

Abstract
There are a number of clinically effective treatments for stress-associated psychiatric diseases, including major depressive disorder (MDD). Nonetheless, many patients exhibit resistance to first-line interventions calling for novel interventions based on pathological mechanisms. Accumulating evidence implicates altered glutamate signaling in MDD pathophysiology, suggesting that modulation of glutamate signaling cascades may offer novel therapeutic potential. Here we report that JHU-083, our recently developed prodrug of the glutaminase inhibitor 6-diazo-5-oxo-l-norleucine (DON) ameliorates social avoidance and anhedonia-like behaviors in mice subjected to chronic social defeat stress (CSDS). JHU-083 normalized CSDS-induced increases in glutaminase activity specifically in microglia-enriched CD11b+ cells isolated from the prefrontal cortex and hippocampus. JHU-083 treatment also reverses the CSDS-induced inflammatory activation of CD11b+ cells. These results support the importance of altered glutamate signaling in the behavioral abnormalities observed in the CSDS model, and identify glutaminase in microglia-enriched CD11b+ cells as a pharmacotherapeutic target implicated in the pathophysiology of stress-associated psychiatric conditions such as MDD.
Subject terms: Pharmacology, Experimental models of disease
Introduction
Major depressive disorder (MDD) is a common and debilitating psychiatric disorder with a high lifetime prevalence, imposing a serious economic burden on society [1]. Although there are a number of clinically effective treatments for MDD, a large segment of patients exhibits resistance to first-line interventions (e.g., selective serotonin reuptake inhibitors; SSRIs), calling for novel interventions based on the pathological mechanisms of MDD [2, 3].
Growing evidence suggests that altered glutamate signaling system is implicated in MDD pathophysiology [4–6]. Several studies have reported increased levels of glutamate in the serum and plasma of MDD patients as well as high concentrations of glutamine, the major synthetic precursor for glutamate production in the brain [7–10]. Increased glutamine-glutamate ratios have also been detected in the cerebrospinal fluid of patients with MDD [7–10]. These findings are supported by recent magnetic resonance spectroscopy (MRS) studies demonstrating increased or altered glutamate content in the brains of MDD patients [11–13]. At the molecular level, expression of ionotropic N-methyl-d-aspartate (NMDA) and α-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid (AMPA) glutamate receptors as well as glutamate transporters have been found to be altered in postmortem MDD brain tissue [4, 14]. Moreover, the NMDA receptor antagonist ketamine and modulators of metabotropic glutamate receptors have recently gained greater attention as potential antidepressant agents [12, 15–18]. Ketamine in particular induces a rapid antidepressant effect in treatment-resistant patients [19], with positive response linked to alterations in extracellular glutamate availability and signaling [12, 15–18], supporting the idea that modulation of glutamate signaling offers novel therapeutic potential for MDD patients.
Complimentary findings from preclinical studies highlight the importance of glial cells in the control of extracellular glutamate and the regulation of glutamatergic neurotransmission [5, 6, 20, 21]. Microglia and astrocytes play critical roles in the glutamate/glutamine metabolic cycle and are key regulators of glutamate release and clearance especially under conditions of neuroinflammation (Miller et al. [22]). For instance, previous studies demonstrated exaggerated release of glutamate by activated immune cells leading to excitotoxicity, neural damage, and impaired excitatory neurotransmission [23, 24]. Although more work is needed to understand the links between neuroinflammatory and glutamatergic mechanisms underlying MDD pathophysiology [25], pharmacological intervention aimed at normalizing inflammation-associated glial glutamate production might alleviate depressive symptoms.
Glutaminase, an enzyme that catalyzes the hydrolysis of glutamine into glutamate, is thought to play a central role in the generation of excitotoxic glutamate in the brain [26, 27]. In addition to synaptic release of glutamate from neurons, glutamate is produced and released from microglia after its conversion from glutamine (Maezawa et al. [28, 29]). In activated microglia culture, expression of glutaminase is elevated, contributing to the overproduction of glutamate [27]. Altered glutamate homeostasis in glial cells downstream of inflammation has been linked to other CNS disease such as multiple sclerosis and brain edema [30, 31], leading researchers to explore glutaminase inhibition as a therapeutic target [32–34]. In fact, 6-diazo-5-oxo-l-norleucine (DON), a glutamine analog that acts as a glutaminase inhibitor, has shown behavioral and disease-modifying efficacy in various models of neuroinflammation including multiple sclerosis, cerebral malaria, brain cancer, viral infection, and HIV-associated neurocognitive disorders [35–40]. Our previous study demonstrating that DON blocks glutamate release from activated microglia [27] suggests that this efficacy is at least partially mediated by normalizing glutamate production, a mechanism of action suggesting therapeutic potential for DON in MDD. We recently developed orally available prodrugs of DON, such as JHU-083 (Ethyl 2-(2-Amino-4-methylpentanamido)-DON), designed to improve CNS penetration for targeting neurological and psychiatric disorders [39].
Herein, we explore the effect of chronic administration of JHU-083 on social avoidance and anhedonia-like behaviors in mice subjected to chronic social defeat stress (CSDS), a well-characterized model of stress-associated psychiatric conditions, including depression [41–47]. We also examine whether CSDS induces glutaminase activity and pro-inflammatory cytokine expression in microglia-enriched CD11b+ cells and non-CD11b+ cells in the brain that may be sensitive to JHU-083 treatment. We report that JHU-083 effectively delivers DON to the mouse brain. We also demonstrate that JHU-083 inhibits CSDS-induced glutaminase activity and cytokine induction in brain CD11b+ cells, alleviating social avoidance and anhedonia-like behavioral abnormalities.
Materials and methods
Animals
Male 7-to 8-week-old C57BL/6J (C57) mice (25–30 g; Jackson Laboratory, Bar Harbor, ME) and 4-to 6-month-old CD-1 retired breeders (35–45 g; Charles River Laboratories, Wilmington, MA) were used for all experiments presented in this study [48]. Mice were housed on a reversed 12-h light/dark cycle, and maintained in a humidity- and temperature-controlled room with water and food available ad libitum. CD-1 mice were singly housed except during social defeats. C57 mice were group housed before starting CSDS and singly housed after CSDS. Behavioral experiments were conducted during the dark cycle. All studies were conducted with approved protocols from the Johns Hopkins University Institutional Animal Care and Use Committee and were in accordance with the NIH guidelines for the Care and Use of Laboratory Animals.
Chronic social defeat stress (CSDS)
C57 male mice were subjected to CSDS according to a previously published method with minor modification [48, 49]. CD-1 mice are larger and more dominant than C57 mice and are commonly used as aggressors in the social defeat of C57 mice [48, 50, 51]. Accordingly, we identified aggressive male CD-1 mice as resident aggressors by a 3 day screening process selecting for CD-1 mice that showed at least one aggressive response to an intruder C57 male within 30 s [48]. Chosen CD-1 mice were then singly housed before CSDS experiments. Intruder male C57 mice were exposed to a novel CD-1 aggressor for 10 min daily. After exposure, C57 mice were maintained for 24 h on the opposite side of a transparent and porous Plexiglas barrier within the home cage of the CD-1 aggressors to enable constant sensory exposure. CSDS was repeated with a new CD-1 aggressor mouse each day for 12 consecutive days. During bouts of exposure to the CD-1 mice, hallmark behavioral signs of CSDS were observed in C57 mice including escape, submissive postures (e.g., defensive upright or supine stance), and freezing. Non-stressed control C57 mice were daily placed in a similar cage, but in the absence of exposure to aggressor CD-1 mice [52].
JHU-083 treatment
JHU-083 (Ethyl 2-(2-Amino-4-methylpentanamido)-DON) was prepared as previously described [39]. For all pharmacokinetic and efficacy studies, JHU-083 was administered per oral (p.o.) at a dose of 1.82 mg/kg (1 mg/kg DON molar equivalent) in 50 mM HEPES-buffered saline. After 12 days of CSDS, C57 mice were orally treated with 1.82 mg/kg JHU-083 or vehicle every other day for 12 days, followed by behavioral assays. This treatment duration was chosen based on chronic dose regimens previously shown to be efficacious in the CSDS model using the antidepressants fluoxetine and imipramine [44, 53, 54]. In these studies, daily treatment for 7–28 days reversed CSDS-induced social avoidance behaviors. However, there are other indications that anhedonia-like behavior dissipates without intervention 29 days after CSDS [42]. Thus, we adopted a 12-day treatment regimen after CSDS to ensure the presence of anhedonia-like behaviors in untreated mice. To examine the acute effect of JHU-083 on behavioral phenotypes in CSDS mice, mice were administered a single dose of JHU-083 (1.82 mg/kg, p.o.) or vehicle one day after exposure to 12 days of CSDS. For all manipulations, care was taken to handle animals gently to minimize stress.
In vivo pharmacokinetics
Adult male C57 mice were administered JHU-083 (1.82 mg/kg, p.o.) by gavage and euthanized 0.083, 0.25, 0.5, 1, 3, or 5 h post-dose (n = 3/time point). Whole brain was extracted and blood was collected by cardiac puncture prior to refrigerated centrifugation (3000×g for 10 min) to obtain plasma. All samples were frozen on dry ice and then stored at −80 °C until bioanalysis.
