

Abstract
The glucocorticoid receptor is an inducible transcription factor which plays important roles in many physiological processes. Upon activation, GR interacts with regulatory elements and modulates the expression of genes. Although GR is widely expressed in multiple tissues, its binding sites within chromatin and the genes it regulates are tissue specific. Many accessory proteins and cofactors are thought to play a role in dictating GR’s function; however, mechanisms involved in targeting GR to specific sites in the genome are not well understood. Here we describe a high-throughput fluorescence-based method to identify factors involved in GR loading at response elements. This screen utilizes a genetically engineered cell line that contains 200 repeats of a glucocorticoid response promoter and expresses GFP-tagged GR. Upon treatment with corticosteroids, GFP–GR forms a steady-state distribution at the promoter array, and its concentration at this focal point can be quantitatively determined. This system provides a novel approach to identify activities important for GR loading at its response element using siRNA libraries to target factors that enhance or inhibit receptor localization.
Keywords: Nuclear receptor, Glucocorticoid receptor, siRNA screen, Chromatin
1. Introduction
The glucocorticoid receptor (GR) belongs to a class of ligand-inducible transcription factors, which control a wide spectrum of important biological processes including metabolism, development, and inflammatory responses. Other members of the nuclear receptor superfamily including the estrogen receptor (ER), progesterone receptor (PR), androgen receptor (AR), and thyroid receptor (TR) also play prominent roles in physiological processes. In the absence of hormone, GR resides in the cytoplasmic compartment of the cell. Once bound to its ligand, the receptor translocates into the nucleus where it binds either to GR response elements (GREs) present on the DNA as a homodimer or to other regulatory sequences through tethering with additional proteins [1]. GR can induce a positive outcome on transcription by recruiting components of the basic transcriptional machinery or can negatively regulate gene expression. Although GR is expressed in most tissues, the genes it regulates are tissue specific [2]. The chromosomal architecture at these response elements has been shown to be an important mediator for nuclear receptor binding at these sites and contributes to the cell-type-specific activity of these regulatory elements. In addition, many accessory proteins and cofactors are thought to play a role in regulating the function of GR.
As a general approach to the identification of factors that affect the ability of GR to bind to GREs, we have conducted an image-based high-throughput screen to identify chromatin modifiers important for GR loading at GREs (Miranda et al., manuscript in preparation). Although our initial screen focused on chromatin modifiers, other siRNA libraries can be screened using the same system. Direct GR binding to response elements in living cells can be visualized using a genetically engineered cell line containing 200 repeats of the glucocorticoid responsive promoter, MMTV, and expressing GFP-tagged GR [3, 4]. Upon treatment with dexamethasone, these cells form a steady-state distribution of GFP–GR at the promoter arrays, and this localized binding appears as a focal structure in the nucleus of hormone-treated cells (Fig. 1). Using previously described computer algorithms [5, 6], these structures can be automatically detected, and the levels of GFP–GR concentrated at the amplified response elements can be quantitatively determined. The imaging data obtained has been previously shown to be comparative to ChIP data for GR loading at this array [6].
Fig. 1.
