

Abstract
Idiopathic pulmonary fibrosis is an extremely aggressive lung disease that develops almost exclusively in older individuals, carries a very poor prognosis, and lacks any truly effective therapies. The current conceptual model is that IPF develops because of an age-related decline in the ability of the lung epithelium to regenerate after injury, largely due to death or senescence of epithelial progenitor cells in the distal airways. This loss of regenerative capacity is thought to initiate a chronic and ineffective wound healing response, characterized by persistent, low-grade lung inflammation and sustained production of collagen and other extracellular matrix materials. Despite recent advances in our understanding of IPF pathobiology, there remains a pressing need to further delineate underlying mechanisms to develop more effective therapies for this disease. In this review, we build the case that many of the manifestations of IPF result from a failure of cells to effectively manage their proteome. We propose that epithelial progenitor cells, as well as immune cells and fibroblasts become functionally impaired, at least in part, because of an accumulation or a loss in the expression of various crucial proteins. Further, we propose that central to this defect is the dysregulation of the ubiquitin proteasome system, which is the major protein degradation system in eukaryotic cells. Lastly, borrowing concepts from other fields, we discuss how targeting the UPS system could be employed as a novel treatment for IPF and perhaps for other fibrotic lung diseases as well.
Keywords: Pulmonary fibrosis, idiopathic pulmonary fibrosis, proteostasis, ubiquitin proteasome system
Introduction.
Pulmonary fibrosis refers to a heterogeneous group of conditions that scar the distal airspaces of the lung. In most cases, pulmonary fibrosis develops as a consequence to some identifiable pulmonary insult, such as a viral infection, exposure to inorganic or organic dusts or treatment with chemotherapeutic agents or thoracic radiation [1, 2]. However, in a significant minority of cases, pulmonary fibrosis develops without any identifiable cause, and in these cases is considered idiopathic in origin. Although several types of idiopathic fibrotic lung conditions are known to exist, by far the most common of these is the highly aggressive respiratory disorder called idiopathic pulmonary fibrosis (IPF).
IPF is often classified as an age-related lung disorder because it develops almost exclusively after 50 years of age and its incidence increases almost exponentially after the seventh decade of life. In the US alone, it is estimated that over 100,000 people are diagnosed with IPF each year, and recent evidence indicates that the incidence of this condition has climbed substantially over the last few decades. In addition to age, various environment factors have been linked to the development of the disease, including chronic cigarette smoking, viral infections and a wide variety of environmental and occupational exposures [2]. A favored conceptual model is that IPF develops from an age-related decline in the ability of lung epithelium to regenerate after injury. Chronic environmental exposures, such as those described above, combined with a genetic susceptibility, are believed to contribute to loss of epithelial regenerative capacity by inducing premature death or senescence of epithelial progenitor cells. As result, these terminal cell fates drive chronic inflammatory and wound healing responses that not only further injure the lung but also severely impede respiratory gas exchange and other respiratory functions [3].
Consistent with the mechanisms described above, there are several groups of cells that are believed to play an important role in the pathobiology of IPF. As just mentioned, the primary cell contributing to disease is the distal epithelial progenitor cell, also known as the type II alveolar (AE2) epithelial cell [4, 5, 6]. Similar to other age-related diseases, AE2 cells adopt many hallmarks of aging tissues, including shortened telomeres, genomic instability, mitochondrial dysfunction and loss of proteostasis [7]. Indeed, the accumulation of misfolded and aggregated proteins is one of the earliest manifestations of epithelial dysfunction in the lung of IPF patients [8]. Moreover, abnormal protein accumulation has also been described in the alveolar epithelium of patients with various other fibrotic lung diseases, suggesting that loss of proteostasis might be a unifying factor contributing to the formation of many fibrotic lung conditions [9]. Although in some rare cases mutations causing the misfolding of surfactant proteins (e.g. surfactant C mutations) have been linked to protein misfolding in IPF, in most cases the cause of protein aggregation is entirely unknown [10, 11, 12].
The second cell type critical to the development of IPF is the immune cell, and in particular the alveolar macrophage [13]. While clinical trials suggest that globally suppressing the immune system is not an effective treatment for IPF, it is still firmly accepted by most experts in the field that inflammatory cells, in particular macrophages, contribute significantly to driving fibrotic responses through their secretion of numerous repair and remodeling proteins [14, 15, 16]. Lastly, the fibroblast also plays a central role in the pathobiology of IPF. These cells undergo a wide range of changes in the IPF lung, including adopting a hyperproliferative and anti-apoptotic phenotype, and produce most, in not all, of the extracellular matrix proteins that comprise pulmonary scar tissues [4, 17, 18]. Although only marginally effective, existing therapies for IPF aim to ameliorate disease by suppressing fibroblast activation and the production of extracellular matrix [19, 20, 21].
Although epithelial cells, immune cells and fibroblasts all play very different roles in the pathogenesis of IPF each population exhibits a remarkable change in their proteome at every stage of disease. In addition to lung epithelial cells accumulating misfolded proteins in IPF, these cells also display reduced levels of various key proteins involved in cell-cell junctional connection, antioxidant defenses and other homeostatic processes such as macroautophagy [22, 23]. Moreover, macrophages and fibroblasts in IPF also display major changes to their proteome. In these cells, proteins levels for numerous pro-inflammatory and pro-fibrotic factors are upregulated, presumably due to increased protein translation. However, regardless of the mechanism contributing to changes in protein levels in IPF, it seems reasonable to assume that targeting processes regulating proteostasis might be effective in the treatment of this disease [24, 25]. In this review, we discuss the major mechanisms by which proteins are targeted for destruction in eukaryotic cells, namely the ubiquitin proteasome system (UPS). We also discuss emerging evidence linking dysfunction of this system to IPF, and propose how targeting components of the UPS could, at some future point, be used as a treatment for this disease.
The importance of maintaining the proteome.