DON bioanalysis
Bioanalysis of plasma and brain tissue to quantify DON concentration was performed as previously described [37, 39]. Briefly, DON was extracted from plasma and brain samples with methanol containing glutamate-d5 (10 μM ISTD) followed by 5 min centrifugation at 16,000×g. Supernatants were aliquoted and dried for 1 h at 45 °C under vacuum. DON was then derivatized by incubation (15 min at 60 °C) with sodium bicarbonate buffer (0.2 M, pH 9.0) and dabsyl chloride (10 mM) in acetone. Derivatized samples were injected and separated on an Agilent 1290 equipped with an Agilent Eclipse plus C18 RRHD 2.1 mm × 100 mm column over a 2.5 min gradient from 20 to 95% acetonitrile + 0.1% formic acid and quantified on an Agilent 6520 QTOF mass spectrometer. Calibration curves over the range of 0.05–50 µM in plasma and brain were constructed from the peak area ratio of the analyte to the internal standard using linear regression with a weighting factor of 1/(nominal concentration). Correlation coefficient of greater than 0.99 was obtained in all analytical runs.
Three-chamber social approach test
A three-chambered apparatus, consisting of 40 cm width × 20 cm height × 26 cm depth with a 12-cm-wide center chamber and 14-cm-wide side chambers, was used for the social approach test as described previously [55–57]. Both side chambers contained a plastic cage in the corner, with a weighted plastic cup placed on top to prevent the subject mouse from climbing. The assay consisted of four sessions. The first session began with 10 min habituation in the center chamber followed by a second 10 min session where the subject mouse could freely explore all three chambers including two side chambers, each with a plastic cage for habituation. In the third session, the mouse was gently confined to the center chamber while a novel intruder C57 male mouse (stranger 1), was placed in one plastic cage in one side chamber. The subject mouse was allowed to freely explore all three chambers for 10 min. Before the last session, the subject mouse was gently guided to the center chamber while another novel intruder C57 male mouse (stranger 2), was placed into another cage in the other side chamber for assessing social novelty recognition. The subject mouse freely explored all three chambers for 10 min. Stranger mice were habituated to the plastic cage for 30 min 24-h before the session day. The positions of the empty cage and stranger 1 cage were alternated between tests to prevent side preference. The plastic cages used in the tests allowed substantial olfactory, auditory, visual and tactile contact between subject mice and stranger mice. Social sniffing was scored from the videotapes for the sum of nose-to-nose sniffing (sniffing or snout contact with the head/neck/mouth area) and nose-to-tail sniffing (sniffing or snout contact with the tail area). Individual movement of the subject mice was analyzed by the researcher who was blinded to the group assignment. The heat maps were generated by Ethovision XT 11.0 (Noldus, Leesburg, VA). The chamber time and total travel distance were analyzed by Topscan 3.0 (CleverSys, Reston, VA) and listed in Supplementary table 1–4.
Sucrose preference test
The sucrose preference test was performed according to a previously described protocol [49, 58]. The mice were singly caged before the test. On day 1, their normal water bottles were replaced with two 50 ml tubes (bottle “A” and bottle “B”) fitted with bottle stoppers containing two-balled sipper tubes. The position of bottles A and B were switched daily to avoid a side bias, and the fluid consumed from each bottle was measured daily. During days 1 and 2, bottles A and B were filled with normal drinking water (W/W). During days 3 and 4, both bottles were filled with a solution of 1.5% sucrose dissolved in drinking water (S/S). On days 5–8, bottle A contained 1.5 % sucrose, and bottle B contained drinking water (S/S). Sucrose preference on each day for each mouse was calculated as 100% x (Vol A/(Vol A + Vol B)) and averaged across the days for a given condition (W/W, S/S, or S/W). The total fluid consumption was also calculated.
Isolation of brain CD11b+ cells
Mice were sacrificed by decapitation and brains were immediately removed. The prefrontal cortex (PFC), hippocampus (HPC), and cerebellum were rapidly dissected on an ice-cold plate. These areas were defined by landmarks and neuroanatomical nomenclature in the atlas of Franklin and Paxinos [59]. PFC was defined as: anteroposterior (AP): +2.57 to +1.53 mm, mediolateral (ML): ±2.75 mm from bregma, dorsoventral (DV): −1.75 to −3.05 mm from the dura, and whole HPC was dissected as: AP: −0.95 to −4.03 mm, ML: ±3.75 mm from bregma, DV: −1.75 to −5.1 mm from the dura according to the atlas. All the brain regions, including whole cerebellum, were bi-lateral dissections. CD11b+ cells were isolated from the prefrontal cortex, hippocampus, and cerebellum homogenates of C57 mice according to a previously described method with minor modification [3]. In brief, the brain tissue was minced in HBSS (cat # 55021C, Sigma-Aldrich, St. Louis, MO, USA) and dissociated with the neural tissue dissociation kits (P) (cat # 130–092–628, MACS Militenyi Biotec, Auburn, CA) according to manufacturer instructions. After passing through a 70 μm cell strainer, resulting homogenates were centrifuged at 300×g for 10 min. Supernatants were removed and cell pellets were resuspended, and myelin was removed by Myelin Removal Beads II (cat # 130-096-733, MACS Militenyi Biotec, Auburn, CA) according to the manufacturer instructions. Myelin-removed cell pellets were resuspended and incubated with CD11b MicroBeads (cat # 130-093-634, MACS Militenyi Biotec, Auburn, CA) for 15 min, loaded on LS columns and separated on a quadroMACS magnet. CD11b+ cells were flushed out from the LS columns, then washed and resuspended in sterile HBSS (cat # 55037C, Sigma-Aldrich, St. Louis, MO). The number of viable cells was determined using a hemacytometer and 0.1% trypan blue staining. Each brain extraction yielded 5 × 105 viable CD11b+ cells. It has been demonstrated that CD11b+ cells isolated from brain homogenates through this antibody-coupled microbeads method are microglia-enriched populations (>95% of isolated cells) [3].
Glutaminase activity
Glutaminase activity measurement in CD11b+ cells and other brain cell fractions was conducted as previously described [27]. Briefly, cells were lysed by sonication in ice-cold potassium phosphate buffer (45 mM, pH 8.2) containing protease inhibitors (cat # 04693116001, Roche). For CD11b+ and non-CD11b+ cells isolated from prefrontal cortex, hippocampus, and cerebellum, lysates were incubated with [3H]-glutamine (0.09 µM, 2.73 µCi) for 180 min at room temperature and the reactions carried out in 50 µl reaction volumes in a 96-well microplate. The reaction was then terminated by addition of imidazole buffer (20 mM, pH 7). 96-well spin columns packed with strong anion ion-exchange resin (cat # 140-1251, Bio-Rad, AG® 1-X2 Resin, 200–400 mesh, chloride form) were used to separate unreacted [3H]-glutamine from [3H]-glutamate. [3H]-glutamate was eluted from the column with 0.1 N HCl and analyzed for radioactivity using Perkin Elmer’s TopCount instrument in conjunction with their 96-well LumaPlates (cat # 6005173). Total protein measurements were carried as per manufacturer’s instructions using BioRad’s Detergent Compatible Protein Assay kit. Counts per minute were converted to fmol and normalized to total protein content. Data are presented as fmol/mg/h.
RNA isolation from CD11b+ cells and quantitative real-time PCR
Total RNA was isolated from purified CD11b+ cells from the cerebrum using RNeasy Mini Kit (QIAGEN). Quantitative real-time PCR (qPCR) was performed using TaqMan manufacturer’s protocol (Applied Biosystems). Briefly, cDNA from isolated CD11b+ cells was prepared by SuperScript® III CellsDirect cDNA Synthesis Kit (Life Technologies) from total RNA in the range of 10–100 ng. Real-time PCR reaction contained diluted cDNA from the synthesis reaction and 200 nM specific forward and reverse TaqMan primers specific to targets of interest (Assay IDs for TNF-α, IL-6, IL-1β, and Glutaminase: Mm00443260_g1, Mm00446190_m1, Mm00434228_m1, and Mm01257297_m1, respectively) (Applied Biosystems). Primers for GAPDH were used to normalize the expression data. The real-time PCR reaction and measurement was carried out with Applied Biosystems PRISM 7900 HT. PCR reaction conditions are as follows: 50 °C, 2 min; 95 °C, 10 min; 50 cycles of 95 °C, 15 s and 60 °C, 1 min, including a dissociation curve at the last step to verify single amplicon in the reaction.
Enzyme-linked immunosorbent assay (ELISA)
Serum were collected from chronic vehicle/JHU-083 treated mice and pro-inflammatory cytokines, including TNF-α (cat # MTA00B, R&D Systems, Minneapolis, MN), IL-1β (cat # MLB00C, R&D Systems, Minneapolis, MN), and IL-6 (cat # M6000B, R&D Systems, Minneapolis, MN), were measured with commercially available ELISA kits according to manufacturer’s protocols.
Statistical analyses
For pharmacokinetic analysis, the area under the curve to the end of sample collection (AUC0–5h), maximum concentration (Cmax), and time to Cmax (Tmax) were calculated by GraphPad Prism (version 7, GraphPad Software, La Jolla, CA). Multiple-group comparisons between mice subjected to CSDS and control mice as well as JHU-083 and vehicle treatment were performed using two-way analyses of variance (ANOVA) (CSDS × drug treatment) followed by the Tukey’s post-hoc test. The Student’s t-test was used in comparing two sets of data. For sucrose preference test analysis, three-way repeated measures ANOVA followed by Bonferroni’s post-hoc comparisons was used. A value of p < 0.05 in two-tailed test was considered statistically significant. All data are presented as the mean ± standard error of the mean (SEM).
Unbiased assessment in experimental procedures
Experiments including behavioral characterization and glutaminase activity measurement were performed by multiple investigators. Investigators conducted these assays without information of the identification of either JHU-083 or vehicle-treated mice.
