

Abstract
The molecular chaperones are central mediators of protein homeostasis. In that role, they engage in widespread protein–protein interactions (PPIs) with each other and with their “client” proteins. Together, these PPIs form the backbone of a network that ensures proper vigilance over the processes of protein folding, trafficking, quality control, and degradation. The core chaperones, such as the heat shock proteins Hsp60, Hsp70, and Hsp90, are widely expressed in most tissues, yet there is growing evidence that the PPIs among them may be re-wired in disease conditions. This possibility suggests that these PPIs, and perhaps not the individual chaperones themselves, could be compelling drug targets. Indeed, recent efforts have yielded small molecules that inhibit (or promote) a subset of inter-chaperone PPIs. These chemical probes are being used to study chaperone networks in a range of models, and the successes with these approaches have inspired a community-wide objective to produce inhibitors for a broader set of targets. In this Review, we discuss progress toward that goal and point out some of the challenges ahead.
Keywords: protein–protein interaction, chemical biology, drug discovery, molecular chaperone, 70 kilodalton heat shock protein (Hsp70), heat shock protein 90 (Hsp90), protein folding, inhibitor, proteostasis, high-throughput screening, small molecule inhibitor
Introduction
Molecular chaperones help ensure protein homeostasis (i.e. proteostasis), playing essential roles in the folding, trafficking, sequestration, and turnover of proteins (1). There are ∼150 genes for molecular chaperones in the human genome, including the heat shock proteins Hsp110, Hsp90, Hsp70, Hsp60, Hsp27, etc. and the associated proteins such as co-chaperones, TCP-1 ring complex (TRiC),2 protein-disulfide isomerases (PDIs), peptidyl-prolyl cis-trans isomerases (PPIases), calnexin/calreticulin, and more (2, 3). Together, the coordinated activity of these factors serves to balance proteostasis and protect cells from protein misfolding and/or aggregation. Other articles in this Review Series cover the structure and function of the individual chaperone families in more detail. Here, we focus on the roles played by chemical probes in understanding their activity (4, 5). For example, our knowledge of Hsp90 biology has benefitted from the availability of chemical inhibitors, which can be applied to cells or organisms to ask how Hsp90 might be involved in a process. In this Review, we briefly introduce how chemical probes are developed and then outline how these ideas are being applied to chaperones.
What is a chemical probe?
One simple definition of a chemical probe is as follows: a small molecule that, at a given concentration, selectively inhibits the function of a biological target (6). It is essential that a chemical probe be selective for the intended target. Otherwise, it is difficult to ascribe its activity in cells or organisms to the function of the intended protein (7). Accordingly, the community of chemists and chemical biologists has developed an intuitive, experimental workflow that can be used to understand whether a molecule might be sufficiently selective to be considered a chemical probe. In 2010, Frye (6) published an influential commentary that coalesced many of these emerging ideas, and this concept has been expanded and extended by others (8, 9). From a pragmatic perspective, a good chemical probe is typically evaluated through a combination of chemical, biochemical, and genetic experiments (Table 1). Often, this process starts with discovery of an active molecule in a high-throughput screen. Then, a medicinal chemistry campaign is used to create analogs that reveal the relationship between the compound's chemical structure and its activity in vitro (e.g. binding and/or functional assays) and in cells (e.g. cell growth, gene expression, or another phenotype). This correlation is typically referred to as a structure–activity relationship (SAR). An important (and sometimes overlooked) product of an SAR campaign is the selection of a negative control molecule, which is structurally similar to the active molecules but does not bind to the target. Finally, this process is often coupled with determination of the solubility, metabolism, permeability, and lifetime of key analogs. Together, these studies provide a chemical and pharmacological basis for understanding how much active compound is present and whether it would be expected to bind to the intended target under those conditions.
Table 1.
Select criteria for consideration of a molecule as a high-quality chemical probe
This list is not inclusive but is intended to provide an overview of the experimental approaches.
| Chemical |
|
| Biochemical |
|
| Genetic |
|
From this starting point, the putative probe and its controls are then evaluated in a series of cell-based experiments that are intended to establish confidence that, at a given concentration, it will primarily bind to the intended target and not others. A classic method to assess selectivity is to immobilize the compound on a bead and determine whether it will preferentially “pull down” the intended target from cell lysates (see Table 1). Often, this experiment is supported by a combination of other assays, including cellular thermal shift assays (10), drug-resistance screens (11), and/or testing of the compound in cells in which the putative target has been knocked down, knocked out, or removed by CRISPRi (12). Together, the results of these experiments are used to assess whether a molecule is selective enough to be considered a chemical probe. Because so many different experiments are needed to understand selectivity, the evaluation process often takes years and involves multiple independent laboratories (typically in both academics and industry). Thus, many groups continue using a probe during its evaluation period, often confirming any results with independent methods. It is also important to note that the criteria needed to define a chemical probe are different from those needed to define a drug/therapeutic, i.e. selectivity is relatively more important for probes, whereas safety is of high value for drugs (13, 14).
Where does one learn whether an existing compound is considered a good chemical probe? On-line resources that collate this information, such as Chemical Probes (www.chemicalprobes.org)3 and Probe Miner (https://probeminer.icr.ac.uk/#/),3 are places to start. In addition, reference tools are available for rooting out the worst, most promiscuous molecules, such as pan-assay interference molecules (15) and protein aggregators (16). PubChem (https://pubchem.ncbi.hlm.nih.gov/) provides another guide, as it can be used to determine other assays in which a molecule has been found to be active (17). Finally, Open Science Probes (https://www.sgc.ffm.uni-frankfurt.de/)3 describes a collection of industry-derived tool molecules that have already undergone extensive validation (18). Together, these resources make it easier for the casual user to become quickly informed about a molecule's suitability for his/her experiment, including using it at the proper concentrations.
Categories of chemical probes
In the case of targets in the chaperone network, it is worth considering two major classes of chemical probes: (i) those that inhibit the enzyme activity of a chaperone, and (ii) those that inhibit protein–protein interactions (PPIs) at the connection between two chaperones. These designations are somewhat arbitrary, but the methods for finding and improving them can be quite different, so a brief review of their characteristics is warranted.
Inhibitors of enzyme activity
The simplest case of a chemical probe is a molecule that binds at an enzyme's active site. The structure of these inhibitors is often based on the enzyme's substrate or product; thus, in a cellular context, it must bind tight enough to compete for the natural ligand (e.g. ATP). In contrast, allosteric inhibitors bind to a distal pocket (i.e. away from the active site) and only indirectly disrupt enzyme function, so they might not be competitive binders. The major target enzymes in the chaperone network are the ATPases, including TriC, Hsp70, Hsp90, and Hsp60. These chaperones use ATP hydrolysis to power conformational motions that are coupled to their function (albeit not directly (19, 20)). Thus, compounds that inhibit nucleotide binding in these proteins would be expected to block chaperone activity.
Inhibitors of PPIs
Not all of the chaperones have enzymatic activity; for example, small heat shock proteins (sHSPs), Spy, trigger factor, clusterin, and prefoldin, are nonenzymatic chaperones that seem to specialize in limiting protein aggregation (21–25). Moreover, even the ATP-utilizing chaperones are assisted by non-enzymatic co-chaperones, which serve as critical adapters between different categories of chaperones (26, 27) and between chaperones and other proteostasis pathways. Thus, PPIs are another possible source of targets for chemical probes.
PPIs are potentially interesting targets because they are often less well conserved than active sites (28); thus, selectivity may be easier to achieve (29). PPIs also tend to be associated with “tuning” activity rather than switching it off, which could be useful when considering the housekeeping roles of some chaperones. Together, these features seem, on the surface, to generate significant opportunities for probe development (30, 31). However, targeting PPIs also comes with a number of important technical hurdles, namely these contacts tend to have a larger buried surface area (BSA) than enzyme-active sites, making it more difficult to identify small, drug-like molecules (less than 500 Da) that are able to block them. Indeed, recent retrospective analyses of ∼200 successful PPI inhibitors have shown that a majority of the most potent ones act on PPIs with relatively small BSA values (<2,000 to 4,000 Å2) (32–34). Moreover, the most “druggable” PPIs also tend to be those with tight affinity (Kd <500 nm), likely because those contacts involve a closely spaced combination of hydrophobic and polar residues that facilitates tight inhibitor binding. Thus, not all PPIs are considered equally “druggable.” If PPI targets are placed into four quadrants based on their BSA and affinity values, then those with weak affinity and large BSA values are usually the most difficult. Conversely, targets with small BSA values and/or tight affinity tend to be more tractable.
Targets in the chaperone network: nodes and edges
The chaperones and co-chaperones are physically linked to each other through a series of protein–protein interactions, existing as a PPI network (35, 36). In this parlance and borrowing from the systems biology lexicon, we term the major chaperones (i.e. Hsp70, Hsp90, Hsp60, and TRiC) as “nodes.” In turn, these nodes are connected by a series of “edges” that represent the PPIs. As will be detailed below, we find these designations useful when considering chemical probes of the chaperone network; specifically, enzyme inhibitors target the ATP-utilizing enzymes of the nodes, whereas PPI inhibitors target the edges.
