alleles, suggesting that it indeed acts as a loss of function mutation. By focussing on the heart we demonstrate that LGR4 is the principle receptor for Rspo3 during coronary stem formation. Given the previous studies with Lgr4 mutant alleles it seems surprising that no cardiac defects have been reported. This may be due to the incomplete penetrance of the phenotype we have observed. Indeed, in the study by Mazerbourg et al. 2004, 40% of Lgr4 null mutants were predicted to die in utero. Although the timing of death was not determined in this work, a later study by Kinzel et al. 2014 revealed that many Lgr4/5 double mutants displayed autolysis at E16.5 and these embryos were excluded from the analysis. It is therefore possible that at E15–16 more severe Lgr4-null mutants begin to die from cardiac complications. This is supported by our previous unpublished observations that around 40% of CAGR3 mutants also died around E15–16.
Why is the cardiac phenotype so variable in Lgr4 mutants? Including this work, there are now at least three independent studies demonstrating that some LGR4 mutants die in utero from unknown complications (Mazerbourg et al., 2004; Kinzel et al., 2014). All three mutants were generated using different methods in different genetic backgrounds and it is unlikely that this variability is due to technical problems or differences in the mouse strains used. Instead RSPO3 may be able to bind to LGR5 and/or LGR6 receptors in the absence of the LGR4 protein and permit the formation of sufficient coronary vessels for survival. Alternatively, a recent study has suggested that, at least in vitro (HAP1 cell line), Rspo3 and Rspo2 can activate β-catenin signaling in the absence of LGR4, 5 and 6 by directly binding to Glypicans or Syndecans (Lebensohn and Rohatgi, 2018). Whether or not this is the case in the developing heart remains to be tested.
On a final note, neither the MYHR3 nor the CRLGR4 mutants displayed outflow tract deformations. This is surprising since deletion of Rspo3 with the Islet-1Cre results in severe outflow tract abnormalities (Cambier et al., 2014). This may be explained by the fact that both the MYHR3 and CRLGR4 mutants only affect RSPO3-mediated signaling in the myocardium, and that somehow this only affects the formation of the right ventricle and not the outflow tract. This is an interesting observation since it points to lineage restrictions within early progenitor populations that are most likely controlled by an RSPO-LGR4 axis.
In conclusion, here we have shown that myocardial specific RSPO3 acts mainly through the LGR4 receptor to promote coronary stem proliferation in the developing heart. These results provide novel insights into the role of RSPO3 and WNT/β-catenin signaling in coronary artery formation that in the long run may be useful for the development of novel regenerative treatments for patients suffering from coronary heart disease.
Supplementary Material
Highlights:
R-spondin3 (Rspo3) is specifically expressed by cardiomyocytes around the csoronary stems
Myocardial-specific deletion of Rspo3 results in coronary artery defects
Leucine Rich Repeat G Protein Coupled Receptor 4 (Lgr4) is the main R-spondin receptor expressed in the heart
Novel CRISPR/Cas9 Lgr4 mutants display coronary artery defects and hypomorphic right ventricles
ACKNOWLEDGEMENTS
We would like to thank the staff of the animal facility for their dedication. We are indebted to Hitoshi Okamoto (Riken Institute, Japan) for providing the Rspo3fl allele. Chen-Leng Cai is supported by grants from the NIH and AHA.
SOURCES OF FUNDING:
This work was supported by grants from the Fondation du France, ARC (SL22020605297) and the ANR (ANR-ADSTEM & ANR-11-LABX-0028–01).
Footnotes
AUTHOR CONTRIBUTIONS
F.D.S. and A.S. designed the project. F.D.S. carried out all experiments, if not otherwise stated. F.M designed the CRISPR/Cas9 construct and E.G. performed the RNAScope analysis. V.V performed the analysis of the Lgr4 mutant kidneys. A.S.R. and F.J.M. provided technical support with various experiments. K.D.W. and A.S.R. provided critical input for experimental design and data analysis. F.D.S. and A.S. wrote the manuscript, and all authors provided editorial input.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES:
- 1).Aisagbonhi O et al. (2011). Experimental myocardial infarction triggers canonical Wnt signaling and endothelial-to-mesenchymal transition. Disease models and Mechanisms 483, 469–483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2).Alfieri CM, Cheek J, Chakraborty S and Yutzey KE (2010). Wnt signaling in heart valve development and osteogenic gene induction. Developmental Biology, 338, 127–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3).Aoki M, Mieda M, Ikeda T, Hamada Y, Nakamura H and Okamoto H (2007) R-spondin3 is required for mouse placental development. Developmental Biology 301, 218–226. [DOI] [PubMed] [Google Scholar]
- 4).Cambier L, Plate M, Sucov HM, and Pashmforoush M (2014). Nkx2–5 regulates cardiac growth through modulation of Wnt signaling by R-spondin3. Development 141, 2959–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5).Carmon KS, Gong X, Lin Q, Thomas A and Liu Q (2011) R-spondins function as ligands of the orphan receptors LGR4 and LGR5 to regulate Wnt/beta-catenin signaling. PNAS 108, 11452–11457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6).de Lau WB, Snel B & Clevers HC (2012). The R-spondin protein family. Genome Biology 13, p.242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7).de Lau W et al. (2011). Lgr5 homologues associate with Wnt receptors and mediate R-spondin signalling, Nature 476, 293–297. [DOI] [PubMed] [Google Scholar]
