

Abstract
Retinyl palmitate (RP), a storage form of vitamin A, is frequently used as a cosmetic ingredient, with more than 700 RP-containing cosmetic products on the U.S. market in 2004. There are concerns for the possible genotoxicity and carcinogenicity of RP when it is exposed to sunlight. To evaluate the photomutagenicity of RP in cells when exposed to ultraviolet A (UVA) light, L5178Y/Tk+/− mouse lymphoma cells were treated with different doses of RP alone/or in the presence of UVA light. Treatment of the cells with RP alone at the dose range of 25–100 μmg/ml did not increase mutant frequencies (MFs) over the negative control, whereas treatment of cells with 1–25 μg/ml RP under UVA light (82.8 mJ/cm2/min for 30 min) produced a dose-dependent mutation induction. The mean induced MF (392 × 10−6) for treatment with 25 mg/ml RP under UVA exposure was about threefold higher than that for UVA alone (122 × 10−6), a synergistic effect. To elucidate the underlying mechanism of action, we examined the mutants for loss of heterozygosity (LOH) at four microsatellite loci spanning the entire chromosome 11, on which the Tk gene is located. The mutational spectrum for the RP + UVA treatment was significantly different from the negative control, but not significantly different from UVA exposure alone. Ninety four percent of the mutants from RP + UVA treatment lost the Tk+ allele, and 91% of the deleted sequences extended more than 6 cM in chromosome length, indicating clastogenic events affecting a large segment of the chromosome. These results suggest that RP is photomutagenic in combination with UVA exposure in mouse lymphoma cells, with a clastogenic mode-of-action.
Keywords: retinyl palmitate, UVA, mouse lymphoma assay, photomutagenicity, loss of heterozygosity, mutant frequency
INTRODUCTION
Vitamin A, a natural retinoid, is required for many biological processes including cell growth, differentiation, and maintenance (Tee, 1992). Retinyl palmitate (all-trans-retinyl palmitate, RP) is a principal storage form of retinol in humans and animals, and it can be enzymatically hydrolyzed back to retinol in vivo (Idson, 1990). Because retinol is thermally unstable and RP is relatively more stable, RP is widely used as a special interest ingredient in cosmetic formulations and was present in more than 700 products available on the U.S. market in 2004 (FDA, 2004). The Cosmetic Ingredient Review Expert Panel, the cosmetic industry’s group for evaluating the safety of cosmetic ingredients, concluded in 1987 that RP is safe as a cosmetic ingredient in the present practices of use and in concentrations up to approximately 10% (Cosmetic Ingredient Review, 1987). These products include skin care and moisturizing preparations, suntan lotions, cosmetic makeup, and lipsticks. The topical application of RP to the skin of humans can produce significant levels of RP both inside and on top of the skin, where it can interact with sunlight (Ihara et al., 1999). The possible toxic effects of RP on skin exposed to sunlight have not been fully investigated. Therefore, RP was referred by the U.S. Food and Drug Administration (FDA) to the National Toxicology Program (NTP) as a high priority compound for phototoxicity and photocarcinogenicity studies, primarily based on its increasingly widespread use on sun-exposed skin and the association between topical application of retinoids and enhancement of photocarcinogenesis (Fu et al., 2002).
The UV radiation that reaches the surface of the earth from the sun is divided into two wavebands, UVA (between 320 and 400 nm) and UVB (between 290 and 320 nm), with visible and infrared light at longer wavelengths (FDA, 1999). Retinyl palmitate shows maximum UV-visible absorption at 326 nm (Tee, 1992). Therefore, UVA may play an important role in the photobiological activity of RP. UVA itself is a carcinogen and mutagen. UVA can induce cutaneous squamous cell carcinoma in mice (de Gruijl et al., 1993; Matsui and DeLeo, 1991) and increase melanoma risk in humans (Swerdlow et al., 1988; Walter et al., 1990; Westerdahl et al., 2000). UVA radiation increases mutant frequency (MF) and micronucleus formation (Phillipson et al., 2002).
In our previous study on the effects of UVA on RP (Cherng et al., 2005), we determined that photoirradiation of RP and its photodecomposition products generated reactive oxygen species (ROS) and resulted in lipid peroxidation. Generally, ROS and lipid peroxidation produce oxidative damage to DNA and result in mutations. Retinyl palmitate and its photodecomposition products in combination with UVA exposure, however, were not mutagenic in Salmonella typhimurium tester strains TA98, TA100, TA102, and TA104 in the presence or absence of S9 activation enzymes and were not photomutagenic in S. typhimurium TA102 when irradiated with UVA. Oxidative damage frequently results in mutations with large chromosomal alterations (Harrington-Brock et al., 2003; Rothfuss et al., 2000; Singh et al., 2005; Takeuchi et al., 1997), which cannot be detected by the S. typhimurium gene mutation test system. Therefore, we hypothesized that mutations generated by the UVA irradiation of RP may be due primarily to large-scale chromosome damage.
To test our hypothesis, we have used the L5178Y mouse lymphoma assay (MLA) to evaluate whether RP can interact with UVA to produce a photomutagenic effect. Unlike the microbial assays, the MLA detects a broad spectrum of genetic damage, including both point mutations and chromosomal mutations. This feature makes the MLA particularly useful for detecting mutational events that result from oxidative DNA damage. We also examined the types of mutations induced by UVA and RP + UVA to understand the underlying mechanism of action for the photomutagenicity of RP in combination with UVA exposure.
MATERIALS AND METHODS
Cells and culture conditions
The L5178Y/Tk+/− −3.7.2C mouse lymphoma cell line was used for the mutation assay. Cells were grown according to the methods described by Chen and Moore (2004). Briefly, the basic medium was Fischer’s medium for leukemic cells of mice with L-glutamine (Quality Biological Inc., Gaithersburg, MD) supplemented with pluronic F68 (0.1%), sodium pyruvate (1 mM), penicillin (100 U/ml), and streptomycin (100 μg/ml). The treatment medium (F5p), growth medium (F10p), and cloning medium (F20p) were the basic medium supplemented with 5%, 10%, and 20% heat-inactivated horse serum, respectively. The cultures were maintained in a humidified incubator with 5% CO2 in air at 37°C. Unless otherwise noted, all culture supplies were purchased from Invitrogen Life Technologies (Carlsbad, CA).