However, one caution in this nomenclature is that the edges should not be considered equivalent. Indeed, the physical connections between chaperones come in a great variety of shapes and sizes. For example, a short peptide sequence from the C terminus of Hsp70, EEVD, binds to the tetratricopeptide repeat (TPR) domain that is present in a family of co-chaperones (Fig. 1), such as CHIP and HOP (37, 38). The EEVD–TPR interaction is of relatively tight affinity (Kd ∼ 0.5 μm), and it involves a small surface area (BSA ∼1,100 Å2) (Fig. 2) (39, 40). By comparison, the interaction between Hsp60 and Hsp10 is weaker (Kd ∼7 μm; Figs. 1 and 2A) and involves a 5-fold bigger contact surface area (BSA ∼5,500 Å2) (41). More globally, we have shown a subset of chaperone PPI structures in Fig. 1 and collated the BSA values from available PDB-deposited structures of chaperone complexes and matched these to measured Kd values in Fig. 2A. Together, this information, although certainly not inclusive, drives home the point that inter-chaperone contacts (“edges”) have quite distinct topologies. For example, the measured Kd values range nearly 6 orders-of-magnitude (from 0.04 to >100 μm), whereas the BSA values can be compact (∼700 Å2 for the Hsp27 system) or large (>20,000 Å2 for the Hsp90–Cdc37 contact).
Figure 1.
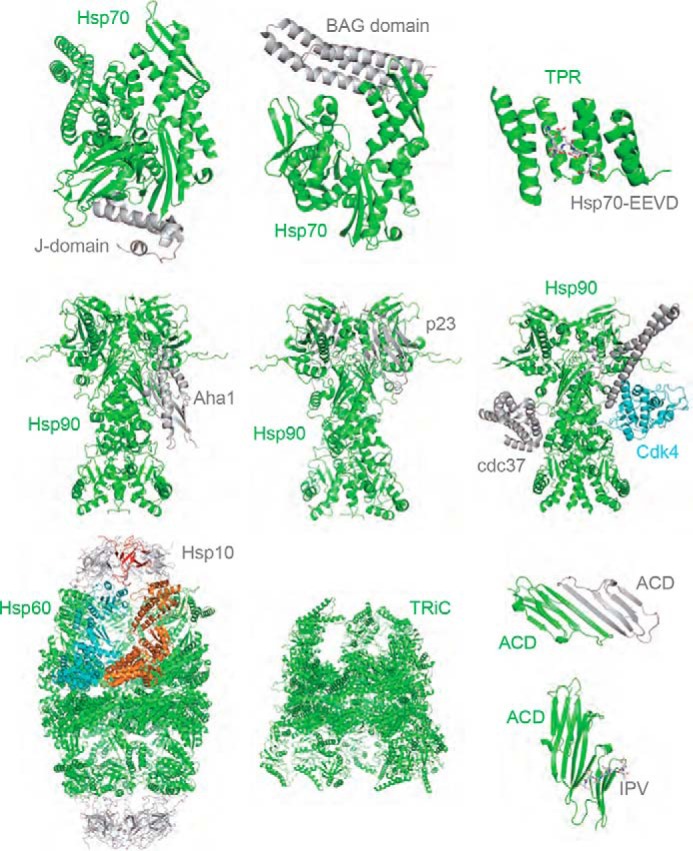
Diversity of PPIs between molecular chaperones. Representative structures of PPIs between chaperones are shown. Hsp70 refers to the nucleotide-binding domain of either the prokaryotic or eukaryotic protein, and ACD is the α-crystallin domain of a small heat shock protein. Monomers of Hsp60 are shown in blue and orange. Please see the citations and PDB codes for information on the exact constructs used: Hsp70-J domain (5NRO); Hsp70–BAG (1HX1); TPR–EEVD (4KBQ); Hsp90–Aha1 (1USU); Hsp90–p23 (2CG9); Hsp90–Cdc37–Cdk4 (5FWP); Hsp60–Hsp10 (4PJ1); TRiC (5GW4); α-crystallin ACD–ACD (2WJ7); and Hsp27 ACD–IPV (4MJH).
Figure 2.

PPIs between chaperones and their binding partners. A, table of BSA and affinity values for PPIs between chaperones. B, categorization of PPIs based on BSA and affinity values. Based on retrospective analyses of PPI inhibitors, certain quadrants are comparatively easier (green), challenging (gray), or difficult (red) to inhibit with drug-like small molecules.
Based on this analysis, some of the chaperone PPIs are expected to be relatively more difficult to inhibit. For example, PPIs with weak affinity (>500 nm) and large BSA values (>4,000 Å2), including Hsp90–p23, Hsp60–Hsp10, and Hsp90–Cdc37, are predicted to be particularly challenging (Fig. 2B). Other contacts, such as the ones between Hsp70–BAG1 and Hsp70-HOP, are predicted to be relatively tractable. Recent examinations of published PPI inhibitors have shown that small molecules (<500 Da) can often be used to inhibit a subset of PPIs, whereas the ones with larger BSA values typically need larger molecules, such as peptides or macrocycles (34). Therefore, it is reasonable to speculate that many different types of chemical scaffolds may be needed to inhibit the full suite of chaperone PPIs. In the following sections, we discuss a few examples of PPI systems that have been successfully targeted, with a focus on Hsp70, Hsp90, Hsp60, and sHSPs. In this discussion, we also comment on the current status of each molecule's ongoing evaluation as a chemical probe, according to the criteria in Table 1.
Inhibitors of the Hsp70 sub-network
Hsp70 is called the “triage” chaperone (42) because it plays keys roles in both protein folding and turnover (43, 44). Hsp70s are composed of a nucleotide-binding domain (NBD) and a substrate-binding domain (SBD) (45). ATP binds in the NBD, and the misfolded clients interact with the SBD. This chaperone is assisted by co-chaperones, including the J-domain containing proteins (JDPs) and nucleotide exchange factors (NEFs). Accordingly, there are at least two conceptual ways of targeting Hsp70: block its ATPase activity or change its PPIs with co-chaperones (46). Molecules targeting the ATP-binding cleft include VER-155008 and apoptozole (Fig. 3) (47, 48), which have been recently reviewed (49). VER-155008 competes with nucleotide for binding, as shown by crystallography (47), and this molecule has been shown to have the anti-proliferative activity expected of an Hsp70 inhibitor in HCT116 cells (50). Similarly, immobilized apoptozole will pull down Hsp70 from A549 (adenocarcinoma) cells (48), and the molecule induces apoptosis in that system. However, Hsp70s, as compared with kinases or Hsp90s, have an unusually tight affinity for ATP (Kd ∼100–500 nm), such that competition for cellular ATP (∼1–10 mm) creates a significant challenge. Covalent versions of VER-155008 have recently been developed (51), which might circumvent this challenge. The next step for these compounds is evaluation in a greater number of biological systems, using the chemical, genetic, and biochemical validation assays in Table 1.
Figure 3.

Selected chemical probes for molecular chaperones. See text for citations and details.
The other way to inhibit Hsp70 is by targeting its PPIs, and one of the first chemical series found to do this were the dihydropyrimidines (Fig. 3; Table 2) (52, 53). These molecules were inspired by the natural product spergualin, and they were found to bind at an interface between bacterial Hsp70 and the JDPs (54). Limited medicinal chemistry efforts (52, 55) showed that, depending on their individual chemical substitution patterns, the dihydropyrimidines either promote or inhibit this PPI (56). A recent crystal structure of Hsp70 bound to a J domain (57) (see Fig. 1) is expected to increase our understanding of how these compounds might work. Still, although the potency and pharmacokinetics of this chemical series remain un-optimized (i.e. EC50 ∼ micromolar), there is reason to be optimistic. For example, immobilized dihydropyrimidines pull down Hsp70 from cell lysates (56), and treatment with analogs, such as MAL3-101 and MAL1-27, has been shown to induce known Hsp70 biomarkers (58). Additional evidence for target engagement comes from studies in which yeast treated with an agonist, SW02, was partially protected from genetic deletion of a JDP (56). Moreover, treatment with MAL1-27 (also called 115-7c) protects against polyglutamine (polyQ) aggregation in multiple models, which mirrors what happens when Hsp70 is overexpressed (59, 60). Finally, acquired resistance to MAL3-101 in rhabdocarcinoma cells was mapped to an hsp70 gene (58). Together, these results provide support for selectivity in cells. The next steps for these molecules include expanded medicinal chemistry efforts to increase their potency and identification of additional negative controls. Given the importance of JDPs to Hsp70 biology (25, 54), this chemical series seems worth careful exploration.
Table 2.