Proteins are highly versatile macromolecules that are involved in essentially every biological process in and outside of a cell. The lifespan of a protein begins with the translation of mRNA on ribosomes and ends with its destruction through one of several degradation mechanisms. Generally speaking, all proteins are synthesized and degraded on a continuous basis, although the rate of turnover of any individual protein can vary significantly from several hours to weeks, or even months to years [25]. While this continuous re-synthesis and degradation process may seem somewhat wasteful, it is absolutely essential to limit the accumulation of damaged or misfolded proteins, and to ensure that only newly synthesized and properly configured proteins are controlling essential functions in a cell [26]. While every cell contains several proteolytic systems to carry out the degradation process, the major mechanism by which intracellular proteins are degraded is through the UPS. Importantly, the UPS system is highly regulated, and contains multiple mechanisms to ensure specificity so that only proteins meant to be degraded are ultimately targeted for destruction [27, 28]. However, this system is not infallible and even small increases or decreases in protein degradation can have important consequence that deleteriously affect the functioning of a cell, and ultimately contribute to the development of disease. Indeed, abnormalities in the UPS have already been linked to a wide range of disorders, including Parkinson’s disease, Alzheimer’s disease, cancer, heart failure and, relevant to this review, various forms of pulmonary fibrosis [29, 30, 31, 32, 33].
The Ubiquitin-Proteasome System
In most mammalian cells, the primary mechanism by which proteins are degraded is through the UPS; this system is estimated to degrade 80–90% of all proteins in eukaryotic cells [28, 34]. In addition to this system, proteins can also be degraded by macroautophagy, which is a multistep process that clears proteins (and other cellular components) by enveloping cellular materials in double membrane vesicles that are then sent to lysosomes for ultimate destruction. Although both processes are important to cells, it is now apparent that no part of the cell is out of reach of the UPS, including the nucleus, mitochondrial and endoplasmic reticulum. Moreover, it is also appreciated that most misfolded, oxidized and damaged proteins are cleared through the UPS rather than through macroautophagy, making the UPS absolutely essential for the health and proper functioning of cells [35, 36].
Protein degradation by the UPS begins with the covalent linkage of ubiquitin, a highly conserved 76 amino acid protein, to a lysine 48 (K48) residue on the substrate protein (Figure 1). Ubiquitination is an ATP-dependent process carried out by three classes of enzymes. In first step, an ubiquitin activating enzyme, also called an E1 enzyme, forms a thio-ester bond with the ubiquitin protein, thereby permitting the binding of ubiquitin to the E2 ubiquitin conjugating enzyme. This second step is followed by the formation of an isopeptide bond between the carboxy-terminus of ubiquitin and a lysine residue on the target protein. Notably, this last step is carried out by the E3 ubiquitin ligase, which has very high specificity for individual substrates. Importantly, for some E3 ligases, the ubiquitin-binding and target protein binding domains exist on different polypeptides, requiring specialized adaptor proteins or cullins to bring these binding sites together [37, 38].
Figure 1:
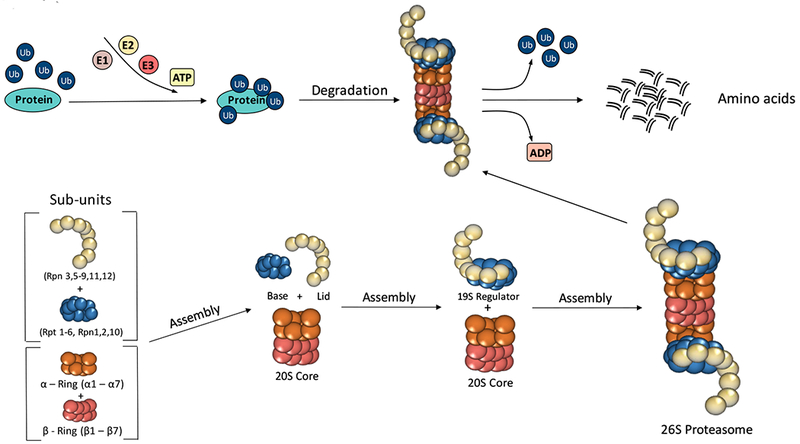
Ubiquitin-Proteasome system. Sub-units of the 26S proteasome are assembled into 19S regulatory and 20S core complex to become the barrel-shaped proteolytic machine. The 26S recognizes ubiquitin-conjugated proteins, a process catalyzed by three ligases known as E1, E2 and E3. Once breakdown ensues, amino acids are released into the cytoplasm and recycled.
As mentioned, E3 ubiquitin ligases have enormous specificity, thereby limiting any indiscriminate targeting of proteins for destruction. To date, many E3 ligases have been identified and they have much more specificity when compared to either E1 or E2 ligases, making them ideal candidates as therapeutic targets. Importantly, E3 ubiquitin ligases can be a single peptide, such Mouse double minute 2 homolog (MDM2), or can be comprised of multiple components as is the case with the SKP1-Cullin 1-F-box (SCF) proteins. Of note, most E3 ligases contain additional linker proteins and collectively fall into one of three main groups: 1) Homologous to the E6-AP C-terminus (HECTs). 2) Really interesting new genes (RINGs) and 3) RING between RING (RBRs) [39, 40, 41, 42].