Cell treatment with RP alone
Retinyl palmitate was purchased from the Sigma (St. Louis, MO). The RP working solution (100×) was prepared just prior to use by dissolving it with anhydrous dimethyl sulfoxide (DMSO). The cells were suspended in 100-mm-diameter tissue culture dishes at a concentration of 6 × 106 cells in 10 ml of treatment medium. Because we focused on the photomutagenicity of RP by UVA, we did not test the mutagenic potential of RP more than 100 μg/ml. One hundred μl of the RP stock solutions at concentrations between 25 and 100 μg/ml were added to the medium, and the cells were incubated for 4 h at 37°C. In all cases, including negative controls (DMSO only) and positive controls [0.1 μg/ml 4-nitroquinoline-1-oxide (4-NQO)], the final concentration of DMSO in the medium was 1%. After treatment, the cells were centrifuged and washed once with fresh medium and then resuspended in growth medium at a density of 3 × 105 cells/ml in 25-cm2 cell culture flasks to begin phenotypic expression.
Cell treatment with UVA alone
The light box, a custom-made 4-lamp unit uses UVA lamps (National Biologics, Twinsburg, OH) (Cherng et al., 2005). The irradiance of light was determined with an Optronics OL754 Spectroradiometer (Optronics Laboratories, Orlando, FL), and the light dose was routinely measured with a Solar Light PMA-2110 UVA detector (Solar Light Inc., Philadelphia, PA). The maximum emission of the UVA was between 340 and 355 nm. The light intensities at wavelengths below 320 nm (UVB light) and above 400 nm (visible light) were about two orders of magnitude lower than the maximum at 340–355 nm. The 6 × 106 cells in 10 ml of treatment medium in 100-mm diameter tissue culture dish were exposed to UVA light at a rate of 82.8 mJ/cm2/min for various times from 5 to 45 min.
Cell treatment with pre-irradiated RP
The pre-irradiated RP was obtained by irradiating the stock RP solution with 82.8 mJ/cm2/min UVA for 30 min immediately before adding the solution to the cell culture to make a final concentration of pre-irradiated RP at 25 μg/ml. The cells were treated with the UVA pre-irradiated RP for 4 h at 37°C.
Cell treatment with RP and UVA.
Cells were treated with different concentrations (1–25 μg/ml) of RP and concomitantly exposed to 82.8 mJ/cm2/min UVA for 30 min. The treated cultures were then incubated with the RP at 37°C for an additional 3.5 h.
The Tk microwell mutation assay
Mutant selection was performed as described previously (Chen and Moore, 2004). Briefly, the cells were counted and the densities were adjusted using fresh medium at approximately 1 and 2 days after exposure. For mutant enumeration, trifluorothymidine (TFT, 3 μg/ml) was added to the cell culture in cloning medium, and cells were seeded into four 96-well flat-bottom microtiter plates using 200 μl per well and a density of 2000 cells/well. For the determination of plating efficiency, approximately 1.6 cells were aliquoted in 200 μl per well into two 96-well flat-bottom microtiter plates. All plates were incubated at 37°C in a humidified incubator with 5% CO2 in air. After 11 days of incubation, colonies were counted and mutant colonies were categorized as small or large. Small colonies are defined as those smaller than 25% of the diameter of the well. Mutant frequencies were calculated using the Poisson distribution. The induced MF was obtained by subtracting the background MF (negative control MF) from the MF in the treatment group. Cytotoxicity was measured using relative total growth (RTG), which includes a measure of growth during treatment, expression, and cloning (Chen and Moore, 2004).
Detection of loss of heterozygosity (LOH) at the Thymidine kinase (Tk1) and other three microsatellite loci spanning the entire chromosome 11 for Tk mutants
Mutant cells were directly taken from TFT-selected plates. Only mutants obtained from the 30-min UVA exposure, the concomitant treatment of 25 μg/ml RP and UVA, and the negative control were isolated and analyzed. The mutants from the RP treatment alone were not analyzed for LOH because RP was not mutagenic under the conditions used (see Results). The mutant cells were washed once with PBS by centrifugation, and cell pellets were quickly frozen and stored at −80°C. Genomic DNA was extracted by digesting the cells in lysis buffer (10 mM Tris-HCl [pH 7.5], 5 mM MgCl2, 1% [v/v] Triton X-100, 1% [v/v] Tween 20) with 200 μg/ml of proteinase K at 60°C for 90 min, followed by inactivation of proteinase K at 95°C for 10 min. The procedure for the polymerase chain reaction (PCR) analysis of LOH at the Tk locus was performed as previously described (Chen et al., 2002a). For PCR analysis of LOH at other loci (D11Mit42, D11Mit29, and D11Mit74 loci; Fig. 1), the amplification reactions were carried out in a total volume of 20 μl using 2×PCR master Mix (Promega, Madison, WI) and pairs of primers described previously (Singh et al., 2005). The thermal cycling conditions were as follows: initial incubation at 94°C for 3 min, 40 cycles of 94°C denaturation for 30 s, 55°C annealing for 30 s, and 72°C extension for 30 s, and a final extension at 72°C for 7 min. The amplification products were scored for the presence of one band (indicating LOH) or two bands (retention of heterozygosity at the given locus) after 2% agarose gel electrophoresis.
FIG. 1.