Subset of reported chaperone inhibitors along with a summary of ongoing studies that have evaluated their suitability as chemical probes
The following values are shown: low micromolar IC/EC50, <1 μm; mid-micromolar IC/EC50, between 1 and 10 μm; and high micromolar IC/EC50, >10 μm. NA is not applicable.
| Target | Chemical series | KD | Muta-genesis | Biochemical assay, IC50 | Cellular assay, EC50 | Pulldown | Biomarker | In vivo efficacy | |
|---|---|---|---|---|---|---|---|---|---|
| Hsp70 | |||||||||
| Hsp70–JDPs | Dihydropyrimidines MAL3-101 (52, 55, 56, 58) | Allosteric | Mid to high μm | Yes | High μm | Mid to high μm | Yes | UPR, protein aggregation | Yes |
| Hsp70–nucleotide-exchange factors | Benzothiazole-rhodacyanines JG-231 (70, 71) | Allosteric | Low μm | Yes | Low μm | Mid nm to low μm | Yes | XIAP, Akt, IAP, CDK4, Raf-1 | Yes (66,74) |
| Hsp70 NBD | YK5 and its analogs (75, 76) | Allosteric | High μm | Yes | ∼7 μm | Low to mid μm | Yes | Her2, Raf-1, Akt | NA |
| Hsp70 NBD | HS-72 (77) | Allosteric | NA | NA | NA | Mid μm | Yes | Her2, Akt | Yes |
| Hsp90 | |||||||||
| C terminus | Novobiocin and its analogs (86, 87, 89–91, 94–96) | Allosteric | NA | Yes | Mid μm | Mid to high μm | Yes | ErbB2, mutant p53 (93), Her2, Akt, Raf-1 (87) | NA |
| Hsp90–cdc37 | Celastrol (100–102) | Allosteric | 1 μm | Yes | ∼10 μm | Mid μm | No | AR, FLT3, EGFR, BCR-ABL, Akt, CDK4 | Yes |
| Hsp60 | |||||||||
| Epolactaene (109) | NA | NA | Yes | >7 μm | Mid μm | Yes | NA | NA | |
| o-Carboranyl phenoxyacetanilide (107) | NA | NA | NA | Low μm | Mid μm | Yes | NA | NA | |
| Gold(III)–porphyrin complexes (105) | NA | ∼3.7 μm | NA | Low μm | Mid μm | Yes | NA | NA | |
| Myrtucommulone (110) | NA | NA | NA | ∼10 μm | Mid μm | Yes | NA | NA | |
| KHS-101 (104) | NA | NA | NA | 14 μm | Mid μm | Yes | Yes | Yes | |
| sHsp | |||||||||
| Hsp27 | Peptide aptamers (115) | NA | NA | Nonenzyme | NA | Yes | p21–Waf1 | Yes | |
| CryAB | Oxysterols ( (117) | Stabilizer | ∼10 μm | Yes | Nonenzyme | NA | No | Protein aggregation | Yes |
The other major category of Hsp70 PPIs is the one with the NEFs, including the BAG family of proteins that bind to the NBD through a conserved BAG domain (61). The NEFs are important mediators of Hsp70 function because they control the release of clients from the complex (62, 63). Thus, blocking NEF binding to Hsp70 would be expected to increase the dwell time of clients in the chaperone complex, favoring their degradation in some cases (63). A series of rhodacyanine-benzothiazoles (Fig. 3; Table 2) have been identified that inhibit this PPI. These molecules were first described by Wadhwa et al. (64) in phenotypic anticancer screens and only later were they found to bind to cytoplasmic and mitochondrial Hsp70 family members in pulldowns. NMR studies showed that the compounds of this series bind in a deep, allosteric pocket on Hsp70 (63, 65). Binding to this site favors the ADP-bound form of Hsp70 and disrupts binding to BAG proteins through a conformational change. As expected from the natural role of the NEFs, treatment of cells with these compounds induces degradation of particularly sensitive “client” proteins, such as FoxM1 (66), Akt (67), RIP1 (68), and inhibitor of apoptosis proteins (69). Medicinal chemistry campaigns (>400 analogs) produced more potent molecules (EC50 ∼30 nm) and inactive controls (JG-258) and allowed initial correlation between in vitro activity and cellular functions (70, 71). Target engagement in cells and animals has also been explored using genetic approaches; for example, overexpression of a point mutant of BAG3 (R480A) that cannot bind to Hsp70s gives a similar phenotype to compound treatment in breast cancer cells (66). In addition, whole-genome CRISPRi studies revealed that knockdown of Hsp70 family members gives rise to compound sensitivity (71). Most recently, JG-231 and other analogs have been characterized in vitro for liver microsome stability and in mice for maximal-tolerated dose and pharmacokinetics (71). This pharmacological information enables use of the compounds in some animal and tissue models. For example, they were used to identify a role for Hsp70–BAG in breast cancer initiation (66), tau homeostasis (72), Dengue viral replication (73), and castration-resistant prostate cancer (74). Together, this type of data, acquired in different laboratories and in different model systems, begins to build confidence in the suitability of the inhibitors as chemical probes. With that being said, the pharmacophore has chemical liabilities that limit its use, including its poor solubility and photosensitivity, so further optimization is needed.
Additional Hsp70 inhibitors are at a comparatively early stage in their evaluation as chemical probes (Fig. 3; Table 2). For example, the compound YK-5 and its analogs were designed to bind to a distinct, allosteric site in Hsp70, and this series has been explored in a series of medicinal chemistry campaigns (75, 76). These molecules have clear SAR; they pull down Hsp70 from lysates, and they have promising anti-proliferative activity in breast cancer models, providing a strong basis for further evaluation. In a quite different approach, the compound HS-72 was discovered in a screen for nucleotide-binding molecules (77). In follow-up studies, binding to Hsp70 was confirmed in vitro and by using pulldowns. Finally, phenotypic screens have identified PES (78) and novolactone (79) as inhibitors of Hsp70. Both of these compounds were found to bind at different allosteric sites in the SBD by structural approaches, and in both cases, the site was confirmed by mutagenesis of the interacting residues. Each of these chemical series (i.e. TK5, HS-72, PES, and novoloactone) is at a different stage of evaluation as a chemical probe, but each holds promise due to their different binding sites and mechanisms-of-action (MoAs) (80).
Inhibitors of the Hsp90 sub-network
Hsp90 is a dimeric chaperone composed of three domains: an N-terminal ATPase domain, a middle region, and a C-terminal dimerization motif. In addition, this protein binds to a number of co-chaperones, including Aha1, p23, and Cdc37 (see Fig. 1). The best-known Hsp90 inhibitors are enzyme inhibitors that bind in the N-terminal domain, such as geldanamycin and its analogs (e.g. 17-AAG) (81). Some of these molecules are clinical candidates (82, 83), and they have been extensively explored for selectivity, including screening against a panel of ATP-binding proteins (84), so they are generally considered to be good chemical probes (85). Indeed, these compounds have been crucial in expanding our knowledge of Hsp90 function, including being used to identify its clients. Molecules of this type have been extensively reviewed (81), so they will not be discussed further.
Alternative ways of inhibiting Hsp90 have also been explored. For example, the natural products novobiocin/coumermycin (86, 87) and sansalvamide A (88) served as inspiration for the development of inhibitors directed against the C-terminal domain (Fig. 3; Table 2). For example, Blagg and co-workers (89–91) and others (92, 93) have synthesized analogs of novobiocin, complete with negative controls (i.e. inactive molecules), and information about the binding site. Treatment with these compounds induces degradation of Hsp90 clients in multiple cancer cell types. In addition, they may disrupt Hsp90 dimerization (94) and co-chaperone interactions (95, 96), suggesting that they are bona fide Hsp90 PPI inhibitors. Similarly, McAlpine and co-workers (88) have produced biotinylated analogs of sansalvamide A and shown that they pull down Hsp90 from cells and disrupt binding to some client proteins. In this way, molecules from these two series are progressing toward becoming chemical probes. Interestingly, treatment with certain analogs of novobiocin and sansalvamide A does not induce a stress response in cells, which is considered a hallmark biomarker of the canonical, competitive Hsp90 inhibitors (85, 97). This finding highlights the importance of using multiple assays for assessing selectivity (see Table 1), as molecules with different MoAs might not always share the same biomarkers.
In addition to these chemical series, a number of other reports have introduced leads toward potential Hsp90 PPI inhibitors (98–102). Although these chemical series, such as celasterol, are relatively early in their analysis as chemical probes, further work may expand the suite of available chemical series for the Hsp90 sub-network.
Inhibitors of the Hsp60 sub-network
Hsp60–Hsp10 and its prokaryotic ortholog, GroEL–GroES, play important roles in protein folding (103). Hsp60 is thought to be located in the mitochondria of eukaryotes, where it helps stabilize client proteins. In addition to their ATPase activity, these systems involve multiple types of PPIs, including interactions between Hsp60 protomers and those between Hsp60 and the regulatory component (i.e. Hsp10; see Fig. 1). This system also likely interacts with the Hsp70 sub-network through direct PPIs. Most of the Hsp60 inhibitors that have been identified thus far originated in phenotypic screens, and only later did the pulldown studies suggest Hsp60 as a potential target. It is not yet clear whether these compounds are enzyme inhibitors (i.e. targeting “nodes”) or whether they disrupt PPIs (i.e. act on “edges”).
The chemical series described as Hsp60 inhibitors thus far are structurally diverse (Fig. 3; Table 2) and include 2-phenothiazole-pyrimidine-2,4-diamines, such as KHS101 (104), gold porphyrins (105), pyrazolo-pyridazines (106), phenoxyacetanilides (107), and the natural products suvanine (108), epolactaene (109), and myrtucommulone (110). Although more work remains to verify the selectivity of these molecules in cells, the striking lack of similarity in these chemical structures is suggestive of different binding sites or MoAs. However, none of these putative Hsp60 inhibitors has yet been subject to extensive medicinal chemistry or the full spectrum of analyses that are needed to give great confidence in their use as probes (see Table 1). Overall, given the central role of Hsp60–Hsp10 in mitochondrial protein quality control (111), it seems worth a greater investment in chemical probe discovery for this system. For example, KHS101 has been shown to disrupt energy metabolism in glioblastoma (104), suggesting a cancer-specific role for Hsp60 in mitochondrial function. These efforts might also benefit from screens focused on finding inhibitors of the folding activity of the prokaryotic GroEL–GroES system using in vitro assays (112).