Once proteins are tagged (ubiquinated) for destruction, they are then delivered to the 26S proteasome, a large, 2.5-MDa molecular protein killing machine, which is extremely well-designed for carrying out proteolysis. Located in the cytoplasm, the 26S proteasome is comprised of a barrel proteolytic core called the 20S particle (core particle, CP), which has ATP-dependent activity, and a regulatory 19S complex (regulatory particle, RP), which recognizes, binds and unwinds substrate proteins before degradation. The 19S RP is divided into two subcomplexes; the lid and the base. The lid consists of nine Rpn subunits and functions to remove ubiquitin molecules, while the base has six regulatory AAA ATPase subunits (Rpt1-Rpt6) and four non-ATP subunits. The AAA family (ATPases associated with various cellular activities) is a large group of ATPases found in all biological kingdoms and is characterized by the presence of one or two conserved ATP-binding domains of a type called the AAA motif. In contrast to the 19S, the 20S CP is made up of duplicating seven α subunits and seven β subunits that form four axially stacked heteroheptameric rings [34]. The outer-alpha rings contain seven subunits (alpha1- alpha7) forming a pore that regulates the entrance and removal of degraded proteins from the proteasome complex. Similarly, the inner-beta rings contain seven subunits (beta1- beta7), and within this site is where protein breakdown occurs, releasing amino acids and small peptide fragments into the cytoplasm. To date, the proteasome has been shown to exhibit three types of protein lysis activities: 1) chymotrypsin-like (CT-L); 2) trypsin-like (T-L) and 3) caspase-like (C-L) [43].
When put together, the proteasome complex is comprised of 33 individual proteins all encoded by different genes [44]. Currently, it remains unclear whether these genes can all be regulated by one master transcription factor or require many distinct regulatory proteins to control gene expression. So far, studies have identified Rpn4 as an important transcription factor of many proteasome genes in various cell systems, and other transcription factors, such as nuclear factor erythroid 2-related factor 1 (Nrf1) appear to also drive gene expression in response to proteasome inhibition [45, 46, 47] (Figure 2). Furthermore, one recent study showed that proteasome biogenesis can be triggered by activating the mechanistic target of rapamycin (mTORC1), a major anabolic factor in mammalian cells. These latter findings suggest that increased protein destruction by the UPS may be important for supplying free amino acids in response to the activation of mTOR-mediated pro-growth pathways. Consistent with this notion, mTORC1 activation is known to be a potent inhibitor of macroautophagy, highlighting the fact that growing cells may be more dependent on the UPS to acquire the amino acids needed for growth and proliferation [48].
Figure 2:
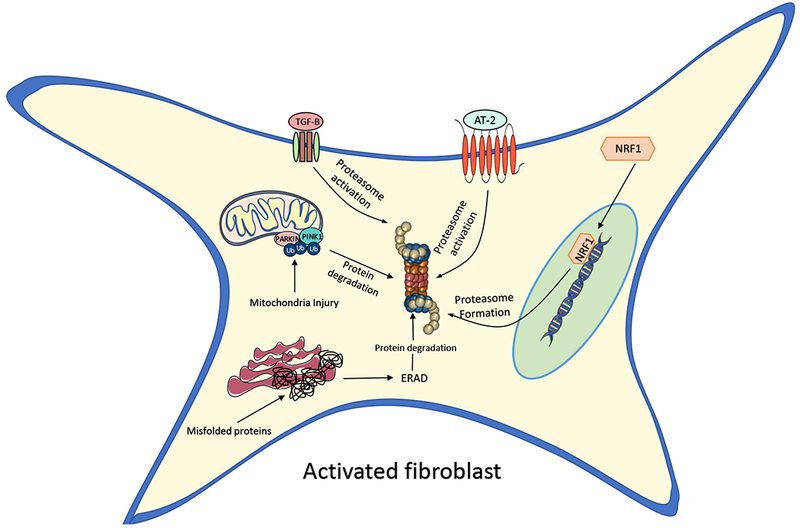
Proposed mechanisms by which the UPS activates in lung fibroblasts during pulmonary fibrosis. Angiotensin 2 (AT-2) and transforming-growth factor beta 1 (TGFβ1) could directly stimulate an increase in proteasome activity. Nuclear receptor factor 1 (NRF1) and other transcriptions can drive the production of proteasomal protein subunits.
The importance of the UPS in mitochondrial homeostasis.
In addition to controlling cytoplasmic protein quality control, the UPS plays a critical role in regulating the proteome of individual organelles. Perhaps no better example of this is in mitochondria, which depend on a constant influx of cytoplasmic proteins [49, 50]. Mitochondria are double-membrane organelles that serve many essential functions, including the generation of ATP and biosynthesis of amino acids, nucleotides and NAD. In addition, mitochondria control cell fate through their release of ROS and other pro- and anti-apoptotic factors, making mitochondrial homeostasis a key determinant of life and death in cells [51].
Mitochondrial dysfunction is now emerging as an important pathogenic player in a wide range of age-related diseases, including IPF. Recently, Bueno et al. provided the first important piece of evidence linking mitochondrial dysfunction to IPF [52]. These investigators showed that large, dysmorphic mitochondrial accumulate in AE2 cells in the IPF lung. They also went on to demonstrate that these findings associated with a marked reduction in levels of PTEN-induced putative kinase 1 (PINK1), a protein important for the clearance of dysfunctional mitochondria from the cytoplasm. Furthermore, targeted deletion of the PINK1 gene in mice was found not only to lead to mitochondrial dysfunction but also to cause protein aggregation in the endoplasmic reticulum (ER) (also known as ER stress) and enhance susceptibility to developing bleomycin-induced pulmonary fibrosis [52]. Together, these finding indicate a mechanistic link among mitochondrial dysfunction, protein aggregation and susceptibility to pulmonary fibrosis in the mouse lung. Since these observations, several groups have shown that various other proteins important for proper functioning of mitochondrial are downregulated in pulmonary fibrosis, including those involved in oxidative phosphorylation (electron transport chain proteins), mitochondrial DNA repair (8-oxoguanine) and anti-oxidant defense (sirtiuns).
Although the mechanisms by which mitochondrial dysfunction causes protein aggregation in IPF are not understood, a mechanistic link between these pathological processes can easily be appreciated if one understands how the mitochondrial proteome is actually formed. Despite containing their own genome, mitochondrial DNA encodes for only a handful of genes (37 to be exact) mostly related to oxidative phosphorylation and the synthesis of tRNAs. This means that the rest of the ~1,200 proteins needed for proper functioning of mitochondria are derived from nuclear DNA, requiring synthesis outside of the organelle and import into the organelle for ultimate usage [53]. As a result, the mitochondria proteome is dependent on the UPS for not only limiting the entry of misfolded proteins but also for degrading misfolded proteins that accumulate after entry into the organelle [22].