Results
JHU-083 delivers DON to the brain after oral administration in mice
JHU-083 (1.82 mg/kg, p.o.) delivered peak brain concentrations (Cmax) of 0.85 nmol/g DON within 0.5 h (Fig. 1a, b). By 5-h post-administration, DON was almost completely cleared from brain and plasma, resulting in respective AUC values of 2.26 and 4.10 h nmol/ml, and a brain/plasma ratio of 0.55 (Fig. 1b). Similar brain concentrations of DON were achieved by repeated administration of 0.6–1.6 mg/kg DON in previous studies demonstrating efficacy in models of CNS diseases associated with excess extracellular glutamate including multiple sclerosis, brain cancer, and sindbus virus infection [36, 38–40, 60]. Thus, administration of JHU-083 (1.82 mg/kg, p.o) was chosen for subsequent experiments in this study.
Fig. 1.

In vivo brain and plasma pharmacokinetics of compound DON following oral administration of JHU-083 in mice. a Chemical structure of JHU-083 is shown. b JHU-083 (1.82 mg/kg, p.o., q.o.d.) exhibited sufficient oral bioavailability and favorable brain penetration for efficacy testing in mice. Data are presented as the mean ± SEM. p.o., per os; q.o.d, every other day; Cmax, maximum concentration observed; Tmax, time at which the maximum concentration is observed; AUC, area under the concentration-time curve; AUC0-5h, AUC up to the last measurable concentration
Chronic treatment with JHU-083 ameliorates social avoidance behavior and anhedonia-like behavior induced by CSDS
Mice were exposed to CSDS and subsequently administered JHU-083 or vehicle every other day for 12 days (Fig. 2a). To examine the effect of JHU-083 treatment on behavioral phenotypes induced by CSDS, we first tested the mice in the three-chambered social approach test, where the social approach of a mouse toward a stranger mouse trapped in a wire cage was measured [55–57]. Control mice exhibited significant preference for exploring a stranger mouse (stranger 1) relative to an empty cage, as measured by the total amount of time spent in each chamber and sniffing each cage (two-tailed Student’s t-test, p = 0.0025, Fig. 3a; p = 0.0007, Fig. 3b). Consistent with previous reports, CSDS resulted in a social avoidance phenotype [61–63]; mice in the CSDS group did not show preference for exploring the stranger mouse relative to the empty cage (two-tailed Student’s t-test, p = 0.1523, Fig. 3a; p = 0.1189, Fig. 3b). Chronic treatment of JHU-083 normalized observed social avoidance behaviors in mice subjected to CSDS (two-tailed Student’s t-test, p = 0.0036, Fig. 3a; p = 0.0035, Fig. 3b), while having no effect on sociability in control mice (two-tailed Student’s t-test, p = 0.0005, Fig. 3a; p = 0.0058, Fig. 3b). When another stranger mouse (stranger 2) was placed into the empty cage, control mice showed significant preference for exploring stranger 2 relative to stranger 1, as measured by the total amount of time spent in each chamber and time spent sniffing (two-tailed Student’s t-test, p = 0.0010, Fig. 3c; p = 0.0444, Fig. 3d). In contrast, mice in the CSDS group did not exhibit preference for exploring stranger 2 relative to stranger 1 (two-tailed Student’s t-test, p = 0.8379, Fig. 3c; p = 0.6016, Fig. 3d). Chronic treatment of JHU-083 had no effect on observed social novelty recognition deficits in mice subjected to CSDS (two-tailed Student’s t-test, p = 0.3834, Fig. 3c; p = 0.8429, Fig. 3d), and had no effect on the social novelty recognition of control mice (two-tailed Student’s t test, p = 0.0016, Fig. 3c; p = 0.0353, Fig. 3d). These effects were unlikely to be related to non-specific changes in locomotion as no alterations were observed in the distance traveled in all chambers during the test phases (Supplementary Table 1–2). These results suggest that chronic treatment with JHU-083 can improve CSDS-induced impairment of social motivation, but has no effect on the altered preference for social novelty induced by CSDS.
Fig. 2.
Experimental timeline for CSDS followed by chronic and acute administration of JHU-083. a Male 7-to 8-week-old C57BL/6J (C57) mice were exposed to chronic social defeat stress (CSDS) for 12 days and were subsequently treated with JHU-083 (1.82 mg/kg, p.o.) or vehicle every other day for 12 days. One day after the completed treatment of JHU-083 or vehicle, behavioral characterization was conducted by social interaction test (SIT) followed by sucrose preference test (SPT). Brain extraction for biochemical experiments was also performed one day after JHU-083 treatment. b To explore acute effects of JHU-083, mice were administered a single dose of JHU-083 (1.82 mg/kg, p.o.) or vehicle one day after exposure to 12 days of CSDS which was followed by behavioral assessments
Fig. 3.

Chronic JHU-083 treatment ameliorates deficits in social behavior and anhedonia-associated behavior induced by CSDS. a, b CSDS impairs sociability assessed by time spent in each chamber and time spent sniffing the wire cage in three-chambered social interaction test. Chronic treatment of JHU-083 (1.82 mg/kg, p.o.) alleviates social avoidance induced by CSDS, but has no effect of JHU-083 on sociability in control mice (Ctr). **p < 0.01, determined by Student’s t-test. Data are presented as the mean ± SEM. c, d CSDS leads to reduced preference for social novelty assessed by measurement of time spent in each chamber and time spent sniffing the stranger 1 and stranger 2 in three-chambered social interaction test. Chronic treatment of JHU-083 (1.82 mg/kg, p.o.) has no effect on altered preference for social novelty induced by CSDS, and none on social novelty recognition in control mice. *p < 0.05, **p < 0.01, determined by Student’s t-test. a–d Representative heat map images (lower panels) represent movements of the CSDS mice that treated with vehicle or JHU-083. e CSDS induced anhedonia-associated behavior reflected by decreased sucrose consumption when given a choice between 1.5% sucrose and water. JHU-083 rescued CSDS-induced sucrose preference deficit, while having no effect on sucrose preference in control mice. CSDS + vehicle vs. CSDS + JHU-083: **p < 0.01, by three-way repeated measures ANOVA followed by Bonferroni’s post-hoc comparisons. a–e Data are presented as the mean ± SEM. Ctr+ vehicle, n = 6; Ctr + JHU-083, n = 6; CSDS + vehicle, n = 11; CSDS + JHU-083, n = 10
Mice subjected to CSDS have repeatedly been shown to exhibit reduced sucrose preference, an effect associated with stress-induced anhedonia [49]. To examine the effect of JHU-083 treatment on anhedonia-like behavior, control and CSDS mice were subjected to an 8-day sucrose preference test. Among all groups of mice, there were no differences of fluid consumption between bottles A and B when filled with either normal drinking water (W/W, days 1 and 2) or 1.5% sucrose solution (S/S, days 3 and 4), suggesting that there was no side preference of solution consumption during test days 1–4. On days 5–8, control mice showed significant preference for the sucrose solution when compared with water consumption (two-tailed Student’s t-test, p < 0.01). Three-way repeated measures ANOVA revealed a CSDS x drug treatment interaction on the sucrose preference (S/W) on days 5–8 (F(1,29) = 5.873, p = 0.022). In agreement with previous studies [49], post-hoc tests indicated that mice in the CSDS group exhibited decreased sucrose solution consumption compared with those in controls (p = 0.0028, 0.0155, and 0.0132 for days 6–8, respectively), with no differences in total fluid intake (Supplementary Figure 1a), suggesting decreased sucrose preference induced by CSDS. Treatment with JHU-083 markedly restored sucrose consumption in the CSDS group (p = 0.0039, 0.0003, 0.0016, and 0.0018 for days 5–8, respectively, Fig. 3e), whereas no change was observed in controls (p = 0.7687, 0.9933, 0.9866, and 0.9871 for days 5–8, respectively), suggesting that JHU-083 can rescue the anhedonia-like behavior phenotype induced by CSDS.
Acute treatment with JHU-083 improves social avoidance behavior, but not anhedonia-like behavior induced by CSDS
Based on observations that a single administration of ketamine can induce a rapid antidepressant effect [19] with a response linked to altered glutamate signaling [12, 15–18], we wanted to explore the acute effects of JHU-083 on behavioral phenotypes in CSDS mice. Mice were administered a single dose of JHU-083 (1.82 mg/kg, p.o.) or vehicle one day after exposure to twelve days of CSDS (Fig. 2b). In the three-chambered social approach test, acute treatment of JHU-083 reversed social avoidance behaviors caused by CSDS (two-tailed Student’s t-test, p = 0.0108, Fig. 4a; p < 0.0001, Fig. 4b), while having no effect on sociability in control mice (two-tailed Student’s t-test, p < 0.0001, Fig. 4a, b). We also found that acute treatment of JHU-083 had no effect on social novelty recognition deficits in mice subjected to CSDS (two-tailed Student’s t-test, p = 0.8606, Fig. 4c; p = 0.2090, Fig. 4d), and had no effect on the social novelty recognition of control mice (two-tailed Student’s t-test, p = 0.0029, Fig. 4c; p = 0.0138, Fig. 4d). These effects were unlikely to be related to non-specific changes in locomotion as no alterations were observed in the distance traveled in all chambers during the test phases (Supplementary Table 3–4). These results suggest that acute treatment with JHU-083 can improve CSDS-induced impairment of social motivation, but has no effect on the altered preference for social novelty induced by CSDS.
Fig. 4.