Inhibitors of the sHSPs
The sHSPs are chaperones that lack enzymatic function; rather, they seem to operate by binding directly to each other and to their client proteins and co-chaperones, such as BAG3 (26). Thus, the only way to inhibit these systems is to target their PPIs. The sHSPs engage in a number of distinct interactions, such as the one between conserved α-crystallin domains (ACDs) that are known to stabilize sHsp dimers (see Fig. 1). Another, nonoverlapping interaction is the one between the ACD and the IXI motif that is found in the C terminus of some sHSPs (113). Finally, the N-terminal domain of some sHSPs also seems to make interactions within larger oligomers (114). Thus, sHSPs are a rich source of PPIs, which could become targets for chemical probes. However, the structural complexity of the system has hindered development of such molecules. Aptamers directed at Hsp27 (HSPB1) (115), diterpenes that seem to bind to Hsp27 (116), and oxysterols, such as compound 29, that bind to the ACD of α-crystallin (HSPB5) (117) have been identified (Fig. 3; Table 2), but their selectivity in cells has not been extensively explored.
Other inhibitors
We have focused this discussion on molecules that target a handful of heat shock proteins (i.e. Hsp70, Hsp90, Hsp60, and sHSPs). However, the proteostasis system includes other chaperones that are not classified as heat shock proteins but could be important targets. For example, Kelly and co-workers (118) have recently described inhibitors of PDIs, including information on target validation, MoA, and medicinal chemistry. These molecules activate the unfolded protein response, so they have promise in improving quality control in the endoplasmic reticulum. Recent efforts are also producing new inhibitors of the FK506-binding protein (FKBP) family of PPIases (119, 120), including the first selective inhibitors of FKBP51 (121). Such molecules might be especially good probes of steroid hormone receptor biology (120). The broader protein quality control field also benefits from the availability of chemical probes that inhibit proteins that are not widely considered to be chaperones, including VCP/p97 (122), the proteasome (123), the Sec61 channel (124), and the integrated stress response (125). Although we lack the space to adequately describe these molecules or evaluate their validation as chemical probes (see Table 1), they collectively serve to provide a wider chemical toolbox for studying proteostasis.
Outlook for the future
Chemical probes are powerful tools for studying and perturbing the chaperone network. Despite the production of probes for a handful of chaperone systems, such as Hsp70 and Hsp90, there is much more work to be done. For example, there are no validated probes for major nodes, such as TRiC. Likewise, hundreds of PPIs (“edges”) lack chemical tools. An optimistic vision for the future is one in which each chaperone node and edge has a well-validated inhibitor. Although this goal is certainly ambitious, there is legitimate reason for hope. New technologies, such as CRISPRi, high content screening, cryo-EM (126), isoelectric-focusing capillary electrophoresis (127), and others, are accelerating the rate of probe discovery and optimization. At the same time, increasingly sophisticated chemical libraries, such as macrocycles (128) and natural product–inspired libraries (31), which tend to be enriched in PPI inhibitors, are being built. It seems likely that these advances will combine to produce additional chemical probes for a wider range of chaperones and their PPIs.
Acknowledgments
We thank our many colleagues in the proteostasis and chemical biology fields, whose combined wisdom we tried to reflect here. We also apologize to those groups whose work we were not able to include, and we thank Dan Schwarz for helpful discussions.
This work was supported by National Institutes of Health Grants NS059690 and AG053619, the Department of Defense, and the Tau Consortium. This is the eighth article in the JBC Reviews series “Molecular chaperones and protein quality control.” The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Please note that the JBC is not responsible for the long-term archiving and maintenance of this site or any other third party hosted site.
- TRiC
- TCP-1 ring complex
- PPI
- protein–protein interaction
- BSA
- buried surface area
- ACD
- α-crystallin domain
- PDB
- Protein Data Bank
- NBD
- nucleotide-binding domain
- SBD
- substrate-binding domain
- JDP
- J-domain containing protein
- NEF
- nucleotide exchange factor
- PDI
- protein-disulfide isomerase
- PPIase
- peptidyl-prolyl cis-trans isomerase
- sHSP
- small heat-hock protein
- TPR
- tetratricopeptide repeat
- MoA
- mechanism-of-action
- SAR
- structure–activity relationship.
References
- 1. Hartl F. U., Bracher A., and Hayer-Hartl M. (2011) Molecular chaperones in protein folding and proteostasis. Nature 475, 324–332 10.1038/nature10317 [DOI] [PubMed] [Google Scholar]
- 2. Hageman J., and Kampinga H. H. (2009) Computational analysis of the human HSPH/HSPA/DNAJ family and cloning of a human HSPH/HSPA/DNAJ expression library. Cell Stress Chaperones 14, 1–21 10.1007/s12192-008-0060-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Kampinga H. H., Hageman J., Vos M. J., Kubota H., Tanguay R. M., Bruford E. A., Cheetham M. E., Chen B., and Hightower L. E. (2009) Guidelines for the nomenclature of the human heat shock proteins. Cell Stress Chaperones 14, 105–111 10.1007/s12192-008-0068-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Brandvold K. R., and Morimoto R. I. (2015) The chemical biology of molecular chaperones–implications for modulation of proteostasis. J. Mol. Biol. 427, 2931–2947 10.1016/j.jmb.2015.05.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Powers E. T., Morimoto R. I., Dillin A., Kelly J. W., and Balch W. E. (2009) Biological and chemical approaches to diseases of proteostasis deficiency. Annu. Rev. Biochem. 78, 959–991 10.1146/annurev.biochem.052308.114844 [DOI] [PubMed] [Google Scholar]
- 6. Frye S. V. (2010) The art of the chemical probe. Nat. Chem. Biol. 6, 159–161 10.1038/nchembio.296 [DOI] [PubMed] [Google Scholar]
- 7. Hu Y., Gupta-Ostermann D., and Bajorath J. (2014) Exploring compound promiscuity patterns and multi-target activity spaces. Comput. Struct. Biotechnol. J. 9, e201401003 10.5936/csbj.201401003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Blagg J., and Workman P. (2017) Choose and use your chemical probe wisely to explore cancer biology. Cancer Cell 32, 9–25 10.1016/j.ccell.2017.06.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Shortt J., Ott C. J., Johnstone R. W., and Bradner J. E. (2017) A chemical probe toolbox for dissecting the cancer epigenome. Nat. Rev. 17, 160–183 10.1038/nrc.2016.148 [DOI] [PubMed] [Google Scholar]
- 10. Martinez Molina D., and Nordlund P. (2016) The cellular thermal shift assay: a novel biophysical assay for in situ drug target engagement and mechanistic biomarker studies. Annu. Rev. Pharmacol. Toxicol. 56, 141–161 10.1146/annurev-pharmtox-010715-103715 [DOI] [PubMed] [Google Scholar]
- 11. Kapoor T. M., and Miller R. M. (2017) Leveraging chemotype-specific resistance for drug target identification and chemical biology. Trends Pharmacol. Sci. 38, 1100–1109 10.1016/j.tips.2017.09.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kampmann M. (2017) Elucidating drug targets and mechanisms of action by genetic screens in mammalian cells. Chem. Commun. 53, 7162–7167 10.1039/C7CC02349A [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Garbaccio R. M., and Parmee E. R. (2016) The impact of chemical probes in drug discovery: a pharmaceutical industry perspective. Cell Chem. Biol. 23, 10–17 10.1016/j.chembiol.2015.11.011 [DOI] [PubMed] [Google Scholar]
- 14. Jackson M. R. (2013) Chemical probe development versus drug development. Methods Mol. Biol. 1053, 1–6 10.1007/978-1-62703-562-0_1 [DOI] [PubMed] [Google Scholar]