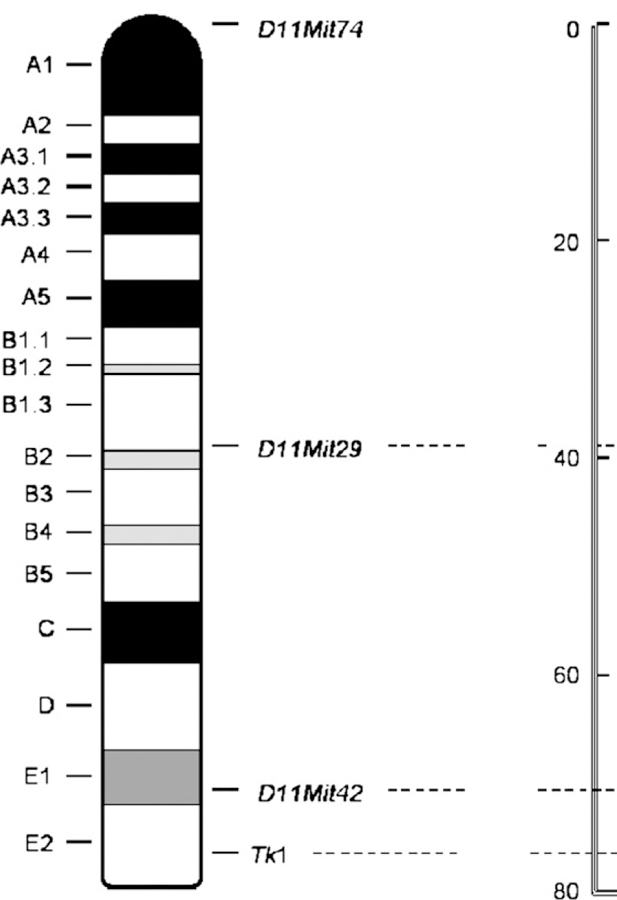
Ideogram of mouse chromosome 11. The loci that were analyzed for LOH (Tk1, D11Mit74, D11Mit29, and D11Mit42) are marked. The right ruler in cM indicates the distance from the top of the chromosome.
Statistical analyses
To evaluate the differences between the treatment groups, one-way analysis of variance (ANOVA) followed by the Student-Newman-Keul test using SigmaStat (SPSS Science, Chicago, IL) was used. Loss of heterozygosity patterns of mutants were compared using the computer program written by Cariello et al. (1994) for the Monte Carlo analysis developed by Adams and Skopek (1987).
RESULTS
The relative total growth (RTG) values, Tk MF, small colony MF, and large colony MF from one representative MLA experiment with treatment of RP, UVA, and RP + UVA are presented in Table 1. These results were replicated in two additional experiments and the data are displayed as mean ± 1 standard deviation in Figures 2 and 3.
TABLE 1.
Toxicity and Mutagenicity of RP, UVA, and RP with UVA in L5178Y/Tk+/− Mouse Lymphoma Cells
| Percent platingefficiency | Total mutantcount | Mutant frequency (MF, ×10−6) | Relative totalgrowth (%) | MF of small colonies (×10−6) | MF of large colonies (×10−6) | ||
|---|---|---|---|---|---|---|---|
| Treatment | Dose | ||||||
| Controla | 1% DMSO | 84 | 35 | 57 | 100 | 26 | 31 |
| RP | 25 μg/ml | 105 | 35 | 46 | 98 | 18 | 27 |
| 50 μg/ml | 92 | 27 | 40 | 89 | 26 | 13 | |
| 100 μg/ml | 96 | 33 | 47 | 77 | 28 | 18 | |
| UVAb | 5 min | 110 | 50 | 63 | 78 | 29 | 34 |
| 10 min | 85 | 55 | 91 | 67 | 46 | 45 | |
| 15 min | 88 | 76 | 125 | 63 | 61 | 64 | |
| 30 min | 94 | 113 | 186 | 52 | 71 | 115 | |
| 45 min | 96 | 156 | 270 | 43 | 125 | 145 | |
| RP + UVAc | 1 μg/ml | 95 | 111 | 180 | 75 | 70 | 110 |
| 2.5 μg/ml | 100 | 115 | 179 | 72 | 81 | 98 | |
| 5 μg/ml | 103 | 151 | 243 | 68 | 137 | 106 | |
| 10 μg/ml | 92 | 156 | 283 | 36 | 183 | 100 | |
| 25 μg/ml | 70 | 175 | 437 | 12 | 287 | 150 | |
| 4-NQOd | 0.1 μg/ml | 58 | 246 | 921 | 25 | 483 | 438 |
In the control group, cells were treated with dimethyl sulfoxide (DMSO) only.
Cells received 82.8 mJ/cm2/min UVA irradiation for different periods.
Cells were concomitantly exposed to different concentrations of RP and 82.8 mJ/cm2/min UVA irradiation for 30 min.
4-NQO was used as a positive control.
FIG. 2.
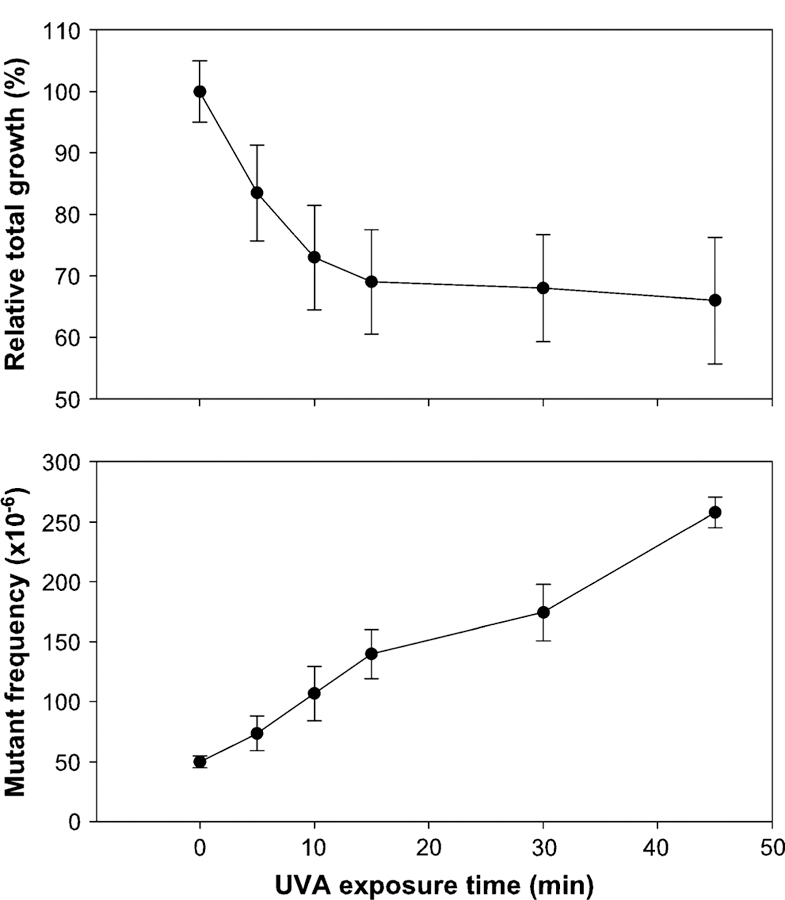
Cytotoxicity and mutagenicity of UVA in mouse lymphoma cells. The cells received 82.8 mJ/cm2/min UVA irradiation for different periods of time. Cytotoxicity (defined as relative total growth) is shown in the top panel, and Tk mutant frequency is displayed in the bottom panel. The data points represent the mean ± 1 standard deviation from three independent experiments.