Acute JHU-083 treatment ameliorates deficits in social behavior, but not anhedonia-associated behavior induced by CSDS. a, b Acute treatment of JHU-083 (1.82 mg/kg, p.o.) one day after CSDS alleviates social avoidance induced by CSDS, but has no effect on sociability in control mice (Ctr). **p < 0.01, determined by Student’s t-test. c, d CSDS leads to reduced preference for social novelty assessed by measurement of time spent in each chamber and time spent sniffing stranger 1 and stranger 2 mouse in the three-chambered social interaction test. Acute treatment of JHU-083 (1.82 mg/kg, p.o.) has no effect on altered preference for social novelty induced by CSDS nor on social novelty recognition in control mice. *p < 0.05, **p < 0.01, determined by Student’s t-test. a–d Representative heat map images (lower panels) represent movements of the CSDS mice treated with vehicle or JHU-083. e CSDS induced anhedonia-associated behavior reflected by decreased sucrose consumption when given a choice between 1.5% sucrose and water. JHU-083 did not rescue CSDS-induced sucrose preference deficit nor have effect on sucrose preference in control mice. CSDS + Vehicle vs. CSDS + JHU-083: **p < 0.01, by three-way repeated measures ANOVA followed by Bonferroni’s post-hoc comparisons. a–e Data are presented as the mean ± SEM. Ctr + vehicle, n = 10; Ctr + JHU-083, n = 10; CSDS + vehicle, n = 11; CSDS + JHU-083, n = 11
To examine the acute effect of JHU-083 treatment on anhedonia-like behavior, control and CSDS mice were subjected to an 8-day sucrose preference test as described above. There was no difference in total fluid intake among groups (Supplementary Figure 1b). In contrast to the treatment effect of chronic administration of JHU-083, a single dose of JHU-083 administered on the day before initiating the 8-day sucrose preference test did not restore sucrose consumption in the CSDS group (p = 0.7378, 0.0819, 0.8775, and 0.6616 for days 5–8, respectively, Fig. 4e), nor affect controls (p = 0.8786, >0.9999, 0.9754, and >0.9999 for days 5–8, respectively).
JHU-083 inhibits increased glutaminase activity in isolated CD11b+ cells, but not other cells in the prefrontal cortex and hippocampus of mice subjected to CSDS
We next explored whether CSDS upregulates glutaminase activity and whether its activity is suppressed by JHU-083. Microglia-enriched CD11b+ cells [3, 64] and non-CD11b+ cells were isolated from specific brain regions including the prefrontal cortex and hippocampus, which are associated with depression-related behavioral phenotypes in the CSDS model [54, 65–67]. Protein was then extracted from these cells and glutaminase activity was measured. These values were compared to those obtained from microglia-enriched CD11b+ cells isolated from the cerebellum. Two-way ANOVA revealed a CSDS x drug treatment interaction on the glutaminase activity in the CD11b+ cells in the prefrontal cortex (F(1,12) = 49.73, p < 0.0001) and hippocampus (F(1,12) = 56.93, p < 0.0001). Post-hoc tests showed that mice exposed to CSDS exhibited a significant increase in glutaminase activity in these brain areas (p < 0.0001, Fig. 5a, b), but not in the cerebellum (p = 0.8964, Fig. 5c). Up-regulated glutaminase activity was completely normalized by JHU-083 treatment in CSDS mice (p < 0.0001, Fig. 5a, b), whereas JHU-083 treatment had no effect in the prefrontal cortex and down-regulated effect in the hippocampus in control mice (p = 0.0781, Fig. 5a; p < 0.0001, Fig. 5b). In non-CD11b+ cells, there was no effect of CSDS or JHU-083 treatment on glutaminase activity in the prefrontal cortex (CSDS x drug treatment interaction (F (1,12) = 0.06303, p = 0.8060), Fig. 5d) or hippocampus (CSDS x drug treatment interaction (F (1,12) = 0.01615, p = 0.9010), Fig. 5e), whereas glutaminase activity slightly increased in the cerebellum (p = 0.0154, Fig. 5f).
Fig. 5.

JHU-083 attenuates CSDS-induced increase in glutaminase activity in CD11b+ cells in the prefrontal cortex and hippocampus, but not in the cerebellum. a–c CSDS induced significant elevation in ex vivo glutaminase activity specifically in CD11b+ cells in the prefrontal cortex (PFC) and hippocampus (HPC), but not in the cerebellum, that was completely normalized by JHU-083 administration (1.82 mg/kg, p.o., q.o.d.). There was no effect of JHU-083 on glutaminase activity in CD11b+ cells harvested from the prefrontal cortex in control mice, whereas down-regulated effect in the hippocampus in control mice (Ctr). d–f There was no effect of CSDS or JHU-083 treatment on glutaminase activity in non-CD11b+ cells in the prefrontal cortex and hippocampus. Expression of glutaminase was slightly increased in the cerebellum by CSDS. Bars represent means of each group in three independent experiments. Ctr + Vehicle vs CSDS + Vehicle: **p < 0.01; CSDS + Vehicle vs CSDS + JHU-083: ##p < 0.01, comparison between groups by two-way ANOVA with Tukey’s post-hoc test. Data are presented as the mean ± SEM
JHU-083 suppresses CSDS-induced upregulation of proinflammatory cytokines in CD11b+ cells
Previous studies have demonstrated that CSDS promotes activation of CD11b+ cells, leading to increased expression of proinflammatory cytokine in the brain, including IL-6, TNF-α, and IL-1β [43, 62, 64]. Two-way ANOVA revealed a CSDS x drug treatment interaction on the mRNA expression of IL-1β (F(1,8) = 5.441, p = 0.0480) and an interaction trend of TNF-α (F(1,8) = 5.103, p = 0.0538), but no interaction effect on IL-6 (F(1,8) = 0.1235, p = 0.7344) in CD11b+ cells. Post-hoc analysis showed that CSDS led to increased expression of IL-1β and TNF-α, but not IL-6 in CD11b+ cells (p = 0.0370, Fig. 6a; p = 0.0472, Fig. 6b; p = 0.9993, Fig. 6c). We also observed that JHU-083 treatment suppressed the CSDS-induced upregulation of IL-1β and TNF-α expression (p = 0.0465, Fig. 6a; p = 0.0483, Fig. 6b), while having no effect on the IL-1β and TNF-α expression in CD11b+ cells isolated from control mice (p > 0.9999, Fig. 6a, b). JHU-083 treatment had no effect on expression of IL-6 in CD11b+ cells isolated from either CSDS mice or control mice (CSDS x drug treatment (F (1,8) = 0.1235, p = 0.7344), Fig. 6c). There was no effect of CSDS or JHU-083 treatment on mRNA expression of glutaminase in CD11b+ cells (Fig. 6d).
Fig. 6.
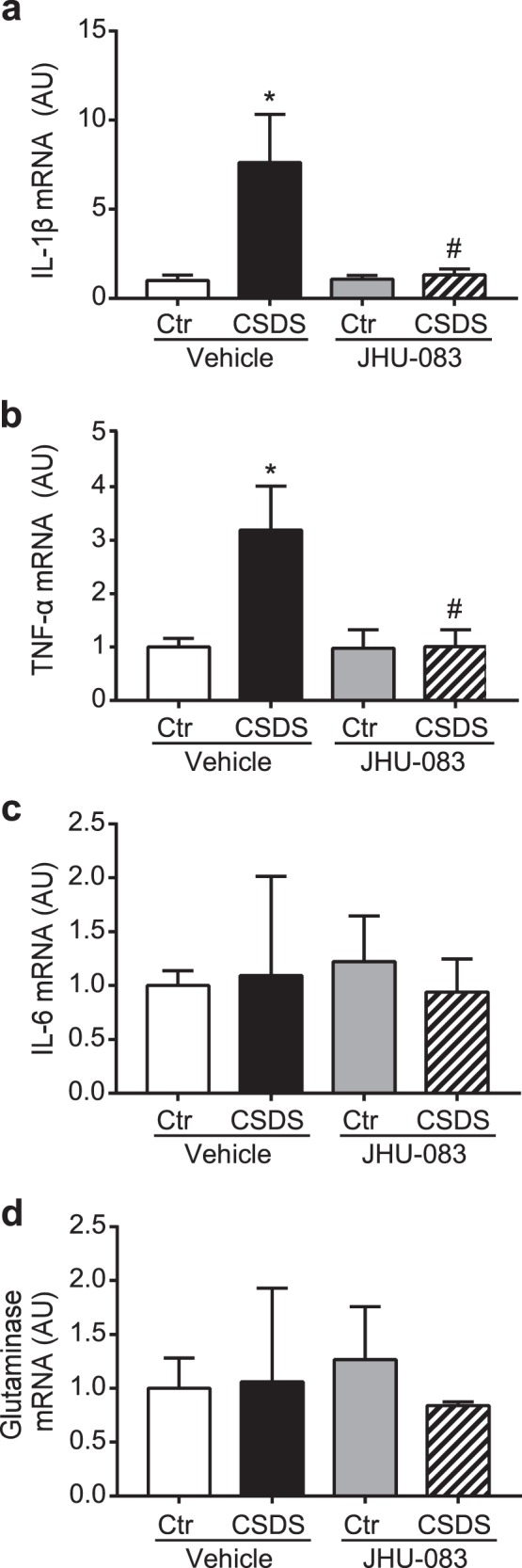
JHU-083 attenuates CSDS-induced inflammatory activation of CD11b+ cells. a–c CSDS induces increased mRNA expression of TNF-α and IL-1β, but not IL-6 in CD11b+ cells assessed by Quantitative real-time PCR (qPCR). Chronic treatment of JHU-083 (1.82 mg/kg, p.o.) suppresses increased TNF-α and IL-1β induced by CSDS, whereas there is no effect of JHU-083 on expression of inflammatory cytokines in CD11b+ cells isolated from control mice (Ctr). d No effect of CSDS or JHU-083 treatment on mRNA expression of glutaminase in CD11b+ cells. Neither CSDS nor chronic treatment of JHU-083 (1.82 mg/kg, p.o.) affected mRNA expression of glutaminase in CD11b+ cells assessed by qPCR. Bars represent means of each group in three independent experiments. Ctr + Vehicle vs CSDS + Vehicle: *p < 0.05; CSDS + Vehicle vs. CSDS + JHU-083: #p < 0.05, comparison between groups by two-way ANOVA with Tukey’s post-hoc test. Data are presented as the mean ± SEM
Previous reports including ours supported DON’s peripheral anti-inflammatory effects with other CNS insults including reducing peripheral lymphocyte proliferation and inhibiting immune cells infiltrating to CNS [35, 36, 38]. Thus, we examined the effect of JHU-083 on peripheral pro-inflammatory cytokines, including TNF-a, IL-6, and IL-1β, the source of which is primarily circulating CD11b+ cells [43, 68]. We found that while CSDS increased the level of TNF-a, IL-6, and IL-1β in serum, chronic administration of JHU-083 dramatically inhibited this increase, suggesting a peripheral anti-inflammatory effect of JHU-083 (Supplementary Figure 2).