- 15. Baell J. B., and Nissink J. W. (2018) Seven year itch: pan-assay interference compounds (PAINS) in 2017-utility and limitations. ACS Chem. Biol. 13, 36–44 10.1021/acschembio.7b00903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Irwin J. J., Duan D., Torosyan H., Doak A. K., Ziebart K. T., Sterling T., Tumanian G., and Shoichet B. K. (2015) An aggregation advisor for ligand discovery. J. Med. Chem. 58, 7076–7087 10.1021/acs.jmedchem.5b01105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Wang Y., Suzek T., Zhang J., Wang J., He S., Cheng T., Shoemaker B. A., Gindulyte A., and Bryant S. H. (2014) PubChem BioAssay: 2014 update. Nucleic Acids Res. 42, D1075–D1082 10.1093/nar/gkt978 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Müller S., Ackloo S., Arrowsmith C. H., Bauser M., Baryza J. L., Blagg J., Böttcher J., Bountra C., Brown P. J., Bunnage M. E., Carter A. J., Damerell D., Dötsch V., Drewry D. H., Edwards A. M., et al. (2018) Donated chemical probes for open science. eLife 7, e34311 10.7554/eLife.34311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Koldewey P., Horowitz S., and Bardwell J. C. (2017) Chaperone-client interactions: non-specificity engenders multifunctionality. J. Biol. Chem. 292, 12010–12017 10.1074/jbc.R117.796862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Chang L., Thompson A. D., Ung P., Carlson H. A., and Gestwicki J. E. (2010) Mutagenesis reveals the complex relationships between ATPase rate and the chaperone activities of Escherichia coli heat shock protein 70 (Hsp70/DnaK). J. Biol. Chem. 285, 21282–21291 10.1074/jbc.M110.124149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Haslbeck M., Franzmann T., Weinfurtner D., and Buchner J. (2005) Some like it hot: the structure and function of small heat shock proteins. Nat. Struct. Mol. Biol. 12, 842–846 10.1038/nsmb993 [DOI] [PubMed] [Google Scholar]
- 22. Horowitz S., Koldewey P., Stull F., and Bardwell J. C. (2018) Folding while bound to chaperones. Curr. Opin. Struct. Biol. 48, 1–5 10.1016/j.sbi.2017.06.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Narayan P., Orte A., Clarke R. W., Bolognesi B., Hook S., Ganzinger K. A., Meehan S., Wilson M. R., Dobson C. M., and Klenerman D. (2011) The extracellular chaperone clusterin sequesters oligomeric forms of the amyloid-β(1–40) peptide. Nat. Struct. Mol. Biol. 19, 79–83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Sörgjerd K. M., Zako T., Sakono M., Stirling P. C., Leroux M. R., Saito T., Nilsson P., Sekimoto M., Saido T. C., and Maeda M. (2013) Human prefoldin inhibits amyloid-β (Aβ) fibrillation and contributes to formation of nontoxic Aβ aggregates. Biochemistry 52, 3532–3542 10.1021/bi301705c [DOI] [PubMed] [Google Scholar]
- 25. Wruck F., Avellaneda M. J., Koers E. J., Minde D. P., Mayer M. P., Kramer G., Mashaghi A., and Tans S. J. (2018) Protein folding mediated by trigger factor and Hsp70: new insights from single-molecule approaches. J. Mol. Biol. 430, 438–449 10.1016/j.jmb.2017.09.004 [DOI] [PubMed] [Google Scholar]
- 26. Rauch J. N., Tse E., Freilich R., Mok S. A., Makley L. N., Southworth D. R., and Gestwicki J. E. (2017) BAG3 is a modular, scaffolding protein that physically links heat shock protein 70 (Hsp70) to the small heat shock proteins. J. Mol. Biol. 429, 128–141 10.1016/j.jmb.2016.11.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Hishiya A., Salman M. N., Carra S., Kampinga H. H., and Takayama S. (2011) BAG3 directly interacts with mutated αB-crystallin to suppress its aggregation and toxicity. PLoS ONE 6, e16828 10.1371/journal.pone.0016828 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Ma B., Elkayam T., Wolfson H., and Nussinov R. (2003) Protein–protein interactions: structurally conserved residues distinguish between binding sites and exposed protein surfaces. Proc. Natl. Acad. Sci. U.S.A. 100, 5772–5777 10.1073/pnas.1030237100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Arkin M. R., and Whitty A. (2009) The road less traveled: modulating signal transduction enzymes by inhibiting their protein–protein interactions. Curr. Opin. Chem. Biol. 13, 284–290 10.1016/j.cbpa.2009.05.125 [DOI] [PubMed] [Google Scholar]
- 30. Arkin M. R., and Wells J. A. (2004) Small-molecule inhibitors of protein–protein interactions: progressing towards the dream. Nat. Rev. Drug Discov. 3, 301–317 10.1038/nrd1343 [DOI] [PubMed] [Google Scholar]
- 31. Dougherty P. G., Qian Z., and Pei D. (2017) Macrocycles as protein–protein interaction inhibitors. Biochem. J. 474, 1109–1125 10.1042/BCJ20160619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Thompson A. D., Dugan A., Gestwicki J. E., and Mapp A. K. (2012) Fine-tuning multiprotein complexes using small molecules. ACS Chem. Biol. 7, 1311–1320 10.1021/cb300255p [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Cesa L. C., Mapp A. K., and Gestwicki J. E. (2015) Direct and propagated effects of small molecules on protein–protein interaction networks. Front. Bioeng. Biotechnol. 3, 119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Ran X., and Gestwicki J. E. (2018) Inhibitors of protein–protein interactions (PPIs): an analysis of scaffold choices and buried surface area. Curr. Opin. Chem. Biol. 44, 75–86 10.1016/j.cbpa.2018.06.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Freilich R., Arhar T., Abrams J. L., and Gestwicki J. E. (2018) Protein–protein interactions in the molecular chaperone network. Acc. Chem. Res. 51, 940–949 10.1021/acs.accounts.8b00036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Rizzolo K., Huen J., Kumar A., Phanse S., Vlasblom J., Kakihara Y., Zeineddine H. A., Minic Z., Snider J., Wang W., Pons C., Seraphim T. V., Boczek E. E., Alberti S., Costanzo M., et al. (2017) Features of the chaperone cellular network revealed through systematic interaction mapping. Cell Rep. 20, 2735–2748 10.1016/j.celrep.2017.08.074 [DOI] [PubMed] [Google Scholar]
- 37. Assimon V. A., Southworth D. R., and Gestwicki J. E. (2015) Specific binding of tetratricopeptide repeat proteins to heat shock protein 70 (Hsp70) and heat shock protein 90 (Hsp90) is regulated by affinity and phosphorylation. Biochemistry 54, 7120–7131 10.1021/acs.biochem.5b00801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. D'Andrea L. D., and Regan L. (2003) TPR proteins: the versatile helix. Trends Biochem. Sci. 28, 655–662 10.1016/j.tibs.2003.10.007 [DOI] [PubMed] [Google Scholar]
- 39. Zhang H., Amick J., Chakravarti R., Santarriaga S., Schlanger S., McGlone C., Dare M., Nix J. C., Scaglione K. M., Stuehr D. J., Misra S., and Page R. C. (2015) A bipartite interaction between Hsp70 and CHIP regulates ubiquitination of chaperoned client proteins. Structure 23, 472–482 10.1016/j.str.2015.01.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Xu Z., Devlin K. I., Ford M. G., Nix J. C., Qin J., and Misra S. (2006) Structure and interactions of the helical and U-box domains of CHIP, the C terminus of HSP70 interacting protein. Biochemistry 45, 4749–4759 10.1021/bi0601508 [DOI] [PubMed] [Google Scholar]
- 41. Parnas A., Nisemblat S., Weiss C., Levy-Rimler G., Pri-Or A., Zor T., Lund P. A., Bross P., and Azem A. (2012) Identification of elements that dictate the specificity of mitochondrial Hsp60 for its co-chaperonin. PLoS ONE 7, e50318 10.1371/journal.pone.0050318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Wickner S., Maurizi M. R., and Gottesman S. (1999) Posttranslational quality control: folding, refolding, and degrading proteins. Science 286, 1888–1893 10.1126/science.286.5446.1888 [DOI] [PubMed] [Google Scholar]
- 43. Mayer M. P. (2013) Hsp70 chaperone dynamics and molecular mechanism. Trends Biochem. Sci. 38, 507–514 10.1016/j.tibs.2013.08.001 [DOI] [PubMed] [Google Scholar]
- 44. Zuiderweg E. R., Hightower L. E., and Gestwicki J. E. (2017) The remarkable multivalency of the Hsp70 chaperones. Cell Stress Chaperones 22, 173–189 10.1007/s12192-017-0776-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Mayer M. P., and Bukau B. (2005) Hsp70 chaperones: cellular functions and molecular mechanism. Cell. Mol. Life Sci. 62, 670–684 10.1007/s00018-004-4464-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Assimon V. A., Gillies A. T., Rauch J. N., and Gestwicki J. E. (2013) Hsp70 protein complexes as drug targets. Curr. Pharm. Des. 19, 404–417 10.2174/138161213804143699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Macias A. T., Williamson D. S., Allen N., Borgognoni J., Clay A., Daniels Z., Dokurno P., Drysdale M. J., Francis G. L., Graham C. J., Howes R., Matassova N., Murray J. B., Parsons R., Shaw T., et al. (2011) Adenosine-derived inhibitors of 78 kDa glucose regulated protein (Grp78) ATPase: insights into isoform selectivity. J. Med. Chem. 54, 4034–4041 10.1021/jm101625x [DOI] [PubMed] [Google Scholar]
- 48. Ko S. K., Kim J., Na D. C., Park S., Park S. H., Hyun J. Y., Baek K. H., Kim N. D., Kim N. K., Park Y. N., Song K., and Shin I. (2015) A small molecule inhibitor of ATPase activity of HSP70 induces apoptosis and has antitumor activities. Chem. Biol. 22, 391–403 10.1016/j.chembiol.2015.02.004 [DOI] [PubMed] [Google Scholar]