FIG. 3.
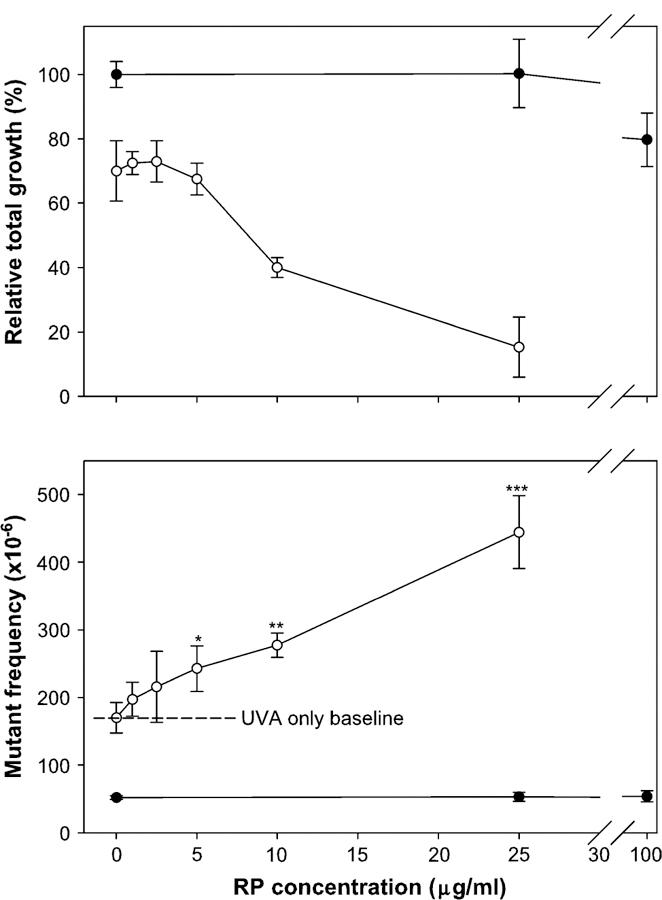
Comparison of cytotoxicity (the top panel) and mutagenicity (the bottom panel) of retinyl palmitate (RP) and RP + UVA in mouse lymphoma cells. The cells were treated with different concentrations of RP in conjunction with 82.8 mJ/cm2/min UVA irradiation for 30 min (open circle) or without UVA irradiation (closed circle). The data points represent the mean ± 1 standard deviation from three independent experiments. The concomitant treatment groups (UVA + RP) with 5, 10, and 25 μg/ml of RP were significantly higher than the UVA alone (*p < 0.05, **p < 0.01, and ***p < 0.001).
A dose-related cytotoxicity and mutagenicity of UVA was observed after the L5178Y/Tk+/− mouse lymphoma cells were exposed to different doses of UVA (Fig. 2). The MFs increased linearly with UVA exposure time. Treatment of cells with RP alone (25, 50, and 100 μg/ml) for 4 h showed little cytotoxicity (the RTG at the highest dose was about 80%) and did not increase the MF (Fig. 3). Treatment of cells with various concentrations of RP concomitantly exposed to 82.8 mJ/cm2/min for 30 min (2.48 J/cm2) UVA, however, resulted in linear dose-dependent increases for both the cytotoxicity and mutagenicity (Fig. 3). Comparing all concomitant treatment groups (UVA + RP) with the UVA alone exposure, the 5, 10, and 25 μg/ml of RP under UVA exposure were significantly different from the UVA alone ( p < 0.05, 0.01, and 0.001, respectively). The MFs for the dose points higher than 25 μg/ml RP in the combination exposure were not determined because of the high cytotoxicity and low plating efficiency. The MFs for the negative control, UVA alone of 30 min, and UVA + RP (25 cg/ml) were 52 ± 3 3 10−6, 174 ± 24 3 10−6, and 444 ± 54 × 10−6, respectively. The induced MF, obtained by subtracting the negative control MF from the total MF in the treated culture, for the concomitant treatment of 25 μg/ml RP and 2.48 J/cm2 UVA (392 3 10−6) was about threefold higher than that for 2.48 J/cm2 UVA exposure alone (122 3 10−6).
To determine the mutagenicity of the photodecomposition products of RP created by UVA irradiation, we pre-irradiated RP with 2.48 J/cm2 in the absence of cells. The pre-irradiated RP then was used to treat the cells at a concentration of 25 μg/ml. The MF for the pre-irradiated RP treatment was higher than that for 25 μg/ml RP treatment alone, but lower than that for 2.48 J/cm2 UVA treatment alone, although there were no statistically significant differences. The MF for the pre-irradiated RP treatment was significantly less than the concomitant cellular exposure of RP and UVA (Fig. 4).
FIG. 4.