- 8).Engleka KA, Gitler AD, Zhang M, Zhou DD, High FA and Epstein JA (2005). Insertion of Cre into the Pax3 locus creates a new allele of Splotch and identifies unexpected Pax3 derivatives. Developmental Biology, 280, 396–406. [DOI] [PubMed] [Google Scholar]
- 9).Glinka A, Dolde C, Kirsch N, Huang Y-L, Kazanskaya O, Ingelfinger D, Boutros M, Cruciat C-M and Niehrs C (2011). LGR4 and LGR5 are R-spondin receptors mediating Wnt/0β-catenin and Wnt/PCP signalling’, EMBO reports 12, 1055–1061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10).Hayashi S & McMahon AP (2002). Efficient Recombination in Diverse Tissues by a Tamoxifen-Inducible Form of Cre: A Tool for Temporally Regulated Gene Activation/Inactivation in the Mouse. Developmental Biology 244, 305–318. [DOI] [PubMed] [Google Scholar]
- 11).Kazanskaya O et al. (2008). The Wnt signaling regulator R-spondin 3 promotes angioblast and vascular development. Development 135, 3655–3664. [DOI] [PubMed] [Google Scholar]
- 12).Kinzel B et al. (2014). Functional roles of Lgr4 and Lgr5 in embryonic gut, kidney and skin development in mice. Developmental Biology 390, 181–190. [DOI] [PubMed] [Google Scholar]
- 13).Lau W De, Peng WC, Gros P and Clevers H (2014). The R-spondin / Lgr5 / Rnf43 module: regulator of Wnt signal strength The R-spondin / Lgr5 / Rnf43 module: regulator of Wnt signal strength. Genes & Development 28, 305–316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14).Lebensohn AM and Rohatgi R (2018). R-spondins can potentiate WNT signaling without LGRs. eLife 7, 1–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15).Lin L et al. (2007). β-Catenin directly regulates Islet1 expression in cardiovascular progenitors and is required for multiple aspects of cardiogenesis. PNAS 104, 9313–9318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16).Mazerbourg S et al. (2004). Leucine-rich repeat-containing, G protein-coupled receptor 4 null mice exhibit intrauterine growth retardation associated with embryonic and perinatal lethality. Mol Endocrinol, 18, 2241–2254. [DOI] [PubMed] [Google Scholar]
- 17).Morita H, Mazerbourg S, Bouley DM, Luo C-W, Kawamura K, Kuwabara Y, Baribault H, Tian H and Hsueh AJW (2004) Neonatal lethality of LGR5 null mice is associated with ankyloglossia and gastrointestinal distension. Molecular and cellular biology, 24, 9736–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18).Mustata RC, Van Loy T, Lefort A, Libert F, Strollo S, Vassart G and Garcia M-I (2011). Lgr4 is required for Paneth cell differentiation and maintenance of intestinal stem cells ex vivo. EMBO reports, 12, 558–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19).Muzumdar MD et al. (2007). A global double-fluorescent Cre reporter mouse. Genesis (New York, N.Y.: 2000) 45, 593–605. [DOI] [PubMed] [Google Scholar]
- 20).Ng W. a, Grupp IL, Subramaniam A, and Robbins J (1991) Cardiac myosin heavy chain mRNA expression and myocardial function in the mouse heart. Circulation research 68, 1742–1750. [DOI] [PubMed] [Google Scholar]
- 21).Red-Horse K et al. (2010). Coronary arteries form by developmental reprogramming of venous cells. Nature 464, 549–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22).Riley PR and Smart N (2011). Vascularizing the heart. Cardiovascular Research 91, 260–268. [DOI] [PubMed] [Google Scholar]
- 23).Rocha AS, Vidal V, Mertz M, Kendall T, Charlet A, Okamoto H and Schedl A (2015). The Angiocrine Factor Rspondin3 Is a Key Determinant of Liver Zonation. Cell Reports. 13, 1757–1764. [DOI] [PubMed] [Google Scholar]
- 24).Scholz B et al. (2016). Endothelial RSPO3 Controls Vascular Stability and Pruning through Non-canonical WNT/Ca(2+)/NFAT Signaling. Developmental cell 36, 79–93. [DOI] [PubMed] [Google Scholar]
- 25).Snippert HJ et al. (2010). Lgr6 Marks Stem Cells in the Hair Follicle That Generate All Cell Lineages of the Skin. Science 327, 1385–1389. [DOI] [PubMed] [Google Scholar]
- 26).Sun Y et al. (2007). Islet 1 is Expressed in Distinct Cardiovascular Lineages, Including Pacemaker and Coronary Vascular Cells. Dev. Biol 304, 286–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27).Tian X et al. (2014). De novo formation of a distinct coronary vascular population in neonatal heart. Science 345, 90–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28).Vidal V et al. (2016). The adrenal capsule is a signaling center controlling cell renewal and zonation through Rspo3. Genes and Development 30, 1389–1394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29).Wilkinson DG (1992). Wholemount in situ hybridization of vertebrate embryos In In situ hybridization, Wilkinson DG, ed. (Oxford:Oxford University Press; ), pp. 75–83. [Google Scholar]
- 30).Wu B et al. (2012). Endocardial Cells Form the Coronary Arteries by Angiogenesis through Myocardial-Endocardial VEGF Signaling. Cell 151, 1083–1096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31).Yan J, Zhang L, Sultana N, Park DS, Shekhar A, Bu L, Hu J, Razzaque S and Cai CL (2015). A murine Myh6MerCreMer knock-in allele specifically mediates temporal genetic deletion in cardiomyocytes after tamoxifen induction, PLoS ONE, 10, 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32).Zhou B et al. (2008) Epicardial progenitors contribute to the cardiomyocyte lineage in the developing heart. Nature 454, 109–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.