- 49. Evans C. G., Chang L., and Gestwicki J. E. (2010) Heat shock protein 70 (hsp70) as an emerging drug target. J. Med. Chem. 53, 4585–4602 10.1021/jm100054f [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Williamson D. S., Borgognoni J., Clay A., Daniels Z., Dokurno P., Drysdale M. J., Foloppe N., Francis G. L., Graham C. J., Howes R., Macias A. T., Murray J. B., Parsons R., Shaw T., Surgenor A. E., et al. (2009) Novel adenosine-derived inhibitors of 70 kDa heat shock protein, discovered through structure-based design. J. Med. Chem. 52, 1510–1513 10.1021/jm801627a [DOI] [PubMed] [Google Scholar]
- 51. Pettinger J., Le Bihan Y. V., Widya M., van Montfort R. L., Jones K., and Cheeseman M. D. (2017) An irreversible inhibitor of HSP72 that unexpectedly targets lysine-56. Angew. Chem. Int. Ed. Engl. 56, 3536–3540 10.1002/anie.201611907 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Fewell S. W., Smith C. M., Lyon M. A., Dumitrescu T. P., Wipf P., Day B. W., and Brodsky J. L. (2004) Small molecule modulators of endogenous and co-chaperone-stimulated Hsp70 ATPase activity. J. Biol. Chem. 279, 51131–51140 10.1074/jbc.M404857200 [DOI] [PubMed] [Google Scholar]
- 53. Huryn D. M., Brodsky J. L., Brummond K. M., Chambers P. G., Eyer B., Ireland A. W., Kawasumi M., Laporte M. G., Lloyd K., Manteau B., Nghiem P., Quade B., Seguin S. P., and Wipf P. (2011) Chemical methodology as a source of small-molecule checkpoint inhibitors and heat shock protein 70 (Hsp70) modulators. Proc. Natl. Acad. Sci. U.S.A. 108, 6757–6762 10.1073/pnas.1015251108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Kampinga H. H., and Craig E. A. (2010) The HSP70 chaperone machinery: J proteins as drivers of functional specificity. Nat. Rev. Mol. Cell Biol. 11, 579–592 10.1038/nrm2941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Wisén S., Androsavich J., Evans C. G., Chang L., and Gestwicki J. E. (2008) Chemical modulators of heat shock protein 70 (Hsp70) by sequential, microwave-accelerated reactions on solid phase. Bioorg. Med. Chem. Lett. 18, 60–65 10.1016/j.bmcl.2007.11.027 [DOI] [PubMed] [Google Scholar]
- 56. Wisén S., Bertelsen E. B., Thompson A. D., Patury S., Ung P., Chang L., Evans C. G., Walter G. M., Wipf P., Carlson H. A., Brodsky J. L., Zuiderweg E. R., and Gestwicki J. E. (2010) Binding of a small molecule at a protein–protein interface regulates the chaperone activity of hsp70–hsp40. ACS Chem. Biol. 5, 611–622 10.1021/cb1000422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Kityk R., Kopp J., and Mayer M. P. (2018) Molecular mechanism of J-domain-triggered ATP hydrolysis by Hsp70 chaperones. Mol. Cell 69, 227–237.e4 10.1016/j.molcel.2017.12.003 [DOI] [PubMed] [Google Scholar]
- 58. Sabnis A. J., Guerriero C. J., Olivas V., Sayana A., Shue J., Flanagan J., Asthana S., Paton A. W., Paton J. C., Gestwicki J. E., Walter P., Weissman J. S., Wipf P., Brodsky J. L., and Bivona T. G. (2016) Combined chemical-genetic approach identifies cytosolic HSP70 dependence in rhabdomyosarcoma. Proc. Natl. Acad. Sci. U.S.A. 113, 9015–9020 10.1073/pnas.1603883113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Walter G. M., Smith M. C., Wisén S., Basrur V., Elenitoba-Johnson K. S., Duennwald M. L., Kumar A., and Gestwicki J. E. (2011) Ordered assembly of heat shock proteins, Hsp26, Hsp70, Hsp90, and Hsp104, on expanded polyglutamine fragments revealed by chemical probes. J. Biol. Chem. 286, 40486–40493 10.1074/jbc.M111.284448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Chafekar S. M., Wisén S., Thompson A. D., Echeverria A., Walter G. M., Evans C. G., Makley L. N., Gestwicki J. E., and Duennwald M. L. (2012) Pharmacological tuning of heat shock protein 70 modulates polyglutamine toxicity and aggregation. ACS Chem. Biol. 7, 1556–1564 10.1021/cb300166p [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Bracher A., and Verghese J. (2015) The nucleotide exchange factors of Hsp70 molecular chaperones. Front. Mol. Biosci. 2, 10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Gowda N. K. C., Kaimal J. M., Kityk R., Daniel C., Liebau J., Öhman M., Mayer M. P., and Andréasson C. (2018) Nucleotide exchange factors Fes1 and HspBP1 mimic substrate to release misfolded proteins from Hsp70. Nat. Struct. Mol. Biol. 25, 83–89 10.1038/s41594-017-0008-2 [DOI] [PubMed] [Google Scholar]
- 63. Young Z. T., Rauch J. N., Assimon V. A., Jinwal U. K., Ahn M., Li X., Dunyak B. M., Ahmad A., Carlson G. A., Srinivasan S. R., Zuiderweg E. R., Dickey C. A., and Gestwicki J. E. (2016) Stabilizing the Hsp70–Tau complex promotes turnover in models of tauopathy. Cell Chem. Biol. 23, 992–1001 10.1016/j.chembiol.2016.04.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Wadhwa R., Sugihara T., Yoshida A., Nomura H., Reddel R. R., Simpson R., Maruta H., and Kaul S. C. (2000) Selective toxicity of MKT-077 to cancer cells is mediated by its binding to the hsp70 family protein mot-2 and reactivation of p53 function. Cancer Res. 60, 6818–6821 [PubMed] [Google Scholar]
- 65. Rousaki A., Miyata Y., Jinwal U. K., Dickey C. A., Gestwicki J. E., and Zuiderweg E. R. (2011) Allosteric drugs: the interaction of antitumor compound MKT-077 with human Hsp70 chaperones. J. Mol. Biol. 411, 614–632 10.1016/j.jmb.2011.06.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Colvin T. A., Gabai V. L., Gong J., Calderwood S. K., Li H., Gummuluru S., Matchuk O. N., Smirnova S. G., Orlova N. V., Zamulaeva I. A., Garcia-Marcos M., Li X., Young Z. T., Rauch J. N., Gestwicki J. E., et al. (2014) Hsp70–Bag3 interactions regulate cancer-related signaling networks. Cancer Res. 74, 4731–4740 10.1158/0008-5472.CAN-14-0747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Koren J. 3rd., Jinwal U. K., Jin Y., O'Leary J., Jones J. R., Johnson A. G., Blair L. J., Abisambra J. F., Chang L., Miyata Y., Cheng A. M., Guo J., Cheng J. Q., Gestwicki J. E., and Dickey C. A. (2010) Facilitating Akt clearance via manipulation of Hsp70 activity and levels. J. Biol. Chem. 285, 2498–2505 10.1074/jbc.M109.057208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Srinivasan S. R., Cesa L. C., Li X., Julien O., Zhuang M., Shao H., Chung J., Maillard I., Wells J. A., Duckett C. S., and Gestwicki J. E. (2018) Heat shock protein 70 (Hsp70) suppresses RIP1-dependent apoptotic and necroptotic cascades. Mol. Cancer Res. 16, 58–68 10.1158/1541-7786.MCR-17-0408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Cesa L. C., Shao H., Srinivasan S. R., Tse E., Jain C., Zuiderweg E. R. P., Southworth D. R., Mapp A. K., and Gestwicki J. E. (2018) X-linked inhibitor of apoptosis protein (XIAP) is a client of heat shock protein 70 (Hsp70) and a biomarker of its inhibition. J. Biol. Chem. 293, 2370–2380 10.1074/jbc.RA117.000634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Li X., Srinivasan S. R., Connarn J., Ahmad A., Young Z. T., Kabza A. M., Zuiderweg E. R., Sun D., and Gestwicki J. E. (2013) Analogs of the allosteric heat shock protein 70 (Hsp70) inhibitor, MKT-077, as anti-cancer agents. ACS Med. Chem. Lett. 4, 1042–1047 10.1021/ml400204n [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Shao H., Li X., Moses M. A., Gilbert L. A., Kalyanaraman C., Young Z. T., Chernova M., Journey S. N., Weissman J. S., Hann B., Jacobson M. P., Neckers L., and Gestwicki J. E. (2018) Exploration of benzothiazole-rhodacyanines as allosteric inhibitors of protein–protein interactions with heat shock protein 70 (Hsp70). J. Med. Chem. 61, 6163–6177 10.1021/acs.jmedchem.8b00583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Abisambra J., Jinwal U. K., Miyata Y., Rogers J., Blair L., Li X., Seguin S. P., Wang L., Jin Y., Bacon J., Brady S., Cockman M., Guidi C., Zhang J., Koren J., et al. (2013) Allosteric heat shock protein 70 inhibitors rapidly rescue synaptic plasticity deficits by reducing aberrant τ. Biol. Psychiatry 74, 367–374 10.1016/j.biopsych.2013.02.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Taguwa S., Maringer K., Li X., Bernal-Rubio D., Rauch J. N., Gestwicki J. E., Andino R., Fernandez-Sesma A., and Frydman J. (2015) Defining Hsp70 subnetworks in dengue virus replication reveals key vulnerability in flavivirus infection. Cell 163, 1108–1123 10.1016/j.cell.2015.10.046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Moses M. A., Kim Y. S., Rivera-Marquez G. M., Oshima N., Watson M. J., Beebe K. E., Wells C., Lee S., Zuehlke A. D., Shao H., Bingman W. E. 3rd., Kumar V., Malhotra S. V., Weigel N. L., Gestwicki J. E., et al. (2018) Targeting the Hsp40/Hsp70 chaperone axis as a novel strategy to treat castration-resistant prostate cancer. Cancer Res. 78, 4022–4035 10.1158/0008-5472.CAN-17-3728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Taldone T., Kang Y., Patel H. J., Patel M. R., Patel P. D., Rodina A., Patel Y., Gozman A., Maharaj R., Clement C. C., Lu A., Young J. C., and Chiosis G. (2014) Heat shock protein 70 inhibitors. 