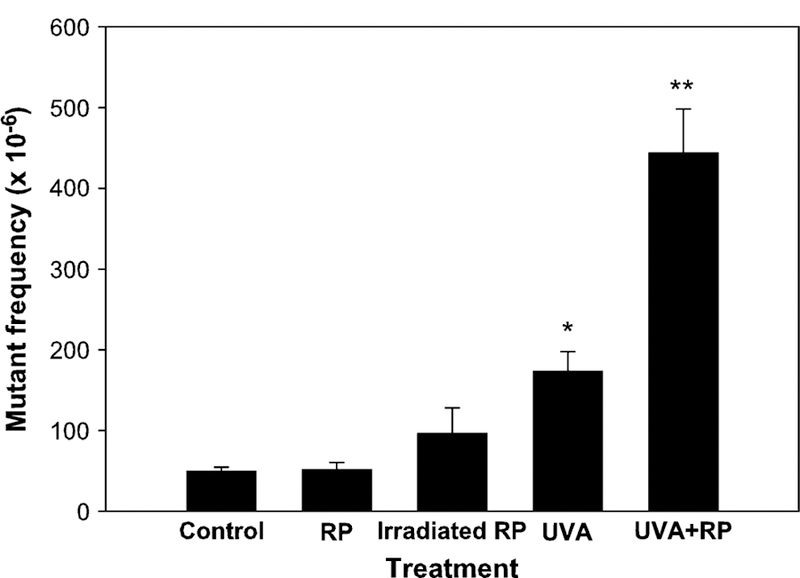
Comparison of the mutant frequencies in the Tk gene of mouse lymphoma cells treated with vehicle control, retinyl palmitate (RP), pre-irradiated RP, UVA, and RP + UVA. The RP concentration used for cell treatment was 25 μg/ml. The pre-irradiated RP was obtained by irradiating RP solution with 82.8 mJ/cm2/min UVA for 30 min immediately before the cell treatment, and then the cells were treated with 25 μg/ml of the pre-irradiated RP for a 4 h incubation. For UVA and UVA + RP treatments, the cell suspensions were irradiated with 82.8 mJ/cm2/min UVA for 30 min during the 4 h incubation. The data points represent the mean ± 1 standard deviation for three independent experiments. An asterisk indicates that the mutant frequency in this treatment is significantly different from those in control or in RP treatment groups ( p < 0.01). Two asterisks indicate that the mutant frequency in this treatment is significantly different from those in all other groups ( p < 0.001).
Loss of heterozygosity analysis of the mutants was conducted using four microsatellite loci spanning the entire chromosome 11 (Fig. 1) to determine the types of the mutations. DNA samples were isolated from 48 large and 48 small mutant colonies for the DMSO negative control, 2.48 J/cm2 UVA treatment alone, and the concomitant treatment of 25 μg/ml RP and 2.48 J/cm2 UVA, respectively. The percentages and MFs of different types of mutations for large and small colonies are displayed in Figure 5. Statistical analysis of the spectra revealed that the mutational spectra induced by UVA or RP + UVA were significantly different from the negative control, respectively, for both large and small colonies, whereas there was no statistical difference between the mutational spectra induced by UVA and RP + UVA treatments for both large and small colonies. A statistical pairwise multiple comparison of these mutational spectra is listed in Table 2. The most common type of mutation for UVA and RP + UVA was the LOH extending to D11Mit42, an alteration of DNA larger than 6 cM, whereas the major type of mutation in the control was non-LOH, indicating intragenic mutations in the Tk gene (Fig. 5).
FIG. 5.
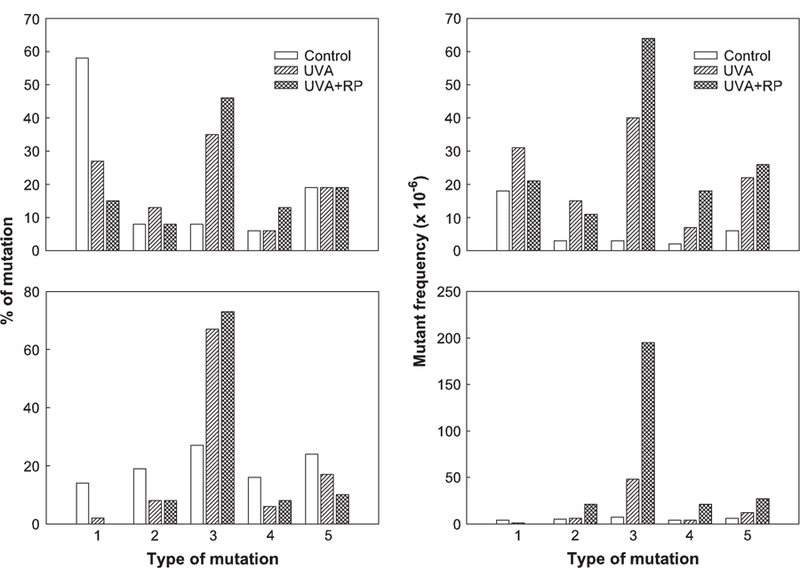
Comparison of percentage (left two panels) and mutant frequencies (right panels) of mutational types of large colonies (top panels) and small colonies (bottom panels) produced in mouse lymphoma cells treated with vehicle, UVA, and UVA + RP. The numbers indicate different types of mutations: 1, non-LOH (loss of heterozygosity); 2, LOH at Tk locus only; 3, LOH extending to D11Mit42 (about 6 cM); 4, LOH extending to D11Mit29 (about 38 cM); and 5, LOH extending to the top of chromosome 11.
TABLE 2.
Statistical Pairwise Multiple Comparison of Mutational Spectra from Control Vehicle, UVA Alone, and RP + UVA-Treated Mouse Lymphoma Cells
| Comparison | p Value | Statistically significant difference |
|---|---|---|
| Control versus UVA (large colony) | 0.013 | Yes |
| Control versus RP + UVA (large colony) | 0.001 | Yes |
| UVA versus RP + UVA (large colony) | 0.448 | No |
| Control versus UVA (small colony) | 0.003 | Yes |
| Control versus RP + UVA (small colony) | 0.001 | Yes |
| UVA versus RP + UVA (small colony) | 0.816 | No |
DISCUSSION
The number of cosmetic retail products containing RP has increased rapidly in the last two decades because of the beneficial effects of RP on the appearance of skin. As a result, a growing number of products containing RP are being applied to sun-exposed skin. The biological consequences of photoactivation of RP by UVA light deserve investigation. Retinoids are effective chemopreventive agents against skin, head and neck, lung, breast, liver, and other forms of cancer (Hansen et al. 2000). However, retinoids administered orally are not effective in preventing skin papillomas and carcinomas caused by UV light (Hill and Grubbs, 1992). The effects of retinoids on experimental animal photocarcinogenesis have been reported (Kligman, 1987; Mikkelsen et al., 1998). Supplementation of diets with vitamin A appeared to enhance UV carcinogenesis in hairless mice (Mikkelsen et al., 1998). To explore the possible photomutagenicity of RP by UVA and its underlying mechanisms, we conducted studies to investigate the genotoxicity of RP in combination with UVA exposure in a previous study (Cherng et al., 2005) and this study.