The best evidence implicating the UPS in mitochondrial protein degradation comes from proteomic analysis of isolated mitochondria showing that over 100 mitochondrial proteins contain ubiquitin binding sites, including many involved in key mitochondrial functions such as ATP production and fatty acids synthesis. Different mechanisms have been described for how the UPS mediates mitochondrial protein degradation. By far, the easiest to understand is how the UPS removes damaged proteins before entry into the organelle. In these scenarios, targeted destruction likely occurs through mechanisms similar to other cytoplasmic proteins; first, they undergo ubiquination by specific ligases and then they are sent to the 26S proteasome for degradation. However, the mechanisms by which proteins are degraded after already arriving in mitochondria is less well understood. Recently, several groups have shown that defective proteins on the outer membrane of mitochondria (OMM) can be polyubiquitinated by E3 ligases and then extracted from the membrane by an AAA-ATPase p97 to be delivered to the proteasome for degradation. One example of this is the OMM protein called Mcl1, a pro-survival protein of the Bcl-2 family. In addition to degrading OMM proteins, several lines of evidence suggest that the UPS can also facilitate destruction of other, more internally located, mitochondrial proteins. Examples of this include endonuclease G, a protein found in the inner mitochondrial space, and two inner mitochondrial proteins called uncoupling proteins 2 and 3. However, the mechanistic details of how proteins within inner mitochondrial compartments are ubiquinated and retro-translocated out of the organelle have not yet been established. That said, recent work indicates that at least 9 of the known 54 human RING-containing E3 ligases are present in mitochondria, indicating that targeting proteins for destruction likely begins within the organelle [54].
As already mentioned, large dysmorphic mitochondria accumulate in the epithelium of the IPF lung. Mitochondrion are unique organelles in that they are constantly undergoing structural changes, including changes in size, shape and position within the cell. Importantly, many of these changes are influenced by the ability of mitochondria to undergo fission (division of a single organelle into two or more independent structures) and fusion (the opposing reaction) events. Notably, myofibroblasts in pulmonary fibrosis have been shown to exhibit an increase in mitochondrial fission events and pharmacologically inhibiting this activity with Astaxanthin has been shown to ameliorate lung parenchymal distortion and collagen deposition by inducing apoptosis [55]. During the fission process, various proteins accumulate on the OMM, such as Drp1 and Fis1, to help mediate the process. Meanwhile, mitochondrial fusion proteins (Mfn1 and Mfn2) are targeted for degradation by the UPS. Conversely, when mitochondrial fusion occurs there is an increase in Mfn1 and Mfn2, and fission proteins are ubiquitinated and targeted for destruction. Interestingly, Radke et al. reported that pharmacological inhibition of the 26S proteasome alone was capable of dramatically increasing mitochondrial fission in cells [56]. Moreover, Tar et al. demonstrated that high levels of Blm10/PA200, a proteasome activator, increased the degradation of mitochondrial fission proteins and augmented mitochondrial fusion events [57, 58]. Together, these observations highlight the critical role of the UPS in regulating mitochondrial dynamics and the potential for targeting this system to ameliorate aggressive fibroblast behaviors.
Proteasome dysfunction in IPF.
The proteome is severely dysregulated in the epithelium of IPF patients. In addition to the accumulation of misfolded and aggregated proteins in the endoplasmic reticulum, it has also been shown that levels of many other important proteins are downregulated in the IPF lung epithelium. This includes cell-cell junctional proteins, various kinases (PTEN) and many other antioxidant and DNA repair proteins, suggesting that restoring the proteome in the IPF lung epithelium will require more than just clearing misfolded proteins [59, 60, 61]. These findings also suggest that functional decline in the lung epithelial in IPF is not just about protein accumulation but also about a global dysregulation of proteostasis. Currently, it is unknown whether activity of the UPS is actually upregulated or downregulated in the epithelium of IPF patients.
Like the epithelium, the proteome of fibroblasts is severely altered in IPF as manifested by both increases and decreases in the expression of many different proteins. Since TGFβ1 secretion represents a crucial element in the pathophysiology of IPF it has been suggested as a possible therapeutic target for the disease [62, 63]. Importantly, levels of many downstream mediators of TGFβ1 are controlled through the UPS [64, 65, 66]. That said, Semren et al. reported that levels of the 19S regulatory Rpn6 subunit were markedly increased in fibroblasts from IPF patients, and that levels of K48 ubiquination were also increased in human lung fibroblasts in response to TGFβ exposure. Consistent with these findings, Baker et al. demonstrated that levels of several proteasome proteins were increased in the lungs of patients with IPF when compared to normal donors and COPD patients [67], although these studies were not performed in isolated fibroblasts. Furthermore, Mutlu et al. reported that Bortezomib prevents bleomycin-induced lung fibrosis in mice. These investigators also showed that these findings were associated with an increase peroxisome proliferator-activated receptor gamma levels in fibroblasts, which is known to repress TGF-β1 production. Altogether, these findings suggest that inhibiting proteasome activity might be effective in the treatment of pulmonary fibrosis by suppressing TGFβ1 signaling and fibroblast activation [68].