2. 2,5′-Thiodipyrimidines, 5-(phenylthio)pyrimidines, 2-(pyridin-3-ylthio)pyrimidines, and 3-(phenylthio)pyridines as reversible binders to an allosteric site on heat shock protein 70. J. Med. Chem. 57, 1208–1224 10.1021/jm401552y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Kang Y., Taldone T., Patel H. J., Patel P. D., Rodina A., Gozman A., Maharaj R., Clement C. C., Patel M. R., Brodsky J. L., Young J. C., and Chiosis G. (2014) Heat shock protein 70 inhibitors. 1. 2,5′-Thiodipyrimidine and 5-(phenylthio)pyrimidine acrylamides as irreversible binders to an allosteric site on heat shock protein 70. J. Med. Chem. 57, 1188–1207 10.1021/jm401551n [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Howe M. K., Bodoor K., Carlson D. A., Hughes P. F., Alwarawrah Y., Loiselle D. R., Jaeger A. M., Darr D. B., Jordan J. L., Hunter L. M., Molzberger E. T., Gobillot T. A., Thiele D. J., Brodsky J. L., Spector N. L., and Haystead T. A. (2014) Identification of an allosteric small-molecule inhibitor selective for the inducible form of heat shock protein 70. Chem. Biol. 21, 1648–1659 10.1016/j.chembiol.2014.10.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Leu J. I., Pimkina J., Frank A., Murphy M. E., and George D. L. (2009) A small molecule inhibitor of inducible heat shock protein 70. Mol. Cell 36, 15–27 10.1016/j.molcel.2009.09.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Hassan A. Q., Kirby C. A., Zhou W., Schuhmann T., Kityk R., Kipp D. R., Baird J., Chen J., Chen Y., Chung F., Hoepfner D., Movva N. R., Pagliarini R., Petersen F., Quinn C., et al. (2015) The novolactone natural product disrupts the allosteric regulation of hsp70. Chem. Biol. 22, 87–97 10.1016/j.chembiol.2014.11.007 [DOI] [PubMed] [Google Scholar]
- 80. Li X., Shao H., Taylor I. R., and Gestwicki J. E. (2016) Targeting allosteric control mechanisms in heat shock protein 70 (Hsp70). Curr. Top. Med. Chem. 16, 2729–2740 10.2174/1568026616666160413140911 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Shrestha L., Patel H. J., and Chiosis G. (2016) Chemical tools to investigate mechanisms associated with HSP90 and HSP70 in disease. Cell Chem. Biol. 23, 158–172 10.1016/j.chembiol.2015.12.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Pacey S., Wilson R. H., Walton M., Eatock M. M., Hardcastle A., Zetterlund A., Arkenau H. T., Moreno-Farre J., Banerji U., Roels B., Peachey H., Aherne W., de Bono J. S., Raynaud F., Workman P., and Judson I. (2011) A phase I study of the heat shock protein 90 inhibitor alvespimycin (17-DMAG) given intravenously to patients with advanced solid tumors. Clin. Cancer Res. 17, 1561–1570 10.1158/1078-0432.CCR-10-1927 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Powers M. V., and Workman P. (2007) Inhibitors of the heat shock response: biology and pharmacology. FEBS Lett. 581, 3758–3769 10.1016/j.febslet.2007.05.040 [DOI] [PubMed] [Google Scholar]
- 84. Nordin B. E., Liu Y., Aban A., Brown H. E., Wu J., Hainley A. K., Rosenblum J. S., Nomanbhoy T. K., and Kozarich J. W. (2015) ATP acyl phosphate reactivity reveals native conformations of Hsp90 paralogs and inhibitor target engagement. Biochemistry 54, 3024–3036 10.1021/acs.biochem.5b00148 [DOI] [PubMed] [Google Scholar]
- 85. Neckers L., Blagg B., Haystead T., Trepel J. B., Whitesell L., and Picard D. (2018) Methods to validate Hsp90 inhibitor specificity, to identify off-target effects, and to rethink approaches for further clinical development. Cell Stress Chaperones 23, 467–482 10.1007/s12192-018-0877-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Anyika M., McMullen M., Forsberg L. K., Dobrowsky R. T., and Blagg B. S. (2016) Development of noviomimetics as C-terminal Hsp90 inhibitors. ACS Med. Chem. Lett. 7, 67–71 10.1021/acsmedchemlett.5b00331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Zhao H., Donnelly A. C., Kusuma B. R., Brandt G. E., Brown D., Rajewski R. A., Vielhauer G., Holzbeierlein J., Cohen M. S., and Blagg B. S. (2011) Engineering an antibiotic to fight cancer: optimization of the novobiocin scaffold to produce anti-proliferative agents. J. Med. Chem. 54, 3839–3853 10.1021/jm200148p [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Vasko R. C., Rodriguez R. A., Cunningham C. N., Ardi V. C., Agard D. A., and McAlpine S. R. (2010) Mechanistic studies of Sansalvamide A-amide: an allosteric modulator of Hsp90. ACS Med. Chem. Lett. 1, 4–8 10.1021/ml900003t [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Ghosh S., Liu Y., Garg G., Anyika M., McPherson N. T., Ma J., Dobrowsky R. T., and Blagg B. S. (2016) Diverging novobiocin anti-cancer activity from neuroprotective activity through modification of the amide tail. ACS Med. Chem. Lett. 7, 813–818 10.1021/acsmedchemlett.6b00224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Burlison J. A., Neckers L., Smith A. B., Maxwell A., and Blagg B. S. (2006) Novobiocin: redesigning a DNA gyrase inhibitor for selective inhibition of hsp90. J. Am. Chem. Soc. 128, 15529–15536 10.1021/ja065793p [DOI] [PubMed] [Google Scholar]
- 91. Yu X. M., Shen G., Neckers L., Blake H., Holzbeierlein J., Cronk B., and Blagg B. S. (2005) Hsp90 inhibitors identified from a library of novobiocin analogues. J. Am. Chem. Soc. 127, 12778–12779 10.1021/ja0535864 [DOI] [PubMed] [Google Scholar]
- 92. Matts R. L., Dixit A., Peterson L. B., Sun L., Voruganti S., Kalyanaraman P., Hartson S. D., Verkhivker G. M., and Blagg B. S. (2011) Elucidation of the Hsp90 C-terminal inhibitor binding site. ACS Chem. Biol. 6, 800–807 10.1021/cb200052x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Marcu M. G., Chadli A., Bouhouche I., Catelli M., and Neckers L. M. (2000) The heat shock protein 90 antagonist novobiocin interacts with a previously unrecognized ATP-binding domain in the carboxyl terminus of the chaperone. J. Biol. Chem. 275, 37181–37186 10.1074/jbc.M003701200 [DOI] [PubMed] [Google Scholar]
- 94. Allan R. K., Mok D., Ward B. K., and Ratajczak T. (2006) Modulation of chaperone function and cochaperone interaction by novobiocin in the C-terminal domain of Hsp90: evidence that coumarin antibiotics disrupt Hsp90 dimerization. J. Biol. Chem. 281, 7161–7171 10.1074/jbc.M512406200 [DOI] [PubMed] [Google Scholar]
- 95. Yun B. G., Huang W., Leach N., Hartson S. D., and Matts R. L. (2004) Novobiocin induces a distinct conformation of Hsp90 and alters Hsp90–cochaperone–client interactions. Biochemistry 43, 8217–8229 10.1021/bi0497998 [DOI] [PubMed] [Google Scholar]
- 96. Ghosh S., Shinogle H. E., Garg G., Vielhauer G. A., Holzbeierlein J. M., Dobrowsky R. T., and Blagg B. S. (2015) Hsp90 C-terminal inhibitors exhibit antimigratory activity by disrupting the Hsp90α/Aha1 complex in PC3-MM2 cells. ACS Chem. Biol. 10, 577–590 10.1021/cb5008713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Wang Y., and McAlpine S. R. (2015) N-terminal and C-terminal modulation of Hsp90 produce dissimilar phenotypes. Chem. Commun. 51, 1410–1413 10.1039/C4CC07284G [DOI] [PubMed] [Google Scholar]
- 98. Zierer B. K., Weiwad M., Rübbelke M., Freiburger L., Fischer G., Lorenz O. R., Sattler M., Richter K., and Buchner J. (2014) Artificial accelerators of the molecular chaperone Hsp90 facilitate rate-limiting conformational transitions. Angew. Chem. Int. Ed. Engl. 53, 12257–12262 10.1002/anie.201406578 [DOI] [PubMed] [Google Scholar]
- 99. Sreeramulu S., Gande S. L., Göbel M., and Schwalbe H. (2009) Molecular mechanism of inhibition of the human protein complex Hsp90–Cdc37, a kinome chaperone-cochaperone, by triterpene celastrol. Angew. Chem. Int. Ed. Engl. 48, 5853–5855 10.1002/anie.200900929 [DOI] [PubMed] [Google Scholar]
- 100. Hieronymus H., Lamb J., Ross K. N., Peng X. P., Clement C., Rodina A., Nieto M., Du J., Stegmaier K., Raj S. M., Maloney K. N., Clardy J., Hahn W. C., Chiosis G., and Golub T. R. (2006) Gene expression signature-based chemical genomic prediction identifies a novel class of HSP90 pathway modulators. Cancer Cell 10, 321–330 10.1016/j.ccr.2006.09.005 [DOI] [PubMed] [Google Scholar]