Retinyl palmitate alone, in concentrations of 25–100 μg/ml, did not increase the MF over control (Fig. 3), which is consistent with our previous study on RP mutagenicity using the S. typhimurium mutation test system (Cherng et al., 2005). In the present study, the combined treatment of RP in concentrations of 1–25 μg/ml with a UVA dose of 2.48 J/cm2 resulted in a significant increase of MFs, in a dose-dependent manner. The induced MF (subtracting the MF of the negative control from the MF observed in the test culture) at the 25 μg/ml RP with UVA exposure was about threefold higher than that for UVA-irradiation alone (Table 1). This is different from our previous studies using the microbial mutation assay (Cherng et al., 2005) in which RP treatment of S. typhimurium TA102 under UVA irradiation was not mutagenic. This inconsistency is most likely related to the fact that combined treatment of RP and UVA causes clastogenicity. Compounds acting primarily by a clastogenic mechanism induce detectable mutagenicity in the MLA but are only weakly mutagenic or nonmutagenic in the microbial assays (Chen et al., 2002b).
In this study, most of the mutants from concomitant treatment of RP and UVA were small colony mutants (66%, at a dose of 25 μg/ml, a MF of 287 of a total MF of 437), whereas the percentage of small colony mutants in the negative control cultures was 46% (a MF of 26 of a total MF of 57). Also, LOH at the Tk locus occurred in 85% in large colony mutants and 100% in small colony mutants (a total of 94% overall) from RP + UVA treatment, indicating that the photomutagenicity of RP by UVA irradiation results from a clastogenic mode of action. In the MLA, compounds that induce point mutations result in a high proportion of large colony Tk mutants and little LOH at the Tk locus, whereas clastogens tend to result in a high proportion of small colony mutants and predominantly LOH at the Tk locus (Applegate et al., 1990; Chen et al., 2002a, 2002b; Harrington-Brock et al., 2003). LOH is an important mutational event in tumorigenesis and is frequently observed in a variety of human cancers at loci that are tumor-suppressor genes. LOH can result from any of several mechanisms, including large deletions, mitotic recombination, and whole chromosome loss (Honma et al., 2001). Generally, depending on the severity of DNA damage of the Tk mutants, LOH will also occur at other loci along chromosome 11 in addition to the Tk gene.
To determine whether RP under UVA irradiation produces mutations through its photodecomposition products or through short-lived products like ROS and lipid peroxides, we irradiated RP for 30 min in the absence of cells, and the resulting pre-irradiated RP reaction mixture then was used to treat cells. The MF for pre-irradiated RP treatment was significantly lower than that for the concomitant exposure to RP and UVA, and it was not significantly different from that for RP treatment or the DMSO control (Fig. 4). This is consistent with our previous results that RP and its identified photodecomposition products did not bind with DNA in the presence of microsomal metabolizing enzymes (Cherng et al., 2005). These results indirectly demonstrate that UVA irradiation of RP produces short-lived species like ROS that damage DNA and result in mutations.
Ultraviolet light causes DNA damage both directly and indirectly. Direct DNA damage is principally by UVB, forming cyclobutane pyrimidine dimers, pyrimidine-pyrimidones, and point mutations. Indirect DNA damage is principally caused by UVA-dependent photoactivation of organic compounds that generates short-lived species (Brendler-Schwaab et al., 2004). Our previous research has shown that irradiation of RP with UVA light can generate ROS and lipid peroxides (Cherng et al., 2005). Photodynamic action results in the production of free radicals, including ROS. Because of their high reactivity, ROS can attack all cellular constituents, including proteins, nucleic acids, and lipids. Hydroxyl radicals can initiate a chain reaction that produces multiple lipid hydroperoxide molecules from a single initial event (Gutteridge and Halliwell, 1990). The chain reaction is in effect amplifying the initial oxidative insult, and the resulting active oxygen species can damage DNA and produce mutations.
UVA itself induces genetic damage in cells via an oxidative stress mechanism (Drobetsky et al., 1995; Kamiya, 2003; Phillipson et al., 2002). In this study, the mutational spectra induced by UVA and RP + UVA are not significantly different ( p = 0.448 in large colonies and p = 0.816 in small colonies; Table 2). This similarity suggests that RP + UVA induces mutations through the same mechanism as UVA, mainly oxidative DNA damage.
Chemicals that absorb UVA and visible light and generate ROS constitute the largest class of photosensitizers. After absorption of UVA light, RP potentially acts as a photosensitizer (Fu et al., 2003). The mutagenicity of photosensitizers probably results from oxidative radicals formed in the photosensitization reaction (Rerko et al., 1992). Several studies using different oxidative agents provide evidence that oxidative DNA damage generated from these agents can lead to different types of mutations, with a large proportion of chromosome mutations that result mainly from chromosome breakage (Harrington-Brock et al., 2003; Nakajima et al., 2002; Rothfuss et al., 2000; Takeuchi et al., 1997). In a recent study on the mutagenicity of the lipid peroxidation product 4-hydroxynonenal (4-HNE) in mouse lymphoma cells (Singh et al., 2005), we found that the major type of mutation was LOH extending to D11Mit42 (see Fig. 1 for the position), the same mutation as currently seen with RP + UVA treatment. The Tk mutants from cells exposed to either of these two compounds are about 60% this type of LOH mutation. A mechanistic pathway initiated by photoirradiation of RP with UVA light leading to induction of chromosome mutations has been suggested and is illustrated in Figure 6.
FIG. 6.