Another protein whose levels changes significantly in pulmonary fibrosis is sirtuin 3 (SIRT3), which is a mitochondrial protein deacetylase that regulates antioxidant responses and mitochondrial homeostasis. For example, Sundaresan et al. reported that levels of SIRT3 were markedly decreased in several major organs developing age-related fibrosis and this associated with a marked downregulation in SIRT3 expression in tissue fibroblasts [69]. Consistent with this, Sosulski et al. showed that SIRT3 expression was reduced in the lungs of old versus young mice, and was also downregulated in two mouse models of pulmonary fibrosis [70]. Moreover, analysis of SIRT3 expression in the lungs of patients with pulmonary fibrosis revealed low SIRT3 staining within the fibrotic regions and decreased SIRT3 expression in human lung fibroblasts was found to promote fibroblast to myofibroblast differentiation [71, 72]. Similarly, reduction in SIRT3 levels has been reported in AE2 cells from mouse and human fibrotic lungs and decreased expression was shown trigger these cells to undergo apoptosis in culture [70, 73]. Notably, in both fibroblasts and alveolar epithelial enforced expression of SIRT3 was found to be protective. Although sirtuins are regulated through diverse types of post-translational modifications, including phosphorylation, methylation and SUMOylation it is also appreciated that ubiquitination remains a vital mechanism for targeting these proteins for degradation. These findings suggest the possibility that increased UPS activity might contribute to reduced SIRT3 in pulmonary fibrosis and that targeting this mechanism could have beneficial effects on both fibroblasts and epithelial cells in IPF.
Although activity of the UPS seems to be increased in pulmonary fibrosis, at least in fibroblasts, the mechanisms contributing to its upregulation remain unknown. One theoretical mechanism could be through the upregulation of angiotensin II (AT-2) (Figure 2). AT-2 is a small peptide that is the main regulatory factor of the renin-angiotension system. However, AT-2 has also been shown to play an important role in driving fibrotic responses in a wide range of tissues, including the lung [74]. Although AT-2 is believe to promote organ fibrosis through stimulating TGFβ1 signaling and extracellular matrix production, Sanders et al. demonstrated that AT-2 can also directly stimulate the proteasome system in a wide range of tissues [75]. Moreover, Uhal et al. recently showed that AT-2 levels were dramatically increased in the mouse lung in response to bleomycin and similar findings have been described in human with pulmonary fibrosis [74]. Together, these findings support the notion that UPS activation might be driven by AT-2 in IPF and that targeting this peptide could attenuate lung fibrosis through multiple avenues [74].
Type I collagen is the principle component of pulmonary scar tissues. Collagen proteins are known to be degraded by the UPS. In addition, extracellular collagen can be degraded by enzymes known as matrix metalloproteinases (MMPs) [76]. Specifically, it is known that MMP1 mediates the degradation of type I collagen in some model systems, functioning as a key element in degradation of pulmonary scars [77]. In this context, studies have reported that TGFβ1 leads this process by upregulating type I collagen, while downregulating MMP1 gene transcription [78]. Interestingly, Goffin et al. showed that inhibition of the UPS with bortezomib enhanced MMP1 production while also decreasing type I collagen synthesis in human lung fibroblast [79]. Similar results have also been described in cardiac fibroblasts using MG132 as the proteasome inhibitor, again suggesting that modifying the UPS could be effective for reducing tissue remodeling in pulmonary fibrosis [80].
Therapeutic targeting of UPS.
There is no doubt that the UPS plays a critical role in the functioning of cells and their adaptation to stress. Moreover, it seems reasonable to assume that fine-tuning this system, particularly in situations where there has been a functional decline in UPS due to genetic mutations, injury or advanced age, could improve cellular function and possibly ameliorate disease. The best example of targeting the UPS as a treatment for disease is in the cancer field in which global inhibitors of the UPS have been shown to be cytotoxic to multiple myeloma cells [81, 82]. However, unlike cancer treatments, cytotoxic outcomes are not desired when treating most other conditions, particularly benign diseases such as pulmonary fibrosis. As such, this makes non-selective inhibitors of the UPS system likely a poor long-term treatment strategy. Thus, ongoing research will be needed to further delineate the proteins whose expression is altered in diseased IPF lung tissues and are also believed to significantly contribute to the pathobiology of disease. Armed with this information, it will be important to determine whether restoring expression of these proteins using more selective approaches can enhance critical cellular functions and undo or dismantle pro-fibrotic responses.
One obvious approach to selectively target the UPS would be to inhibit the activity of individual E3 ligases responsible for targeting specific proteins for destruction (see Table 1). Since E3 ligases responsible for targeting most proteins have not been identified the first step will be to catalog the E3 ligases responsible for controlling protein expression in relevant cell populations in the lung. Once achieved, the next step will be to develop small molecules capable of augmenting or inhibiting the activity of specific E3 ligases, like has recently been done for specific E3 ligases in the cancer field [83, 84, 85]. Alternatively, one could also develop small molecules that bind preferentially to specific substrates or to their ancillary proteins rather than to an E3 ligase. However, this theoretical approach is also limited at this time due to the absence of critical information related to the key structural interactions between E3 ligases and their substrate proteins. Finally, if these approaches are employed we will need to pay careful attention to the effects on other tissues to avoid unwanted extra-pulmonary side effects. That said, the lung is unique in that drugs could theoretically be administered through inhalation in order to minimize unwanted systemic side effects.
Table 1.
Fine-tuning the ubiquitin-proteasome system to treat pulmonary fibrosis
| Cell type | Target protein for UPS degradation | E3 ubiquitin-ligase | Drug |
|---|---|---|---|
| AEC | |||
| ↓PINK1 ↓SIRT3 ↓PGC1α ↑MMP7 / MMP1 ↑CTGF ↑PDGF ↑CXCL12 |
Parkin1 SKP22 RNF343 / PARIS4 / Fbxw75 Unidentified Smurf26 c-Cbl / Cbl-b7 WWP1 / ITCH (AIP4)8 |
LRPPRC CG-12 / SKP2-C25 ------ ------ Heclin ------ 1,4-Naphthoquinones |
|
| Macrophage | |||
| ↑TGF-β ↑Galectin-3 |
Smad9 TRIMs10 |
SIS3 HCl | |
| Fibroblast | |||
| ↑UCP2 ↑NOX4 ↑TGF-β |
Unidentified STUB1 (CHIP)11 Smad9 |
----- ------ SIS3 HCl |
Conclusion.