- 101. Zhang T., Li Y., Yu Y., Zou P., Jiang Y., and Sun D. (2009) Characterization of celastrol to inhibit hsp90 and cdc37 interaction. J. Biol. Chem. 284, 35381–35389 10.1074/jbc.M109.051532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Zhang T., Hamza A., Cao X., Wang B., Yu S., Zhan C. G., and Sun D. (2008) A novel Hsp90 inhibitor to disrupt Hsp90/Cdc37 complex against pancreatic cancer cells. Mol. Cancer Ther. 7, 162–170 10.1158/1535-7163.MCT-07-0484 [DOI] [PubMed] [Google Scholar]
- 103. Bukau B., Weissman J., and Horwich A. (2006) Molecular chaperones and protein quality control. Cell 125, 443–451 10.1016/j.cell.2006.04.014 [DOI] [PubMed] [Google Scholar]
- 104. Polson E. S., Kuchler V. B., Abbosh C., Ross E. M., Mathew R. K., Beard H. A., da Silva B., Holding A. N., Ballereau S., Chuntharpursat-Bon E., Williams J., Griffiths H. B. S., Shao H., Patel A., Davies A. J., et al. (2018) KHS101 disrupts energy metabolism in human glioblastoma cells and reduces tumor growth in mice. Sci. Transl. Med. 10, eaar2718 10.1126/scitranslmed.aar2718 [DOI] [PubMed] [Google Scholar]
- 105. Hu D., Liu Y., Lai Y. T., Tong K. C., Fung Y. M., Lok C. N., and Che C. M. (2016) Anticancer gold(III) porphyrins target mitochondrial chaperone Hsp60. Angew. Chem. Int. Ed. Engl. 55, 1387–1391 10.1002/anie.201509612 [DOI] [PubMed] [Google Scholar]
- 106. Alagramam K. N., Gopal S. R., Geng R., Chen D. H., Nemet I., Lee R., Tian G., Miyagi M., Malagu K. F., Lock C. J., Esmieu W. R., Owens A. P., Lindsay N. A., Ouwehand K., Albertus F., et al. (2016) A small molecule mitigates hearing loss in a mouse model of Usher syndrome III. Nat. Chem. Biol. 12, 444–451 10.1038/nchembio.2069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Ban H. S., Shimizu K., Minegishi H., and Nakamura H. (2010) Identification of HSP60 as a primary target of o-carboranylphenoxyacetanilide, an HIF-1α inhibitor. J. Am. Chem. Soc. 132, 11870–11871 10.1021/ja104739t [DOI] [PubMed] [Google Scholar]
- 108. Cassiano C., Monti M. C., Festa C., Zampella A., Riccio R., and Casapullo A. (2012) Chemical proteomics reveals heat shock protein 60 to be the main cellular target of the marine bioactive sesterterpene suvanine. Chembiochem 13, 1953–1958 10.1002/cbic.201200291 [DOI] [PubMed] [Google Scholar]
- 109. Nagumo Y., Kakeya H., Shoji M., Hayashi Y., Dohmae N., and Osada H. (2005) Epolactaene binds human Hsp60 Cys442 resulting in the inhibition of chaperone activity. Biochem. J. 387, 835–840 10.1042/BJ20041355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Wiechmann K., Müller H., König S., Wielsch N., Svatoš A., Jauch J., and Werz O. (2017) Mitochondrial chaperonin HSP60 is the apoptosis-related target for myrtucommulone. Cell Chem. Biol. 24, 614–623.e6 10.1016/j.chembiol.2017.04.008 [DOI] [PubMed] [Google Scholar]
- 111. Cappello F., Conway de Macario E., Marasà L., Zummo G., and Macario A. J. (2008) Hsp60 expression, new locations, functions and perspectives for cancer diagnosis and therapy. Cancer Biol. Ther. 7, 801–809 10.4161/cbt.7.6.6281 [DOI] [PubMed] [Google Scholar]
- 112. Chapman E., Farr G. W., Furtak K., and Horwich A. L. (2009) A small molecule inhibitor selective for a variant ATP-binding site of the chaperonin GroEL. Bioorg. Med. Chem. Lett. 19, 811–813 10.1016/j.bmcl.2008.12.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Delbecq S. P., Jehle S., and Klevit R. (2012) Binding determinants of the small heat shock protein, αB-crystallin: recognition of the 'IxI' motif. EMBO J. 31, 4587–4594 10.1038/emboj.2012.318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Mainz A., Peschek J., Stavropoulou M., Back K. C., Bardiaux B., Asami S., Prade E., Peters C., Weinkauf S., Buchner J., and Reif B. (2015) The chaperone αB-crystallin uses different interfaces to capture an amorphous and an amyloid client. Nat. Struct. Mol. Biol. 22, 898–905 10.1038/nsmb.3108 [DOI] [PubMed] [Google Scholar]
- 115. Rérole A. L., Gobbo J., De Thonel A., Schmitt E., Pais de Barros J. P., Hammann A., Lanneau D., Fourmaux E., Deminov O. N., Micheau O., Lagrost L., Colas P., Kroemer G., and Garrido C. (2011) Peptides and aptamers targeting HSP70: a novel approach for anticancer chemotherapy. Cancer Res. 71, 484–495 10.1158/0008-5472.CAN-10-1443 [DOI] [PubMed] [Google Scholar]
- 116. Faiella L., Piaz F. D., Bisio A., Tosco A., and De Tommasi N. (2012) A chemical proteomics approach reveals Hsp27 as a target for proapoptotic clerodane diterpenes. Mol. Biosyst. 8, 2637–2644 10.1039/c2mb25171j [DOI] [PubMed] [Google Scholar]
- 117. Makley L. N., McMenimen K. A., DeVree B. T., Goldman J. W., McGlasson B. N., Rajagopal P., Dunyak B. M., McQuade T. J., Thompson A. D., Sunahara R., Klevit R. E., Andley U. P., and Gestwicki J. E. (2015) Pharmacological chaperone for α-crystallin partially restores transparency in cataract models. Science 350, 674–677 10.1126/science.aac9145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Plate L., Cooley C. B., Chen J. J., Paxman R. J., Gallagher C. M., Madoux F., Genereux J. C., Dobbs W., Garza D., Spicer T. P., Scampavia L., Brown S. J., Rosen H., Powers E. T., Walter P., et al. (2016) Small molecule proteostasis regulators that reprogram the ER to reduce extracellular protein aggregation. eLife 5, e15550 10.7554/eLife.15550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Holt D. A., Luengo J. I., Yamashita D. S., Oh H.-J., Konialian A. L., Yen H.-K., Rozamus L. W., Brandt M., Bossard M. J., Levy M. A., Eggelston D. S., Liang J., Schultz L. W., Stout T. J., and Clardy J. (1993) Design, synthesis and kinetic evaluation of high-affinity FKBP ligands and the X-ray crystal structure of their complexes with FKBP12. J. Am. Chem. Soc. 115, 9925–9938 10.1021/ja00075a008 [DOI] [Google Scholar]
- 120. De Leon J. T., Iwai A., Feau C., Garcia Y., Balsiger H. A., Storer C. L., Suro R. M., Garza K. M., Lee S., Kim Y. S., Chen Y., Ning Y. M., Riggs D. L., Fletterick R. J., Guy R. K., et al. (2011) Targeting the regulation of androgen receptor signaling by the heat shock protein 90 cochaperone FKBP52 in prostate cancer cells. Proc. Natl. Acad. Sci. U.S.A. 108, 11878–11883 10.1073/pnas.1105160108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Gaali S., Feng X., Hähle A., Sippel C., Bracher A., and Hausch F. (2016) Rapid, structure-based exploration of pipecolic acid amides as novel selective antagonists of the FK506-binding protein 51. J. Med. Chem. 59, 2410–2422 10.1021/acs.jmedchem.5b01355 [DOI] [PubMed] [Google Scholar]
- 122. Anderson D. J., Le Moigne R., Djakovic S., Kumar B., Rice J., Wong S., Wang J., Yao B., Valle E., Kiss von Soly S., Madriaga A., Soriano F., Menon M. K., Wu Z. Y., Kampmann M., et al. (2015) Targeting the AAA ATPase p97 as an approach to treat cancer through disruption of protein homeostasis. Cancer Cell 28, 653–665 10.1016/j.ccell.2015.10.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Crawford L. J., Walker B., and Irvine A. E. (2011) Proteasome inhibitors in cancer therapy. J. Cell Commun. Signal. 5, 101–110 10.1007/s12079-011-0121-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Mackinnon A. L., Paavilainen V. O., Sharma A., Hegde R. S., and Taunton J. (2014) An allosteric Sec61 inhibitor traps nascent transmembrane helices at the lateral gate. eLife 3, e01483 10.7554/eLife.01483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Sidrauski C., Tsai J. C., Kampmann M., Hearn B. R., Vedantham P., Jaishankar P., Sokabe M., Mendez A. S., Newton B. W., Tang E. L., Verschueren E., Johnson J. R., Krogan N. J., Fraser C. S., Weissman J. S., et al. (2015) Pharmacological dimerization and activation of the exchange factor eIF2B antagonizes the integrated stress response. eLife 4, e07314 10.7554/eLife.07314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Verba K. A., Wang R. Y., Arakawa A., Liu Y., Shirouzu M., Yokoyama S., and Agard D. A. (2016) Atomic structure of Hsp90–Cdc37–Cdk4 reveals that Hsp90 traps and stabilizes an unfolded kinase. Science 352, 1542–1547 10.1126/science.aaf5023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Ouimet C. M., Dawod M., Grinias J., Assimon V. A., Lodge J., Mapp A. K., Gestwicki J. E., and Kennedy R. T. (2018) Protein cross-linking capillary electrophoresis at increased throughput for a range of protein–protein interactions. Analyst 143, 1805–1812 10.1039/C7AN02098H [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Villar E. A., Beglov D., Chennamadhavuni S., Porco J. A. Jr, Kozakov D., Vajda S., and Whitty A. (2014) How proteins bind macrocycles. Nat. Chem. Biol. 10, 723–731 10.1038/nchembio.1584 [DOI] [PMC free article] [PubMed] [Google Scholar]