Discussion
In the present study, we demonstrate that chronic treatment of JHU-083, a prodrug of the glutaminase inhibitor DON, reversed social avoidance and anhedonia-like behaviors in mice subjected to CSDS, and normalized glutaminase hyperactivity and pro-inflammatory cytokine induction specifically in CD11b+ cells in the brain. These results provide novel pharmacological evidence supporting an emerging link between glutamatergic dysfunction and neuroinflammation in the pathology of MDD [22].
It is known that currently approved antidepressants, such as imipramine and fluoxetine, that act on monoaminergic systems require chronic administration before efficacy is observed in the CSDS model [69–71]. Many reports have showed that CSDS induces molecular and circuit abnormalities including alterations in BDNF-TrkB signaling, ΔFosB signaling, and prefrontal cortex-amygdala circuitry [63, 65, 72], consistent with studies of MDD patients [73–76]. Importantly, CSDS mice also respond to the rapid-acting antidepressant ketamine [47, 77, 78], which is known to be efficacious even in treatment-resistant patients and to depend on alterations in glutamate homeostasis [79–81]. Thus, while CSDS mice are not exclusively a model of treatment-resistant depression, they do exhibit predictive validity for one of the few pharmacotherapies effective at treating refractory depression through a mechanism distinct from monoaminergic agents. In addition to the chronic treatment effect of JHU-083 in CSDS model, our results demonstrated that acute treatment with JHU-083 rescues social avoidance induced by CSDS. By similarly targeting glutamate signaling with JHU-083, the antidepressant response achieved in CSDS mice may translate to efficacy in clinical scenarios similar to those that respond to ketamine, possibly including treatment-resistance. Collectively, CSDS-induced behavioral abnormalities may be relevant to human psychopathology and thus JHU-083 may have clinical benefit as an antidepressant with a novel mechanism of action.
Our results also suggest that chronic treatment with JHU-083 improves CSDS-induced impairment of social motivation, but has no effect on altered preference for social novelty induced by CSDS. Recent studies suggest that these two behavioral domains are subserved by distinct neural circuits [82] and are dissociable through modulation of glutamatergic function [83]. Thus, JHU-083 may act to specifically normalize social motivation, the mechanisms of which warrant future investigation. It should also be noted that, although our experimental paradigm for assessing sociability and social novelty is a well-established standardized approach [55–57], conspecific mouse vs non-animate object comparisons may more precisely reflect novel social vs. non-social object preferences during the “sociability” session [84].
Despite the recent focus on aberrant glutamate homeostasis in MDD and stress-related disorders, to the best of our knowledge, this is the first study demonstrating that social stress elevates glutaminase activity in the prefrontal cortex and hippocampus. Consistent with high expression of glutaminase in neurons and astrocytes [85, 86], we observed glutaminase activity is higher in non-CD11b+ cells compared with CD11b+ cells at baseline. Surprisingly, however, CSDS was found to specifically elevate glutaminase activity in microglia-enriched CD11b+ cells, but not in non-CD11b+ cells. In addition, our DON prodrug was found to completely normalize the glutaminase activity in CD11b+ cells, while having no effect on non-CD11b+ cells. Consistently, recent studies demonstrated the importance of microglia-associated mechanisms for stress-induced behavioral outcomes in mice models [64, 87, 88]. These results support our hypothesis that specific upregulation of glutaminase activity in these immune cells in the prefrontal cortex and hippocampus, critical for mood regulation, may contribute to the observed depression-associated phenotypes in CSDS mice, although the mechanism by which CSDS selectively activates glutaminase in CD11b+ cells remains elusive. It should be noted, however, that brain CD11b+ cells contain not only microglia, but also other cells such as perivascular macrophages and infiltrating monocytes [64]. Thus, the importance of microglial glutaminase activity in CSDS-induced behaviors should be investigated at a more detailed mechanistic level. It is also important to consider that glutaminase activity in other brain regions such as the amygdala and nucleus accumbens which are also crucial for mood regulation [65, 89–91] may be involved in behavioral phenotypes in the CSDS model. However, these individual brain regions are too small to yield sufficient numbers of CD11b+ cells to produce enough glutaminase protein to generate a detectable signal in our activity assay.
In addition to altered glutamate signaling in MDD, clinical evidence from postmortem brain studies, neuroimaging studies, as well as studies using cerebrospinal fluid and blood, demonstrate that altered inflammation signaling is, at least in part, involved in MDD pathology [92–94]. Many groups have reported similar peripheral and CNS inflammatory alteration in the CSDS model, including microglia activation, monocyte trafficking, and increased pro-inflammatory cytokine levels [43, 62, 64]. Based on the idea that aberrant glutamate signaling and neuroinflammation may converge to affect synaptic function and behavioral abnormalities relevant to depression [5, 6, 22], Haroon et al. proposed that stress-induced inflammation may exaggerate release and inhibit clearance of glutamate from microglia and astrocytes, resulting in an increase of extracellular glutamate that ultimately leads to synaptic dysfunction underlying depressive phenotypes [22]. In agreement with this hypothesis, our results showed that CSDS simultaneously induces an increase of proinflammatory cytokines IL-1β and TNF-α and augment glutaminase activity in CD11b+ cells, effects which are suppressed by JHU-083. Nonetheless, the downstream mechanisms by which normalization of glutaminase activity in CD11b+ cells ameliorate CSDS pathology are still unclear. Further investigation to address whether the effect of JHU-083 on glutamate production precedes or follows its effect on inflammatory cytokine production will be important to understand the mechanisms underlying the antidepressant effect of JHU-083. It should also be noted that although these experiments were performed in male mice, sex differences associated with stress and MDD have been reported [43, 95], warranting examination of sex-specific social stress effects on glutaminase activity and JHU-083 efficacy.
Taken together, our findings provide proof-of-principle that pharmacological intervention targeting glial glutaminase activity may be useful for development of a novel MDD treatment. Given that many clinical trials testing glutamate modulators have failed to show beneficial effects in humans [96–98], further studies are needed to better understand the mechanisms underlying the potential antidepressant effect of JHU-083.
Electronic supplementary material
Acknowledgements
We are grateful for the commitment and involvement of Shota Iwaoka, Tomonori Matsushita, and Tiffany Green. We thank Dr. Hironori Kuga for advice on statistical analysis. We also thank Dr. Yan Jouroukhin and Chantelle Terrillion for technical support in the behavior tests that were performed at the Behavioral Core of the School of Medicine of Johns Hopkins University. We thank Mr. Kamal Abdi for providing support in animal maintenance throughout the study. This research was supported by multiple grants including R01DA041208 (A.K.), R21AT008547 (A.K.), P50MH094268 (A.K.), Catalyst award (A.K.), The Brain and Behavior Research Foundation (A.K., A.S.), P30MH075673 (B.S.S.), R01CA193895 (B.S.S.), Bloomberg Kimmel Institute for Cancer Immunotherapy (B.S.S.), R25MH080661 Pilot award (X.Z.), Postdoctoral Fellowship in Pharmacology/Toxicology from the PhRMA foundation (M.T.N.), and TEDCO Maryland Innovation Initiative grant award (B.S.S.) as well as the Institute of Organic Chemistry and Biochemistry of the Academy of Sciences of the Czech Republic, v.v.i (RVO 61388963).
Competing interests
The authors declare no competing interests.
Footnotes
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
These authors contributed equally: Xiaolei Zhu, Michael T. Nedelcovych.
Change history
3/13/2019
The original version of this Article omitted the following from the Acknowledgements: Under a license agreement between Dracen Pharmaceuticals, Inc. and the Johns Hopkins University, the University, Dr. Slusher, Dr. Rais, and Mr. Alt are entitled to royalty distributions related to technology used in the research described in this publication. In addition, Dr. Slusher, Dr. Majer and Dr. Rais are founders of and hold equity in Dracen Pharmaceuticals, Inc. This arrangement has been reviewed and approved by the Johns Hopkins University in accordance with its conflict of interest policies.
Contributor Information
Barbara S. Slusher, Phone: +1-410-614-0662, Email: bslusher@jhmi.edu
Atsushi Kamiya, Phone: +1-410-502-0060, Email: akamiya1@jhmi.edu.
Electronic supplementary material
Supplementary Information accompanies this paper at (10.1038/s41386-018-0177-7).