Proteins are highly versatile macromolecules that are involved in essentially every biological process in and outside cells. However, organ homeostasis is not just dependent on the production of proteins but also on precise control of their degradation. It is now appreciated that the UPS is the major degradation system to minimize the toxic effects of damaged or misfolded proteins in cells. Although just a theoretically possibility in the lung, targeting the UPS is emerging as a valid approach for restoring health to cells and ameliorating the severity of disease. Because IPF is a disease characterized by loss of proteostasis, it seems reasonable that UPS targeted approaches may one day be effective in treating this disease. Until then, future studies are clearly needed to better define and establish the mechanisms by which protein dysregulation contributes to the onset and progression of this deadly disease.
References
- 1.Wolters PJ, Collard HR, Jones KD. Pathogenesis of idiopathic pulmonary fibrosis. Annu Rev Pathol. 2014;9:157–79. doi: 10.1146/annurev-pathol-012513-104706. PubMed PMID: ; PubMed Central PMCID: PMCPMC4116429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.King TE Jr, Pardo A, Selman M. Idiopathic pulmonary fibrosis. Lancet (London, England). 2011. December 3;378(9807):1949–61. doi: 10.1016/s0140-6736(11)60052-4. PubMed PMID: ; eng. [DOI] [PubMed] [Google Scholar]
- 3.Fernandez IE, Eickelberg O. New cellular and molecular mechanisms of lung injury and fibrosis in idiopathic pulmonary fibrosis. The Lancet. 2012;380(9842):680–688. doi: 10.1016/s0140-6736(12)61144-1. [DOI] [PubMed] [Google Scholar]
- 4.Sakai N, Tager AM. Fibrosis of Two: Epithelial Cell-Fibroblast Interactions in Pulmonary Fibrosis. Biochim Biophys Acta. 2013. July;1832(7):911–21. doi: 10.1016/j.bbadis.2013.03.001. PubMed PMID: ; PubMed Central PMCID: PMCPMC4041487. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kasper M, Barth K. Potential contribution of alveolar epithelial type I cells to pulmonary fibrosis. Biosci Rep. 2017. December 22;37(6). doi: 10.1042/BSR20171301. PubMed PMID: ; PubMed Central PMCID: PMCPMC5696455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kulkarni T, de Andrade J, Zhou Y, et al. Alveolar epithelial disintegrity in pulmonary fibrosis. Am J Physiol Lung Cell Mol Physiol. 2016. August 1;311(2):L185–91. doi: 10.1152/ajplung.00115.2016. PubMed PMID: ; PubMed Central PMCID: PMCPMC5005273. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kim SJ, Cheresh P, Jablonski RP, et al. The Role of Mitochondrial DNA in Mediating Alveolar Epithelial Cell Apoptosis and Pulmonary Fibrosis. Int J Mol Sci. 2015. September 7;16(9):21486–519. doi: 10.3390/ijms160921486. PubMed PMID: ; PubMed Central PMCID: PMCPMC4613264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Romero F, Summer R. Protein Folding and the Challenges of Maintaining Endoplasmic Reticulum Proteostasis in Idiopathic Pulmonary Fibrosis. Ann Am Thorac Soc. 2017. November;14(Supplement_5):S410–s413. doi: 10.1513/AnnalsATS.201703-207AW. PubMed PMID: ; PubMed Central PMCID: PMCPMC5711273. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.McElvaney NG, Greene CM. Mechanisms of protein misfolding in conformational lung diseases. Current molecular medicine. 2012. August;12(7):850–9. PubMed PMID: ; eng. [DOI] [PubMed] [Google Scholar]
- 10.Tanjore H, Cheng DS, Degryse AL, et al. Alveolar epithelial cells undergo epithelial-to-mesenchymal transition in response to endoplasmic reticulum stress. J Biol Chem. 2011. September 2;286(35):30972–80. doi: 10.1074/jbc.M110.181164. PubMed PMID: ; PubMed Central PMCID: PMCPMC3162456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang Y, Kuan PJ, Xing C, et al. Genetic defects in surfactant protein A2 are associated with pulmonary fibrosis and lung cancer. American journal of human genetics. 2009. January;84(1):52–9. doi: 10.1016/j.ajhg.2008.11.010. PubMed PMID: ; PubMed Central PMCID: PMCPMC2668050. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.van Moorsel C, van der Vis J, van Oosterhout M, et al. Mutations in SFTPC, SFTPA2 and TERT explain 60% of familial pulmonary fibrosis and correlate to specific disease phenotypes. European Respiratory Journal. 2011;38(Suppl 55). [Google Scholar]
- 13.Misharin AV, Morales-Nebreda L, Reyfman PA, et al. Monocyte-derived alveolar macrophages drive lung fibrosis and persist in the lung over the life span. The Journal of experimental medicine. 2017. August 7;214(8):2387–2404. doi: 10.1084/jem.20162152. PubMed PMID: ; PubMed Central PMCID: PMCPMC5551573. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Byrne AJ, Maher TM, Lloyd CM. Pulmonary Macrophages: A New Therapeutic Pathway in Fibrosing Lung Disease? Trends in molecular medicine. 2016. April;22(4):303–316. doi: 10.1016/j.molmed.2016.02.004. PubMed PMID: ; eng. [DOI] [PubMed] [Google Scholar]