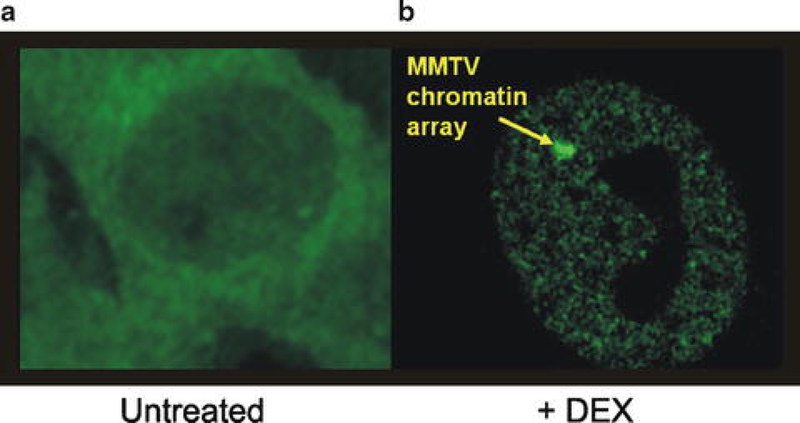
Cells stably expressing GFP–GR and containing 200 copies of the MMTV promoter were treated either with the vehicle (untreated) or with dexamethasone for 30 min. Cells were then imaged using the Opera Imaging System (PerkinElmer). In the untreated cells (a), GFP–GR is localized to the cytoplasm. Upon treatment with dexamethasone (b), GFP–GR localizes to the nucleus and forms on the array of MMTV repeats
In this chapter we provide a detailed protocol that can be used to screen for factors that affect GR loading at its response elements (Fig. 2). This screen can be adapted to other nuclear receptors and/or transcription factors. Using multiple robotic systems (High-Throughput Imaging Facility, NCI), cells are reverse transfected with a siRNA library targeting 300 different chromatin modifiers. Forty-eight hours after transfection cells are treated with either dexamethasone (DEX) or the antagonist RU486. RU486 is a type II antagonist that induces GR to translocate into the nucleus but causes a decrease in GR loading at the array [6–8]. Cells transfected with a scrambled siRNA are treated with RU486 as a control for decreases in array loading (Fig. 3a). Cells transfected with siRNA to GAPDH are used to determine transfection efficiency (Fig. 3b, c). After treatment with the corticoid steroids for 30 min, cells are fixed and nuclei are stained with a fluorescent dye. Cells are then imaged using a high-throughput imaging system and images are analyzed using previously defined algorithms. Although we screened for factors that affect GR loading at a response element, other parameters, such as screening for factors that are involved in translocation of GR or GR turnover, can be examined with this system by incorporating modifications to the image analysis algorithms.
Fig. 2.
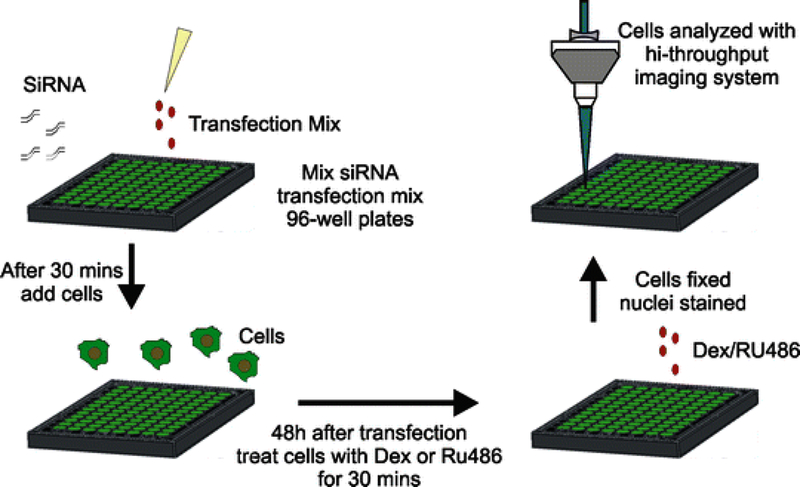
Scheme of the siRNA screening procedure. Briefly, 3,617 cells are transfected with siRNA for 48 h. Cells are then treated with corticoid steroids for 30 min and then fixed with paraformaldehyde. Nuclei are stained and then cells are imaged using a high-throughput imaging system
Fig. 3.

Controls used in screen. (a) Upon treatment of cells with RU486 or dexamethasone (DEX), GR translocates into the nucleus. However, the fraction of arrays formed by GFP–GR binding to the MMTV repeats is lower in cells treated with RU486 than cells treated with DEX. (b) Immunofluorescence for GAPDH in cells transfected with nontargeting siRNA or cells transfected with siGAPDH. (c) Quantification of total cell intensity for GAPDH immunofluorescence. Numbers are an average from six 96-well plates with four wells per plate containing either nontargeting siRNA or siRNA to GAPDH
2. Materials
2.1. Cell Culture
A genetically engineered mouse mammary cell line containing 200 repeats of the glucocorticoid responsive promoter, MMTV, and expressing GFP-tagged GR (3,617 cells) [3, 4] (see Note 1).