References
- 1.Kessler RC, Berglund P, Demler O, Jin R, Koretz D, Merikangas KR, et al. The epidemiology of major depressive disorder: results from the National Comorbidity Survey Replication (NCS-R) JAMA. 2003;289:3095–105. doi: 10.1001/jama.289.23.3095. [DOI] [PubMed] [Google Scholar]
- 2.Mathew SJ, Manji HK, Charney DS. Novel drugs and therapeutic targets for severe mood disorders. Neuropsychopharmacology. 2008;33:2080–92. doi: 10.1038/sj.npp.1301652. [DOI] [PubMed] [Google Scholar]
- 3.Sharma K, Schmitt S, Bergner CG, Tyanova S, Kannaiyan N, Manrique-Hoyos N, et al. Cell type- and brain region-resolved mouse brain proteome. Nat Neurosci. 2015;18:1819–31. doi: 10.1038/nn.4160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Krystal JH, Sanacora G, Duman RS. Rapid-acting glutamatergic antidepressants: the path to ketamine and beyond. Biol Psychiatry. 2013;73:1133–41. doi: 10.1016/j.biopsych.2013.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Murrough JW, Abdallah CG, Mathew SJ. Targeting glutamate signalling in depression: progress and prospects. Nat Rev Drug Discov. 2017;16:472–86. doi: 10.1038/nrd.2017.16. [DOI] [PubMed] [Google Scholar]
- 6.Reus GZ, de Moura AB, Silva RH, Resende WR, Quevedo J (2017). Resilience dysregulation in major depressive disorder: focus on glutamatergic imbalance and microglial activation. Curr Neuropharmacol. 2018;16:297-307. [DOI] [PMC free article] [PubMed]
- 7.Hashimoto K, Bruno D, Nierenberg J, Marmar CR, Zetterberg H, Blennow K, et al. Abnormality in glutamine-glutamate cycle in the cerebrospinal fluid of cognitively intact elderly individuals with major depressive disorder: a 3-year follow-up study. Transl Psychiatry. 2016;6:e744. doi: 10.1038/tp.2016.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kim JS, Schmid-Burgk W, Claus D, Kornhuber HH. Increased serum glutamate in depressed patients. Arch Psychiatr Nervenkr. 1982;232:299–304. doi: 10.1007/BF00345492. [DOI] [PubMed] [Google Scholar]
- 9.Levine J, Panchalingam K, Rapoport A, Gershon S, McClure RJ, Pettegrew JW. Increased cerebrospinal fluid glutamine levels in depressed patients. Biol Psychiatry. 2000;47:586–93. doi: 10.1016/S0006-3223(99)00284-X. [DOI] [PubMed] [Google Scholar]
- 10.Umehara H, Numata S, Watanabe SY, Hatakeyama Y, Kinoshita M, Tomioka Y, et al. Altered KYN/TRP, Gln/Glu, and Met/methionine sulfoxide ratios in the blood plasma of medication-free patients with major depressive disorder. Sci Rep. 2017;7:4855. doi: 10.1038/s41598-017-05121-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Haroon E, Fleischer CC, Felger JC, Chen X, Woolwine BJ, Patel T, et al. Conceptual convergence: increased inflammation is associated with increased basal ganglia glutamate in patients with major depression. Mol Psychiatry. 2016;21:1351–7. doi: 10.1038/mp.2015.206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Milak MS, Proper CJ, Mulhern ST, Parter AL, Kegeles LS, Ogden RT, et al. A pilot in vivo proton magnetic resonance spectroscopy study of amino acid neurotransmitter response to ketamine treatment of major depressive disorder. Mol Psychiatry. 2016;21:320–7. doi: 10.1038/mp.2015.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Taylor R, Neufeld RW, Schaefer B, Densmore M, Rajakumar N, Osuch EA, et al. Functional magnetic resonance spectroscopy of glutamate in schizophrenia and major depressive disorder: anterior cingulate activity during a color-word Stroop task. NPJ Schizophr. 2015;1:15028. doi: 10.1038/npjschz.2015.28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kristiansen LV, Meador-Woodruff JH. Abnormal striatal expression of transcripts encoding NMDA interacting PSD proteins in schizophrenia, bipolar disorder and major depression. Schizophr Res. 2005;78:87–93. doi: 10.1016/j.schres.2005.06.012. [DOI] [PubMed] [Google Scholar]
- 15.Jaso BA, Niciu MJ, Iadarola ND, Lally N, Richards EM, Park M, et al. Therapeutic modulation of glutamate receptors in major depressive disorder. Curr Neuropharmacol. 2017;15:57–70. doi: 10.2174/1570159X14666160321123221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lener MS, Niciu MJ, Ballard ED, Park M, Park LT, Nugent AC, et al. Glutamate and gamma-aminobutyric acid systems in the pathophysiology of major depression and antidepressant response to ketamine. Biol Psychiatry. 2017;81:886–97. doi: 10.1016/j.biopsych.2016.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Quiroz JA, Tamburri P, Deptula D, Banken L, Beyer U, Rabbia M, et al. Efficacy and safety of basimglurant as adjunctive therapy for major depression: a randomized clinical trial. JAMA Psychiatry. 2016;73:675–84. doi: 10.1001/jamapsychiatry.2016.0838. [DOI] [PubMed] [Google Scholar]
- 18.Sanacora G, Zarate CA, Krystal JH, Manji HK. Targeting the glutamatergic system to develop novel, improved therapeutics for mood disorders. Nat Rev Drug Discov. 2008;7:426–37. doi: 10.1038/nrd2462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zarate CA, Jr., Singh JB, Carlson PJ, Brutsche NE, Ameli R, Luckenbaugh DA, et al. A randomized trial of an N-methyl-D-aspartate antagonist in treatment-resistant major depression. Arch General Psychiatry. 2006;63:856–64. doi: 10.1001/archpsyc.63.8.856. [DOI] [PubMed] [Google Scholar]
- 20.Miller AH, Raison CL. The role of inflammation in depression: from evolutionary imperative to modern treatment target. Nat Rev Immunol. 2016;16:22–34. doi: 10.1038/nri.2015.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sanacora G, Banasr M. From pathophysiology to novel antidepressant drugs: glial contributions to the pathology and treatment of mood disorders. Biol Psychiatry. 2013;73:1172–9. doi: 10.1016/j.biopsych.2013.03.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Haroon E, Miller AH, Sanacora G. Inflammation, glutamate, and glia: a trio of trouble in mood disorders. Neuropsychopharmacology. 2017;42:193–215. doi: 10.1038/npp.2016.199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pitt D, Werner P, Raine CS. Glutamate excitotoxicity in a model of multiple sclerosis. Nat Med. 2000;6:67–70. doi: 10.1038/71555. [DOI] [PubMed] [Google Scholar]
- 24.Werner P, Pitt D, Raine CS. Multiple sclerosis: altered glutamate homeostasis in lesions correlates with oligodendrocyte and axonal damage. Ann Neurol. 2001;50:169–80. doi: 10.1002/ana.1077. [DOI] [PubMed] [Google Scholar]
- 25.Miller AH, Maletic V, Raison CL. Inflammation and its discontents: the role of cytokines in the pathophysiology of major depression. Biol Psychiatry. 2009;65:732–41. doi: 10.1016/j.biopsych.2008.11.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Maezawa I, Jin LW. Rett syndrome microglia damage dendrites and synapses by the elevated release of glutamate. J Neurosci. 2010;30:5346–56. doi: 10.1523/JNEUROSCI.5966-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Thomas AG, O’Driscoll CM, Bressler J, Kaufmann W, Rojas CJ, Slusher BS. Small molecule glutaminase inhibitors block glutamate release from stimulated microglia. Biochem Biophys Res Commun. 2014;443:32–36. doi: 10.1016/j.bbrc.2013.11.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Potter MC, Figuera-Losada M, Rojas C, Slusher BS. Targeting the glutamatergic system for the treatment of HIV-associated neurocognitive disorders. J NeuroImmune Pharmacol. 2013;8:594–607. doi: 10.1007/s11481-013-9442-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Takeuchi H, Jin S, Wang J, Zhang G, Kawanokuchi J, Kuno R, et al. Tumor necrosis factor-alpha induces neurotoxicity via glutamate release from hemichannels of activated microglia in an autocrine manner. J Biol Chem. 2006;281:21362–8. doi: 10.1074/jbc.M600504200. [DOI] [PubMed] [Google Scholar]
- 30.Jayakumar AR, Rao KV, Murthy Ch R, Norenberg MD. Glutamine in the mechanism of ammonia-induced astrocyte swelling. Neurochem Int. 2006;48:623–8. doi: 10.1016/j.neuint.2005.11.017. [DOI] [PubMed] [Google Scholar]
- 31.Kostic M, Zivkovic N, Stojanovic I. Multiple sclerosis and glutamate excitotoxicity. Rev Neurosci. 2013;24:71–88. doi: 10.1515/revneuro-2012-0062. [DOI] [PubMed] [Google Scholar]