- 15.Young LR, Gulleman PM, Short CW, et al. Epithelial-macrophage interactions determine pulmonary fibrosis susceptibility in Hermansky-Pudlak syndrome. JCI insight. 2016. October 20;1(17):e88947. doi: 10.1172/jci.insight.88947. PubMed PMID: ; PubMed Central PMCID: PMCPMC5070955. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rossi G, Cavazza A, Spagnolo P, et al. The role of macrophages in interstitial lung diseases: Number 3 in the Series “Pathology for the clinician” Edited by Peter Dorfmuller and Alberto Cavazza. Eur Respir Rev. 2017. September 30;26(145). doi: 10.1183/16000617.0009-2017. PubMed PMID: ; eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Moore MW, Herzog EL. Regulation and Relevance of Myofibroblast Responses in Idiopathic Pulmonary Fibrosis. Curr Pathobiol Rep. 2013. September;1(3):199–208. doi: 10.1007/s40139-013-0017-8. PubMed PMID: ; PubMed Central PMCID: PMCPMC4334480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kendall RT, Feghali-Bostwick CA. Fibroblasts in fibrosis: novel roles and mediators. Front Pharmacol. 2014;5:123. doi: 10.3389/fphar.2014.00123. PubMed PMID: ; PubMed Central PMCID: PMCPMC4034148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Robalo-Cordeiro C, Campos P, Carvalho L, et al. Idiopathic pulmonary fibrosis in the era of antifibrotic therapy: Searching for new opportunities grounded in evidence. Revista portuguesa de pneumologia. 2017. Sep-Oct;23(5):287–293. doi: 10.1016/j.rppnen.2017.05.005. PubMed PMID: ; eng. [DOI] [PubMed] [Google Scholar]
- 20.Fraser E, Hoyles RK. Therapeutic advances in idiopathic pulmonary fibrosis. Clinical medicine (London, England). 2016. February;16(1):42–51. doi: 10.7861/clinmedicine.16-1-42. PubMed PMID: ; eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lagares D, Grasberger P, Probst C, et al. Therapeutic Targeting of Fibroblast Durotaxis: A Novel Class of Anti-Fibrotic Therapies for IPF. C17. FASCINATING MECHANISMS IN LUNG FIBROSIS. American Thoracic Society International Conference Abstracts: American Thoracic Society; 2016. p. A4582–A4582. [Google Scholar]
- 22.Livnat-Levanon N, Glickman MH. Ubiquitin–Proteasome System and mitochondria — Reciprocity. Biochimica et Biophysica Acta (BBA) - Gene Regulatory Mechanisms. 2011. 2011/02/01/;1809(2):80–87. doi: 10.1016/j.bbagrm.2010.07.005. [DOI] [PubMed] [Google Scholar]
- 23.Guzy RD, Li L, Smith C, et al. Pulmonary fibrosis requires cell-autonomous mesenchymal fibroblast growth factor (FGF) signaling. J Biol Chem. 2017. June 23;292(25):10364–10378. doi: 10.1074/jbc.M117.791764. PubMed PMID: ; PubMed Central PMCID: PMCPMC5481550. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kim KK, Sisson TH, Horowitz JC. Fibroblast growth factors and pulmonary fibrosis: it’s more complex than it sounds. J Pathol. 2017. January;241(1):6–9. doi: 10.1002/path.4825. PubMed PMID: ; PubMed Central PMCID: PMCPMC5499705. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hinkson IV, Elias JE. The dynamic state of protein turnover: It’s about time. Trends in cell biology. 2011. May;21(5):293–303. doi: 10.1016/j.tcb.2011.02.002. PubMed PMID: ; eng. [DOI] [PubMed] [Google Scholar]
- 26.Rothman S How is the balance between protein synthesis and degradation achieved? Theoretical biology & medical modelling. 2010. June 23;7:25. doi: 10.1186/1742-4682-7-25. PubMed PMID: ; PubMed Central PMCID: PMCPMC2909984. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Glickman MH, Ciechanover A. The ubiquitin-proteasome proteolytic pathway: destruction for the sake of construction. Physiological reviews. 2002. April;82(2):373–428. doi: 10.1152/physrev.00027.2001. PubMed PMID: ; eng. [DOI] [PubMed] [Google Scholar]
- 28.Hershko A, Ciechanover A. The ubiquitin system. Annual review of biochemistry. 1998;67:425–79. doi: 10.1146/annurev.biochem.67.1.425. PubMed PMID: ; eng. [DOI] [PubMed] [Google Scholar]
- 29.Hong L, Huang HC, Jiang ZF. Relationship between amyloid-beta and the ubiquitin-proteasome system in Alzheimer’s disease. Neurological research. 2014. March;36(3):276–82. doi: 10.1179/1743132813y.0000000288. PubMed PMID: ; eng. [DOI] [PubMed] [Google Scholar]
- 30.Ihara Y, Morishima-Kawashima M, Nixon R. The ubiquitin-proteasome system and the autophagic-lysosomal system in Alzheimer disease. Cold Spring Harbor perspectives in medicine. 2012. August 1;2(8). doi: 10.1101/cshperspect.a006361. PubMed PMID: ; PubMed Central PMCID: PMCPMC3405832. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Salome M, Campos J, Keeshan K. TRIB2 and the ubiquitin proteasome system in cancer. Biochemical Society transactions. 2015. October;43(5):1089–94. doi: 10.1042/bst20150103. PubMed PMID: ; eng. [DOI] [PubMed] [Google Scholar]
- 32.Day SM, Divald A, Wang P, et al. Impaired assembly and post-translational regulation of 26S proteasome in human end-stage heart failure. Circulation Heart failure. 2013. May;6(3):544–9. doi: 10.1161/circheartfailure.112.000119. PubMed PMID: ; PubMed Central PMCID: PMCPMC3864674. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fessart D, Martin-Negrier ML, Claverol S, et al. Proteomic remodeling of proteasome in right heart failure. Journal of molecular and cellular cardiology. 2014. January;66:41–52. doi: 10.1016/j.yjmcc.2013.10.015. PubMed PMID: ; eng. [DOI] [PubMed] [Google Scholar]