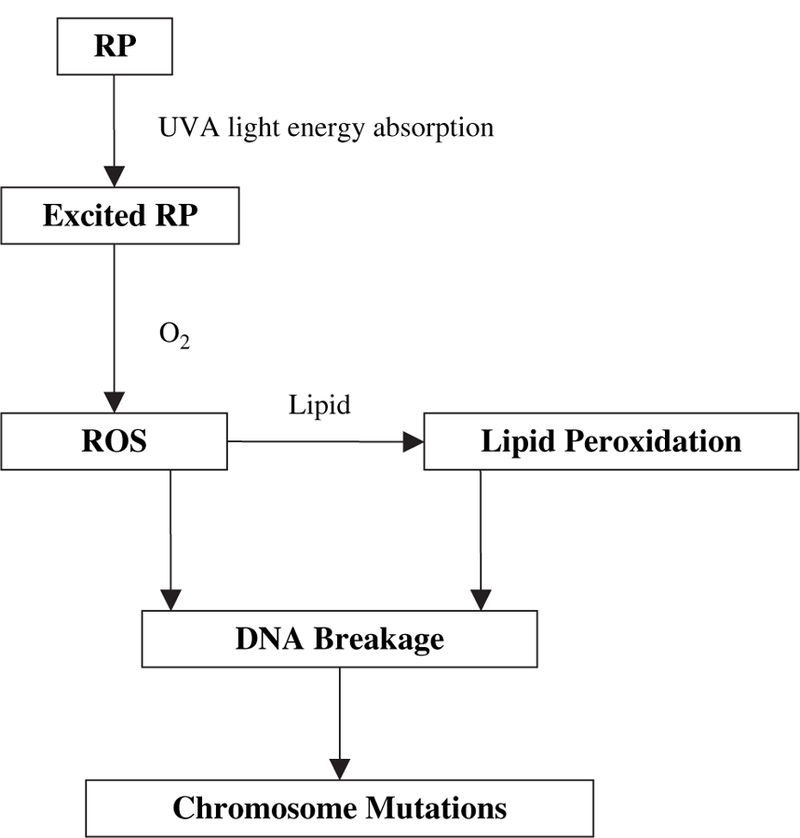
The proposed mechanistic pathway for chromosome mutations induced by combined exposure of RP and UVA in cells.
In summary, a combined treatment of RP and UVA irradiation produced a clearly synergistic photomutagenic effect at the heterozygous Tk locus in mouse lymphoma cells. Most of the Tk mutants induced by the concomitant treatment were the result of LOH, indicating a clastogenic mode of action. Oxidative DNA lesions are likely responsible for the photomutagenicity from the combined exposure of RP and UVA irradiation. These in vitro results suggest that the biological consequences of concomitant exposure to RP and UVA warrants further investigation.
ACKNOWLEDGMENTS
This research was partially supported by appointments (N.M., L.C.) to the Postgraduate Research Program at the National Center for Toxicological Research (NCTR), administered by the Oak Ridge Institute for Science and Education through an interagency agreement between the U.S. Department of Energy and the U.S. Food and Drug Administration.
The views presented in this article do not necessarily reflect those of the U.S. Food and Drug Administration.
REFERENCES
- Adams WT, and Skopek TR (1987). Statistical test for the comparison of samples from mutational spectra. J. Mol. Biol 194, 391–396. [DOI] [PubMed] [Google Scholar]
- Applegate ML, Moore MM, Broder CB, Burrell A, Juhn G, Kasweck KL, Lin PF, Wadhams A, and Hozier JC (1990). Molecular dissection of mutations at the heterozygous thymidine kinase locus in mouse lymphoma cells. Proc. Natl. Acad. Sci. U.S.A 87, 51–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brendler-Schwaab S, Czich A, Epe B, Gocke E, Kaina B, Muller L, Pollet D, and Utesch D (2004). Photochemical genotoxicity: Principles and test methods. Report of a GUM task force. Mutat. Res 566, 65–91. [DOI] [PubMed] [Google Scholar]
- Cariello NF, Piegorsch WW, Adams WT, and Skopek TR (1994). Computer program for the analysis of mutational spectra: Application to p53 mutations. Carcinogenesis 15, 2281–2285. [DOI] [PubMed] [Google Scholar]
- Chen T, Harrington-Brock K, and Moore MM (2002a). Mutant frequencies and loss of heterozygosity induced by N-ethyl-N-nitrosourea in the thymidine kinase gene of L5178Y/TK+/−−3.7.2C mouse lymphoma cells. Mutagenesis 17, 105–109. [DOI] [PubMed] [Google Scholar]
- Chen T, Harrington-Brock K, and Moore MM (2002b). Mutant frequency and mutational spectra in the Tk and Hprt genes of N-ethyl-N-nitrosourea-treated mouse lymphoma cells. Environ. Mol. Mutagen 39, 296–305. [DOI] [PubMed] [Google Scholar]
- Chen T, and Moore MM (2004). Screening for chemical mutagens using the mouse lymphoma assay. In Optimization in Drug Discovery: In-vitro Methods (Yan Z and Caldwell GW, eds.), pp. 337–352. Humana Press, Totowa, New Jersey. [Google Scholar]
- Cherng SH, Xia Q, Blankenship LR, Freeman JP, Wamer WG, Howard PC, and Fu PP (2005). Photodecomposition of retinyl palmitate in ethnaol by UVA light-formation of photodecomposition products, reactive oxygen species, and lipid peroxides. Chem. Res. Toxicol 18, 129–138. [DOI] [PubMed] [Google Scholar]
- Cosmetic Ingredient Review. (1987). Final report on the safety assessment of retinyl palmitate and retinol. J. Am. Coll. Toxicol 6, 279–320. [Google Scholar]
- de Gruijl FR, Sterenborg HJ, Forbes PD, Davies RE, Cole C, Kelfkens G, van Weelden H, Slaper H, and van der Leun JC (1993). Wavelength dependence of skin cancer induction by ultraviolet irradiation of albino hairless mice. Cancer Res 53, 53–60. [PubMed] [Google Scholar]
- Drobetsky EA, Turcotte J, and Chateauneuf A (1995). A role for ultraviolet A in solar mutagenesis. Proc. Natl. Acad. Sci. U.S.A 92, 2350–2354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FDA (U.S. Food and Drug Administration). (1999). Sunscreen drug products for over-the-counter human use; Final Monograph. Fed. Register 64, 27666–27693. [PubMed] [Google Scholar]
- FDA (U.S. Food and Drug Administration). (2004). The U.S. Food and Drug Administration’s Voluntary Cosmetics Registration Program www.fda.gov.