- 32.Gaisler-Salomon I, Miller GM, Chuhma N, Lee S, Zhang H, Ghoddoussi F, et al. Glutaminase-deficient mice display hippocampal hypoactivity, insensitivity to pro-psychotic drugs and potentiated latent inhibition: relevance to schizophrenia. Neuropsychopharmacol. 2009;34:2305–22. doi: 10.1038/npp.2009.58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Huang Y, Zhao L, Jia B, Wu L, Li Y, Curthoys N, et al. Glutaminase dysregulation in HIV-1-infected human microglia mediates neurotoxicity: relevant to HIV-1-associated neurocognitive disorders. J Neurosci. 2011;31:15195–204. doi: 10.1523/JNEUROSCI.2051-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Seltzer MJ, Bennett BD, Joshi AD, Gao P, Thomas AG, Ferraris DV, et al. Inhibition of glutaminase preferentially slows growth of glioma cells with mutant IDH1. Cancer Res. 2010;70:8981–7. doi: 10.1158/0008-5472.CAN-10-1666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gordon EB, Hart GT, Tran TM, Waisberg M, Akkaya M, Kim AS, et al. Targeting glutamine metabolism rescues mice from late-stage cerebral malaria. Proc Natl Acad Sci USA. 2015;112:13075–80. doi: 10.1073/pnas.1516544112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Manivannan S, Baxter VK, Schultz KL, Slusher BS, Griffin DE. Protective effects of glutamine antagonist 6-diazo-5-oxo-l-norleucine in mice with alphavirus encephalomyelitis. J Virol. 2016;90:9251–62. doi: 10.1128/JVI.01045-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nedelcovych MT, Tenora L, Kim BH, Kelschenbach J, Chao W, Hadas E, et al. N-(Pivaloyloxy)alkoxy-carbonyl prodrugs of the glutamine antagonist 6-diazo-5-oxo-l-norleucine (DON) as a potential treatment for HIV associated neurocognitive disorders. J Med Chem. 2017;60:7186–98. doi: 10.1021/acs.jmedchem.7b00966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Potter MC, Baxter VK, Mathey RW, Alt J, Rojas C, Griffin DE, et al. Neurological sequelae induced by alphavirus infection of the CNS are attenuated by treatment with the glutamine antagonist 6-diazo-5-oxo-l-norleucine. J Neurovirol. 2015;21:159–73. doi: 10.1007/s13365-015-0314-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rais R, Jancarik A, Tenora L, Nedelcovych M, Alt J, Englert J, et al. Discovery of 6-diazo-5-oxo-l-norleucine (DON) prodrugs with enhanced CSF delivery in monkeys: a potential treatment for glioblastoma. J Med Chem. 2016;59:8621–33. doi: 10.1021/acs.jmedchem.6b01069. [DOI] [PubMed] [Google Scholar]
- 40.Shijie J, Takeuchi H, Yawata I, Harada Y, Sonobe Y, Doi Y, et al. Blockade of glutamate release from microglia attenuates experimental autoimmune encephalomyelitis in mice. Tohoku J Exp Med. 2009;217:87–92. doi: 10.1620/tjem.217.87. [DOI] [PubMed] [Google Scholar]
- 41.Francis TC, Chandra R, Gaynor A, Konkalmatt P, Metzbower SR, Evans B, et al. (2017). Molecular basis of dendritic atrophy and activity in stress susceptibility. Mol Psychiatry. 2017;22:1512-19. [DOI] [PMC free article] [PubMed]
- 42.Krishnan V, Han MH, Graham DL, Berton O, Renthal W, Russo SJ, et al. Molecular adaptations underlying susceptibility and resistance to social defeat in brain reward regions. Cell. 2007;131:391–404. doi: 10.1016/j.cell.2007.09.018. [DOI] [PubMed] [Google Scholar]
- 43.McKim DB, Weber MD, Niraula A, Sawicki CM, Liu X, Jarrett BL, et al. Microglial recruitment of IL-1beta-producing monocytes to brain endothelium causes stress-induced anxiety. Mol Psychiatry. 2018;23:1421–31. doi: 10.1038/mp.2017.64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Berton O, McClung CA, Dileone RJ, Krishnan V, Renthal W, Russo SJ, et al. Essential role of BDNF in the mesolimbic dopamine pathway in social defeat stress. Science. 2006;311:864–8. doi: 10.1126/science.1120972. [DOI] [PubMed] [Google Scholar]
- 45.Nestler EJ, Hyman SE. Animal models of neuropsychiatric disorders. Nat Neurosci. 2010;13:1161–9. doi: 10.1038/nn.2647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wohleb ES, Hanke ML, Corona AW, Powell ND, Stiner LM, Bailey MT, et al. beta-Adrenergic receptor antagonism prevents anxiety-like behavior and microglial reactivity induced by repeated social defeat. J Neurosci. 2011;31:6277–88. doi: 10.1523/JNEUROSCI.0450-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yang C, Ren Q, Qu Y, Zhang JC, Ma M, Dong C, et al. Mechanistic target of rapamycin-independent antidepressant effects of (R)-ketamine in a social defeat stress model. Biol Psychiatry. 2018;83:18–28. doi: 10.1016/j.biopsych.2017.05.016. [DOI] [PubMed] [Google Scholar]
- 48.Golden SA, Covington HE, 3rd, Berton O, Russo SJ. A standardized protocol for repeated social defeat stress in mice. Nat Protoc. 2011;6:1183–91. doi: 10.1038/nprot.2011.361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Roybal K, Theobold D, Graham A, DiNieri JA, Russo SJ, Krishnan V, et al. Mania-like behavior induced by disruption of CLOCK. Proc Natl Acad Sci USA. 2007;104:6406–11. doi: 10.1073/pnas.0609625104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ito N, Hirose E, Ishida T, Hori A, Nagai T, Kobayashi Y, et al. Kososan, a Kampo medicine, prevents a social avoidance behavior and attenuates neuroinflammation in socially defeated mice. J Neuroinflamm. 2017;14:98. doi: 10.1186/s12974-017-0876-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kinsey SG, Bailey MT, Sheridan JF, Padgett DA, Avitsur R. Repeated social defeat causes increased anxiety-like behavior and alters splenocyte function in C57BL/6 and CD-1 mice. Brain Behav Immun. 2007;21:458–66. doi: 10.1016/j.bbi.2006.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Veerakumar A, Challis C, Gupta P, Da J, Upadhyay A, Beck SG, et al. Antidepressant-like effects of cortical deep brain stimulation coincide with pro-neuroplastic adaptations of serotonin systems. Biol Psychiatry. 2014;76:203–12. doi: 10.1016/j.biopsych.2013.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Browne CA, Falcon E, Robinson SA, Berton O, Lucki I. Reversal of stress-induced social interaction deficits by buprenorphine. Int J Neuropsychopharmacol. 2018;21:164–74. doi: 10.1093/ijnp/pyx079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Tsankova NM, Berton O, Renthal W, Kumar A, Neve RL, Nestler EJ. Sustained hippocampal chromatin regulation in a mouse model of depression and antidepressant action. Nat Neurosci. 2006;9:519–25. doi: 10.1038/nn1659. [DOI] [PubMed] [Google Scholar]
- 55.Kaidanovich-Beilin O, Lipina T, Vukobradovic I, Roder J, Woodgett JR (2011). Assessment of social interaction behaviors. JoVE. 2011;48:e2473. [DOI] [PMC free article] [PubMed]
- 56.Nadler JJ, Moy SS, Dold G, Trang D, Simmons N, Perez A, et al. Automated apparatus for quantitation of social approach behaviors in mice. Genes Brain Behav. 2004;3:303–14. doi: 10.1111/j.1601-183X.2004.00071.x. [DOI] [PubMed] [Google Scholar]
- 57.Zhan Y, Paolicelli RC, Sforazzini F, Weinhard L, Bolasco G, Pagani F, et al. Deficient neuron-microglia signaling results in impaired functional brain connectivity and social behavior. Nat Neurosci. 2014;17:400–6. doi: 10.1038/nn.3641. [DOI] [PubMed] [Google Scholar]
- 58.Cao X, Li LP, Wang Q, Wu Q, Hu HH, Zhang M, et al. Astrocyte-derived ATP modulates depressive-like behaviors. Nat Med. 2013;19:773–7. doi: 10.1038/nm.3162. [DOI] [PubMed] [Google Scholar]
- 59.Paxinos G, Franklin KBJ. Paxinos and Franklin’s the mouse brain in stereotaxic coordinates. 4th edn. Amsterdam: Elsevier/Academic Press; 2013. [Google Scholar]
- 60.Baxter VK, Glowinski R, Braxton AM, Potter MC, Slusher BS, Griffin DE. Glutamine antagonist-mediated immune suppression decreases pathology but delays virus clearance in mice during nonfatal alphavirus encephalomyelitis. Virology. 2017;508:134–49. doi: 10.1016/j.virol.2017.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Anacker C, Scholz J, O’Donnell KJ, Allemang-Grand R, Diorio J, Bagot RC, et al. Neuroanatomic differences associated with stress susceptibility and resilience. Biol Psychiatry. 2016;79:840–9. doi: 10.1016/j.biopsych.2015.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hodes GE, Pfau ML, Leboeuf M, Golden SA, Christoffel DJ, Bregman D, et al. Individual differences in the peripheral immune system promote resilience versus susceptibility to social stress. Proc Natl Acad Sci USA. 2014;111:16136–41. doi: 10.1073/pnas.1415191111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wook Koo J, Labonte B, Engmann O, Calipari ES, Juarez B, Lorsch Z, et al. Essential role of mesolimbic brain-derived neurotrophic factor in chronic social stress-induced depressive behaviors. Biol Psychiatry. 2016;80:469–78. doi: 10.1016/j.biopsych.2015.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Wohleb ES, Patterson JM, Sharma V, Quan N, Godbout JP, Sheridan JF. Knockdown of interleukin-1 receptor type-1 on endothelial cells attenuated stress-induced neuroinflammation and prevented anxiety-like behavior. J Neurosci. 2014;34:2583–91. doi: 10.1523/JNEUROSCI.3723-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Hultman R, Mague SD, Li Q, Katz BM, Michel N, Lin L, et al. Dysregulation of prefrontal cortex-mediated slow-evolving limbic dynamics drives stress-induced emotional pathology. Neuron. 2016;91:439–52. doi: 10.1016/j.neuron.2016.05.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kumar S, Hultman R, Hughes D, Michel N, Katz BM, Dzirasa K. Prefrontal cortex reactivity underlies trait vulnerability to chronic social defeat stress. Nat Commun. 2014;5:4537. doi: 10.1038/ncomms5537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Zhang RX, Han Y, Chen C, Xu LZ, Li JL, Chen N, et al. EphB2 in the medial prefrontal cortex regulates vulnerability to stress. Neuropsychopharmacology. 2016;41:2541–56. doi: 10.1038/npp.2016.58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Menard C, Pfau ML, Hodes GE, Kana V, Wang VX, Bouchard S, et al. Social stress induces neurovascular pathology promoting depression. Nat Neurosci. 2017;20:1752–60. doi: 10.1038/s41593-017-0010-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Beitia G, Garmendia L, Azpiroz A, Vegas O, Brain PF, Arregi A. Time-dependent behavioral, neurochemical, and immune consequences of repeated experiences of social defeat stress in male mice and the ameliorative effects of fluoxetine. Brain Behav Immun. 2005;19:530–9. doi: 10.1016/j.bbi.2004.11.002. [DOI] [PubMed] [Google Scholar]