Complete medium: Dulbecco’s Modified Eagle’s Medium (DMEM) supplemented with 10 % fetal calf serum, sodium pyruvate, nonessential amino acids, 2 mM glutamine, penicillin/streptomycin, and 5 μg/mL tetracycline to repress expression of the tet-regulated fusion proteins is used for maintenance of cells.
0.25 % trypsin.
Phosphate-buffered saline (PBS).
Hemacytometer.
2.2. siRNA Screening
DharmaFECT 2 transfection reagent (Dharmacon) (see Note 2).
JANUS Automated Workstation (PerkinElmer 8-tip MPD).
Multidrop Combi (Thermo Scientific).
MatriPlate 96-well glass bottom (Matrical Bioscience; cat. No. MGB096-1-1-LG-BC).
DMEM.
A siRNA library (Dharmacon siGENOME siRNA pools).
Control siRNAs including a siRNA targeting GAPDH (Dharmacon ON-TARGETplus pool; cat. No. D-001830-20-05) and a nontargeting siRNA control (Dharmacon ON-TARGETplus control nontargeting pool; cat. No. D-001810-10-05).
Inducible medium: DMEM growth medium supplemented with 10 % charcoal–dextran-treated serum, sodium pyruvate, nonessential amino acids, 2 mM glutamine, and penicillin/streptomycin (see Note 3).
2.3. Treatment of Cells with Dexamethasone/RU486 and Fixation
100 μM dexamethasone (DEX) in ethanol (store at −20 °C).
100 μM RU486 in ethanol (store at −20 °C).
Inducible medium.
PBS.
12 % paraformaldehyde (w/v) in PBS.
Bio Tek liquid handler (BioTek Instruments EL406).
2.4. Staining of the Nuclear Compartment
PBS.
DRAQ5 (BioStatus Limited; 5 mM; cat. No. DR50200) or DAPI diluted 1:5,000 in PBS.
2.5. Immunofluorescence Labeling of GAPDH
GAPDH antibody.
Texas Red dye conjugated AffiniPure donkey anti-mouse IgG (H + L) antibody.
PBS.
0.5 % Triton X-100 in PBS.
5 % bovine serum albumin (w/v) in PBS. Filter through a 0.45 μM filter.
0.5 % Tween 20 in PBS.
2.6. Imaging and Imaging Analysis
3. Methods
3.1. Cell Culture
For maintenance, grow 3,617 cells in DMEM supplemented with 10 % fetal calf serum, sodium pyruvate, nonessential amino acids, 2 mM glutamine, penicillin/streptomycin, and 5 μg/mL tetracycline at 37 °C and 5 % CO2.
Passage the cells using standard tissue culture practices. Cells are usually split 1:6 every 2 days.
3.2. siRNA Screening
Dissolve all the siRNAs to a final concentration of 500 nM with siRNA buffer provided by the manufacturer. Manufacturer provides siRNAs in a 96-well plate.
Aliquot 100 μL/well of each siRNA into individual 96-well plates (see Note 4). At least four wells should contain nontargeting siRNA and two wells should contain siRNA against GAPDH as controls (see Note 5). We use the JANUS Automated Workstation for all siRNA pipetting to avoid any errors. Each well contains a pool of four siRNAs per gene. Small library screens are done independently at least three times with two technical replicates each time.
Prepare a transfection mix: Dilute DharmaFECT 2 in DMEM to a concentration of 0.1 μL/10 μL (see Note 6). DMEM should be at room temperature.
Using the JANUS automated system, take 10 μL from the siRNA daughter plate and add to the 96-well glass bottom MatriPlate.
Then take 10 μL of the transfection mix and add to the siRNA on the MatriPlate 96-well glass bottom. Mixing of transfection mix and siRNA is done by pipetting up and down five times. Incubate at room temperature for at least 30 min to allow transfection complexes to form.
Meanwhile, wash flasks containing the cells three times with PBS. Add the appropriate volume of 0.25 % trypsin and incubate the cells at room temperature until they detach from the bottom of the flask (approximately 1–2 min). Suspend the cells in inducible medium. Determine the concentration by counting the cells using a hemacytometer. Dilute cells in the inducible medium to a concentration of 10,000 cells/80 μL.
After transfection complexes have formed, add 80 μL of cells per each well of the 96-well glass bottom MatriPlate using the Multidrop Combi.