- Fu PP, Cheng SH, Coop L, Xia Q, Culp SJ, Tolleson WH, Wamer WG, and Howard PC (2003). Photoreaction, phototoxicity, and photocarcinogenicity of retinoids. J. Environ. Sci. Health, C, Environ. Carcinog. Ecotoxicol. Rev C21, 165–197. [DOI] [PubMed] [Google Scholar]
- Fu PP, Howard PC, Culp SG, Xia Q, Webb PJ, Blankenship LR, Wamer WG, and Bucher JR (2002). Do topically applied skin creams containing retinyl palmitate affect the photocarcinogenecity of simulated solar light? J. Food Drug Anal 10, 262–268. [Google Scholar]
- Gutteridge JM, and Halliwell B (1990). The measurement and mechanism of lipid peroxidation in biological systems. Trends Biochem. Sci 15, 129–135. [DOI] [PubMed] [Google Scholar]
- Hansen LA, Sigman CC, Andreola F, Ross SA, Kelloff GJ and De Luca LM (2000). Retinoids in chemoprevention and differentiation therapy. Carcinogenesis 21, 1271–1279. [PubMed] [Google Scholar]
- Harrington-Brock K, Collard DD, and Chen T (2003). Bromate induces loss of heterozygosity in the thymidine kinase gene of L5178Y/Tk+/−−3.7.2C mouse lymphoma cells. Mutat. Res 537, 21–28. [DOI] [PubMed] [Google Scholar]
- Hill DL, and Grubbs CJ (1992). Retinoids and cancer prevention. Annu. Rev. Nutr 12, 161–181. [DOI] [PubMed] [Google Scholar]
- Honma M, Momose M, Sakamoto H, Sofuni T, and Hayashi M (2001). Spindle poisons induce allelic loss in mouse lymphoma cells through mitotic non-disjunction. Mutat. Res 493, 101–114. [DOI] [PubMed] [Google Scholar]
- Idson B (1990). Vitamins in cosmetics, an update I. Overview and Vitamin A. Drug Cosmet. Ind 146, 26–91. [Google Scholar]
- Ihara H, Hashizume N, Hirase N, and Suzue R (1999). Esterification makes retinol more labile to photolysis. J. Nutr. Sci. Vitaminol. (Tokyo) 45, 353–358. [DOI] [PubMed] [Google Scholar]
- Kamiya H (2003). Mutagenic potentials of damaged nucleic acids produced by reactive oxygen/nitrogen species: Approaches using synthetic oligonucleotides and nucleotides: Survey and summary. Nucleic Acids Res 31, 517–531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kligman LH (1987). Retinoic acid and photocarcinogenesis—A controversy. Photodermatology 4, 88–101. [PubMed] [Google Scholar]
- Matsui MS, and DeLeo VA (1991). Longwave ultraviolet radiation and promotion of skin cancer. Cancer Cells 3, 8–12. [PubMed] [Google Scholar]
- Mikkelsen S, Berne B, Staberg B, and Vahlquist A (1998). Potentiating effect of dietary vitamin A on photocarcinogenesis in hairless mice. Carcinogenesis 19, 663–666. [DOI] [PubMed] [Google Scholar]
- Nakajima M, Takeuchi T, Ogino K, and Morimoto K (2002). Lack of direct involvement of 8-hydroxy-2#-deoxyguanosine in hypoxanthine-guanine phosphoribosyltransferase mutagenesis in V79 cells treated with N,N#-bis(2-hydroxyperoxy-2-methoxyethyl)-1,4,5,8-naphthalenetetracarboxylic-diimide (NP-III) or riboflavin. Jpn. J. Cancer Res 93, 247–252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillipson RP, Tobi SE, Morris JA, and McMillan TJ (2002). UV-A induces persistent genomic instability in human keratinocytes through an oxidative stress mechanism. Free Radic. Biol. Med 32, 474–480. [DOI] [PubMed] [Google Scholar]
- Rerko RM, Clay ME, Antunez AR, Oleinick NL, and Evans HH (1992). Photofrin II photosensitization is mutagenic at the tk locus in mouse L5178Y cells. Photochem. Photobiol 55, 75–80. [DOI] [PubMed] [Google Scholar]
- Rothfuss A, Merk O, Radermacher P, and Speit G (2000). Evaluation of mutagenic effects of hyperbaric oxygen (HBO) in vitro. II. Induction of oxidative DNA damage and mutations in the mouse lymphoma assay. Mutat. Res 471, 87–94. [DOI] [PubMed] [Google Scholar]
- Singh SP, Chen T, Chen L, Mei N, McLain E, Samokyszyn V, Thaden JJ, Moore MM, and Zimniak P (2005). Mutagenic effects of 4-hydroxynonenal triacetate, a chemically protected form of the lipid peroxidation product 4-hydroxynonenal, as assayed in L5178Y/Tk+/−mouse lymphoma cells. J. Pharmacol. Exp. Ther 313, 855–861. [DOI] [PubMed] [Google Scholar]
- Swerdlow AJ, English JS, MacKie RM, O’Doherty CJ, Hunter JA, Clark J, and Hole DJ (1988). Fluorescent lights, ultraviolet lamps, and risk of cutaneous melanoma. B. M. J 297, 647–650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeuchi T, Matsugo S, and Morimoto K (1997). Mutagenicity of oxidative DNA damage in Chinese hamster V79 cells. Carcinogenesis 18, 2051–2055. [DOI] [PubMed] [Google Scholar]
- Tee ES (1992). Carotenoids and retinoids in human nutrition. Crit. Rev. Food Sci. Nutr 31, 103–163. [DOI] [PubMed] [Google Scholar]
- Walter SD, Marrett LD, From L, Hertzman C, Shannon HS, and Roy P (1990). The association of cutaneous malignant melanoma with the use of sunbeds and sunlamps. Am. J. Epidemiol 131, 232–243. [DOI] [PubMed] [Google Scholar]
- Westerdahl J, Ingvar C, Masback A, Jonsson N, and Olsson H (2000). Risk of cutaneous malignant melanoma in relation to use of sunbeds: Further evidence for UV-A carcinogenicity. Br. J. Cancer 82, 1593–1599. [DOI] [PMC free article] [PubMed] [Google Scholar]