- 70.Cao JL, Covington HE, 3rd, Friedman AK, Wilkinson MB, Walsh JJ, Cooper DC, et al. Mesolimbic dopamine neurons in the brain reward circuit mediate susceptibility to social defeat and antidepressant action. J Neurosci. 2010;30:16453–8. doi: 10.1523/JNEUROSCI.3177-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Vialou V, Robison AJ, Laplant QC, Covington HE, 3rd, Dietz DM, et al. DeltaFosB in brain reward circuits mediates resilience to stress and antidepressant responses. Nat Neurosci. 2010;13:745–52. doi: 10.1038/nn.2551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Vialou V, Bagot RC, Cahill ME, Ferguson D, Robison AJ, Dietz DM, et al. Prefrontal cortical circuit for depression- and anxiety-related behaviors mediated by cholecystokinin: role of DeltaFosB. J Neurosci. 2014;34:3878–87. doi: 10.1523/JNEUROSCI.1787-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Jiang H, Chen S, Li C, Lu N, Yue Y, Yin Y, et al. The serum protein levels of the tPA-BDNF pathway are implicated in depression and antidepressant treatment. Transl Psychiatry. 2017;7:e1079. doi: 10.1038/tp.2017.43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Kong L, Chen K, Tang Y, Wu F, Driesen N, Womer F, et al. Functional connectivity between the amygdala and prefrontal cortex in medication-naive individuals with major depressive disorder. J Psychiatry Neurosci. 2013;38:417–22. doi: 10.1503/jpn.120117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Reinhart V, Bove SE, Volfson D, Lewis DA, Kleiman RJ, Lanz TA. Evaluation of TrkB and BDNF transcripts in prefrontal cortex, hippocampus, and striatum from subjects with schizophrenia, bipolar disorder, and major depressive disorder. Neurobiol Dis. 2015;77:220–7. doi: 10.1016/j.nbd.2015.03.011. [DOI] [PubMed] [Google Scholar]
- 76.Teyssier JR, Ragot S, Chauvet-Gelinier JC, Trojak B, Bonin B. Activation of a DeltaFOSB dependent gene expression pattern in the dorsolateral prefrontal cortex of patients with major depressive disorder. J Affect Disord. 2011;133:174–8. doi: 10.1016/j.jad.2011.04.021. [DOI] [PubMed] [Google Scholar]
- 77.Bagot RC, Cates HM, Purushothaman I, Vialou V, Heller EA, Yieh L, et al. Ketamine and imipramine reverse transcriptional signatures of susceptibility and induce resilience-specific gene expression profiles. Biol Psychiatry. 2017;81:285–95. doi: 10.1016/j.biopsych.2016.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Yang C, Shirayama Y, Zhang JC, Ren Q, Yao W, Ma M, et al. R-ketamine: a rapid-onset and sustained antidepressant without psychotomimetic side effects. Transl Psychiatry. 2015;5:e632. doi: 10.1038/tp.2015.136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Li CT, Chen MH, Lin WC, Hong CJ, Yang BH, Liu RS, et al. The effects of low-dose ketamine on the prefrontal cortex and amygdala in treatment-resistant depression: a randomized controlled study. Hum Brain Mapp. 2016;37:1080–90. doi: 10.1002/hbm.23085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Murrough JW, Burdick KE, Levitch CF, Perez AM, Brallier JW, Chang LC, et al. Neurocognitive effects of ketamine and association with antidepressant response in individuals with treatment-resistant depression: a randomized controlled trial. Neuropsychopharmacology. 2015;40:1084–90. doi: 10.1038/npp.2014.298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Singh JB, Fedgchin M, Daly EJ, De Boer P, Cooper K, Lim P, et al. A double-blind, randomized, placebo-controlled, dose-frequency study of intravenous ketamine in patients with treatment-resistant depression. Am J Psychiatry. 2016;173:816–26. doi: 10.1176/appi.ajp.2016.16010037. [DOI] [PubMed] [Google Scholar]
- 82.Soden ME, Miller SM, Burgeno LM, Phillips PEM, Hnasko TS, Zweifel LS. Genetic isolation of hypothalamic neurons that regulate context-specific male social behavior. Cell Rep. 2016;16:304–13. doi: 10.1016/j.celrep.2016.05.067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Mesic I, Guzman YF, Guedea AL, Jovasevic V, Corcoran KA, Leaderbrand K, et al. Double dissociation of the roles of metabotropic glutamate receptor 5 and oxytocin receptor in discrete social behaviors. Neuropsychopharmacology. 2015;40:2337–46. doi: 10.1038/npp.2015.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Chung W, Choi SY, Lee E, Park H, Kang J, Choi Y, et al. Social deficits in IRSp53 mutant mice improved by NMDAR and mGluR5 suppression. Nat Neurosci. 2015;18:435–43. doi: 10.1038/nn.3927. [DOI] [PubMed] [Google Scholar]
- 85.Mingote S, Chuhma N, Kalmbach A, Thomsen GM, Wang Y, Mihali A, et al. Dopamine neuron dependent behaviors mediated by glutamate cotransmission. eLife. 2017;6:e27566. [DOI] [PMC free article] [PubMed]
- 86.Cardona C, Sanchez-Mejias E, Davila JC, Martin-Rufian M, Campos-Sandoval JA, Vitorica J, et al. Expression of Gls and Gls2 glutaminase isoforms in astrocytes. Glia. 2015;63:365–82. doi: 10.1002/glia.22758. [DOI] [PubMed] [Google Scholar]
- 87.Chalmers SA, Wen J, Shum J, Doerner J, Herlitz L, Putterman C. CSF-1R inhibition attenuates renal and neuropsychiatric disease in murine lupus. Clin Immunol. 2017;185:100–8. doi: 10.1016/j.clim.2016.08.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Han J, Harris RA, Zhang XM. An updated assessment of microglia depletion: current concepts and future directions. Mol Brain. 2017;10:25. doi: 10.1186/s13041-017-0307-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Hamilton PJ, Burek DJ, Lombroso SI, Neve RL, Robison AJ, Nestler EJ, et al. Cell-type-specific epigenetic editing at the Fosb gene controls susceptibility to social defeat stress. Neuropsychopharmacology. 2018;43:272–84. doi: 10.1038/npp.2017.88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Heshmati M, Aleyasin H, Menard C, Christoffel DJ, Flanigan ME, Pfau ML, et al. Cell-type-specific role for nucleus accumbens neuroligin-2 in depression and stress susceptibility. Proc Natl Acad Sci USA. 2018;115:1111–6. doi: 10.1073/pnas.1719014115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Mineur YS, Fote GM, Blakeman S, Cahuzac EL, Newbold SA, Picciotto MR. Multiple nicotinic acetylcholine receptor subtypes in the mouse amygdala regulate affective behaviors and response to social stress. Neuropsychopharmacology. 2016;41:1579–87. doi: 10.1038/npp.2015.316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Raison CL, Rutherford RE, Woolwine BJ, Shuo C, Schettler P, Drake DF, et al. A randomized controlled trial of the tumor necrosis factor antagonist infliximab for treatment-resistant depression: the role of baseline inflammatory biomarkers. JAMA Psychiatry. 2013;70:31–41. doi: 10.1001/2013.jamapsychiatry.4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Setiawan E, Wilson AA, Mizrahi R, Rusjan PM, Miler L, Rajkowska G, et al. Role of translocator protein density, a marker of neuroinflammation, in the brain during major depressive episodes. JAMA Psychiatry. 2015;72:268–75. doi: 10.1001/jamapsychiatry.2014.2427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Shelton RC, Claiborne J, Sidoryk-Wegrzynowicz M, Reddy R, Aschner M, Lewis DA, et al. Altered expression of genes involved in inflammation and apoptosis in frontal cortex in major depression. Mol Psychiatry. 2011;16:751–62. doi: 10.1038/mp.2010.52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Vargish GA, Pelkey KA, Yuan X, Chittajallu R, Collins D, Fang C, et al. Co-activation of VEGF and NMDA receptors promotes synaptic targeting of AMPA receptors. Mol Psychiatry. 2017;22:1. doi: 10.1038/mp.2016.245. [DOI] [PubMed] [Google Scholar]
- 96.Doble A. The role of excitotoxicity in neurodegenerative disease: implications for therapy. Pharmacol Ther. 1999;81:163–221. doi: 10.1016/S0163-7258(98)00042-4. [DOI] [PubMed] [Google Scholar]
- 97.Kostandy BB. The role of glutamate in neuronal ischemic injury: the role of spark in fire. Neurol Sci. 2012;33:223–37. doi: 10.1007/s10072-011-0828-5. [DOI] [PubMed] [Google Scholar]
- 98.Lau A, Tymianski M. Glutamate receptors, neurotoxicity and neurodegeneration. Pflug Arch. 2010;460:525–42. doi: 10.1007/s00424-010-0809-1. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.