Incubate the plates for 48 h at 37 °C and 5 % CO2 before treating the cells with DEX or RU486.
3.3. Treatment of Cells with DEX/RU486 and Fixation
Dilute 100 μM of DEX stock solution and 100 μM of RU486 stock solution in inducible medium so that the final concentration is 200 nM.
Using the Multidrop Combi, add 100 μL of the diluted RU486 (final concentration is 100 nM) to the control wells and 100 μL of the diluted DEX (final concentration is 100 nM) to every other well of the 96-well glass bottom MatriPlate. Incubate the cells at 37 °C for 30 min.
Using the BioTek liquid handler, add 100 μL of 12 % paraformaldehyde to each well (see Note 7). Incubate plate at room temperature for 15 min.
Wells are then washed six times with 200 μL of PBS using the BioTek liquid handler. Plates can be stored at this time at 4 °C in PBS.
3.4. Staining of the Nuclear Compartment
Using the BioTek liquid handler, the liquid is aspirated from the wells.
Then 100 μL of DRAQ5 (diluted 1:5,000 in PBS) is added to each well. Plates are incubated at room temperature for 15 min.
Wells are then washed six times with 200 μL of PBS using the BioTek liquid handler (plates can be stored at this time at 4 °C in PBS wrapped in aluminum foil).
3.5. Immunofluorescence Labeling of GAPDH
In order to confirm that the transfections worked, control cells transfected with siRNA against GAPDH should be included on every plate. Changes in the levels of GAPDH protein upon knock-down can be observed by conducting immunofluorescence using an antibody against GAPDH (Fig. 3b, c). The levels of GAPDH in cells transfected with nontargeting siRNA are then compared with wells transfected with the siRNA targeting GAPDH. Levels of cellular GAPDH can be quantified using the Acapella software. Under our conditions we observe a 50 % knockdown of GAPDH (see Note 8):
For control wells that contained siRNA to GAPDH or nontargeting siRNA, remove the PBS and add 100 μL of 0.5 % Triton X-100 to each well to permeabilize the cells. Incubate the plates on ice for 10 min.
Then wash the wells twice with 100 μL of 5 % BSA.
To block the cells, add 100 μL of 5 % BSA and incubate for 20 min at room temperature. Then remove the blocking solution.
Dilute the GAPDH antibody 1:1,000 in 5 % BSA. Then add 100 μL of antibody to each well and incubate for 1–2 h at room temperature.
Wash the wells three times with 0.5 % Tween 20.
Dilute the Texas Red donkey anti-mouse antibody 1:400 in 5 % BSA.
Remove the 0.5 % Tween 20 from the wells and add 100 μL of the diluted antibody.
Incubate at room temperature for 1 h.
Then wash the wells six times with 200 μL of PBS. Plates can be stored at 4 °C for about 1 week in PBS wrapped in aluminum foil.
3.6. Imaging and Imaging Analysis
The Opera High Content Screening System is used to image the plates. Twelve fields per well should be imaged with five z-stacks 1 μM apart.
Three exposures are imaged using the following wavelengths: 488 nM (images GFP–GR), 561 nM (images GAPDH immunofluorescence), and 640 nM (images DRAQ5).
Nuclear segmentation is then done using the Acapella imaging software as per manufacturer’s instructions. Levels of GAPDH in the cells can also be analyzed using Acapella.
The tandem array of genes with GFP–GR is detected using previously described algorithms [5, 6].
3.7. Secondary Screen
To confirm the results from the primary screen, a secondary screen is conducted using siRNA against the top 20 hits from the primary screen. This time ON-TARGETplus siRNA SMART pools (Dharmacon) is used. These are siRNA that have been chemically modified to reduce off-target effects. The screen is conducted exactly as described for the primary screen except that the secondary screen is performed independently at least four times with three technical replicates each time, in order to produce solid statistical data.
3.8. Deconvolution of siRNA
After completion of the secondary screen, the siRNA pools are separated into individual siRNAs and re-screened in order to further eliminate off-target effects. For targets that were confirmed by the secondary screen, the screen is repeated using each of the individual siRNAs from the SMART siRNA pools. If more than two siRNAs per target from the SMART pools give the same effect, then it is less likely that the effect is an off-target effect. In this screen the individual siRNAs are randomly placed on a 96-well plate (nine wells per siRNA) to account for variation due to well position. This screen is conducted independently for two times, with three technical replicates each time.
3.9. Other Potential Validation Experiments
Many other types of experiments can be conducted in order to validate the siRNA screen. Immunofluorescence can be performed using antibodies raised against the top hits to confirm that the target localizes to the array of response elements. Chromatin immunoprecipitation (ChIP) studies to examine GR binding can be done in cells in which a top hit has been knocked down, in order to confirm the loss of binding at the array using a different method. RNA can be isolated from cells transfected with siRNA from a top hit, and qPCR can be conducted on the cDNA to confirm knockdown of the siRNA target.
4. Notes
In order to conduct the screen using other receptors or transcription factors, new cell lines would have to be engineered to contain an array of response elements specific to the factor being analyzed. That factor would also have to be tagged with a fluorescent tag.
Transfection reagents should be stored at 4 °C and kept on ice while working with them in order to prevent a decrease in it efficiency.
Antibiotics can be used with 3,617 cells, as it does not affect the health of these cells during transfection. This keeps the cells from becoming contaminated during the transfection process. However, it should be noted that the health of other cell types may be affected by the presence of antibiotics in the medium during the transfection procedure.
Diluted siRNA stocks should be aliquoted into several individual plates and stored at −80 °C to avoid more than two freeze–thaw cycles. This will help maintain the integrity of the siRNA for an extended period of time.
Control siRNAs should be included on all plates. This includes nontargeting siRNA (for both DEX or RU486 treatments) and siRNA to GAPDH.
If transfections are done in other cell types, different transfection reagents should be tested and concentrations optimized.
Fresh paraformaldehyde is in PBS usually prepared before each experiment. However, we find that using 16 % paraformaldehyde (Electron Microscopy Sciences cat. No. 15710) diluted to 12 % on the day of the experiments works well.
It should be noted that optimization of the system should be conducted carefully. Optimal transfection conditions can cause a decrease in array formation. Therefore, a fine balance needs to be found between transfection conditions and cell health.
Acknowledgements
This research was supported, in part, by the Intramural Research Program of the National Institutes of Health, National Cancer Institute, Center for Cancer Research. T. B. M. was supported, in part, by a National Institute of General Medical Sciences Pharmacological Research and Training Fellowship.
References
- 1.Biddie SC, Hager GL (2009) Glucocorticoid receptor dynamics and gene regulation. Stress 12:193–205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wiench M, Miranda TB, Hager GL (2011) Control of nuclear receptor function by local chromatin structure. FEBS J 278: 2211–2230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kramer P, Fragoso G, Pennie WD et al. (1999) Transcriptional state of the mouse mammary tumor virus promoter can effect topological domain size in vivo. J Biol Chem 274:28590–28597 [DOI] [PubMed] [Google Scholar]
- 4.Walker D, Htun H, Hager GL (1999) Using inducible vectors to study intracellular trafficking of GFP-tagged steroid/nuclear receptors in living cells. Methods 19:386–393 [DOI] [PubMed] [Google Scholar]
- 5.Voss TC, Schiltz RL, Sung MH et al. (2011) Dynamic exchange at regulatory elements during chromatin remodeling underlies assisted loading mechanism. Cell 146:544–554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Voss TC, John S, Hager GL (2006) Single cell analysis of glucocorticoid receptor action reveals that stochastic post-chromatin association mechanisms regulate ligand-specific transcription. Mol Endocrinol 20:2641–2655 [DOI] [PubMed] [Google Scholar]
- 7.Schulz M, Eggert M, Baniahmad A et al. (2002) RU486-induced glucocorticoid receptor agonism is controlled by the receptor N terminus and by corepressor binding. J Biol Chem 277: 26238–26243 [DOI] [PubMed] [Google Scholar]
- 8.Szapary D, Huang Y, Simons SS Jr (1999) Opposing effects of corepressor and coactivators in determining the dose–response curve of agonists, and residual agonist activity of antagonists, for glucocorticoid receptor-regulated gene expression. Mol Endocrinol 13:2108–2121 [DOI] [PubMed] [Google Scholar]