- 34.Budenholzer L, Cheng CL, Li Y, et al. Proteasome Structure and Assembly. J Mol Biol. 2017. November 10;429(22):3500–3524. doi: 10.1016/j.jmb.2017.05.027. PubMed PMID: ; PubMed Central PMCID: PMCPMC5675778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wang C, Wang X. The interplay between autophagy and the ubiquitin-proteasome system in cardiac proteotoxicity. Biochim Biophys Acta. 2015. February;1852(2):188–94. doi: 10.1016/j.bbadis.2014.07.028. PubMed PMID: ; PubMed Central PMCID: PMCPMC4277934. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nam T, Han JH, Devkota S, et al. Emerging Paradigm of Crosstalk between Autophagy and the Ubiquitin-Proteasome System. Molecules and Cells. 2017. December 31;40(12):897–905. doi: 10.14348/molcells.2017.0226. PubMed PMID: ; PubMed Central PMCID: PMCPMC5750708. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pickart CM. Mechanisms underlying ubiquitination. Annual review of biochemistry. 2001;70:503–33. doi: 10.1146/annurev.biochem.70.1.503. PubMed PMID: ; eng. [DOI] [PubMed] [Google Scholar]
- 38.Kleiger G, Mayor T. Perilous journey: a tour of the ubiquitin-proteasome system. Trends in cell biology. 2014. June;24(6):352–9. doi: 10.1016/j.tcb.2013.12.003. PubMed PMID: ; PubMed Central PMCID: PMCPMC4037451. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Metzger MB, Pruneda JN, Klevit RE, et al. RING-type E3 ligases: master manipulators of E2 ubiquitin-conjugating enzymes and ubiquitination. Biochim Biophys Acta. 2014. January;1843(1):47–60. doi: 10.1016/j.bbamcr.2013.05.026. PubMed PMID: ; PubMed Central PMCID: PMCPMC4109693. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kamadurai HB, Qiu Y, Deng A, et al. Mechanism of ubiquitin ligation and lysine prioritization by a HECT E3. eLife. 2013. August 8;2:e00828. doi: 10.7554/eLife.00828. PubMed PMID: ; PubMed Central PMCID: PMCPMC3738095. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Spratt DE, Walden H, Shaw GS. RBR E3 ubiquitin ligases: new structures, new insights, new questions. Biochemical Journal. 2014. March 15;458(3):421–37. doi: 10.1042/bj20140006. PubMed PMID: ; PubMed Central PMCID: PMCPMC3940038. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Riley BE, Lougheed JC, Callaway K, et al. Structure and function of Parkin E3 ubiquitin ligase reveals aspects of RING and HECT ligases. Nature Communications. 2013;4:1982. doi: 10.1038/ncomms2982. PubMed PMID: ; PubMed Central PMCID: PMCPMC3709503. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wolf DH, Hilt W. The proteasome: a proteolytic nanomachine of cell regulation and waste disposal. Biochim Biophys Acta. 2004. November 29;1695(1–3):19–31. doi: 10.1016/j.bbamcr.2004.10.007. PubMed PMID: ; eng. [DOI] [PubMed] [Google Scholar]
- 44.Gu ZC, Enenkel C. Proteasome assembly. Cell Mol Life Sci. 2014. December;71(24):4729–45. doi: 10.1007/s00018-014-1699-8. PubMed PMID: ; eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Shirozu R, Yashiroda H, Murata S. Identification of minimum Rpn4-responsive elements in genes related to proteasome functions. Febs Letters. 2015. April 2;589(8):933–40. doi: 10.1016/j.febslet.2015.02.025. PubMed PMID: ; eng. [DOI] [PubMed] [Google Scholar]
- 46.Radhakrishnan SK, Lee CS, Young P, et al. Transcription factor Nrf1 mediates the proteasome recovery pathway after proteasome inhibition in mammalian cells. Mol Cell. 2010. April 9;38(1):17–28. doi: 10.1016/j.molcel.2010.02.029. PubMed PMID: ; PubMed Central PMCID: PMCPMC2874685. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sotzny F, Schormann E, Kuhlewindt I, et al. TCF11/Nrf1-Mediated Induction of Proteasome Expression Prevents Cytotoxicity by Rotenone. Antioxid Redox Signal. 2016. December 1;25(16):870–885. doi: 10.1089/ars.2015.6539. PubMed PMID: ; eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhang Y, Manning BD. mTORC1 signaling activates NRF1 to increase cellular proteasome levels. Cell cycle (Georgetown, Tex). 2015;14(13):2011–7. doi: 10.1080/15384101.2015.1044188. PubMed PMID: ; PubMed Central PMCID: PMCPMC4613906. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bragoszewski P, Turek M, Chacinska A. Control of mitochondrial biogenesis and function by the ubiquitin–proteasome system. Open Biol. 2017. April;7(4). doi: 10.1098/rsob.170007. PubMed PMID: ; PubMed Central PMCID: PMCPMC5413908. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lehmann G, Udasin RG, Ciechanover A. On the linkage between the ubiquitin-proteasome system and the mitochondria. Biochemical and Biophysical Research Communications. 2016. 2016/04/22/;473(1):80–86. doi: 10.1016/j.bbrc.2016.03.055. [DOI] [PubMed] [Google Scholar]
- 51.Friedman JR, Nunnari J. Mitochondrial form and function. Nature. 2014. January 16;505(7483):335–43. doi: 10.1038/nature12985. PubMed PMID: ; PubMed Central PMCID: PMCPMC4075653. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bueno M, Lai YC, Romero Y, et al. PINK1 deficiency impairs mitochondrial homeostasis and promotes lung fibrosis. J Clin Invest. 2015. February 2;125(2):521–38. doi: 10.1172/jci74942. PubMed PMID: ; PubMed Central PMCID: PMCPMC4319413. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Allen JF, de Paula WB. Mitochondrial genome function and maternal inheritance. Biochemical Society transactions. 2013. October;41(5):1298–304. doi: 10.1042/bst20130106. PubMed PMID: ; eng. [DOI] [PubMed] [Google Scholar]
- 54.Li W, Bengtson MH, Ulbrich A, et al. Genome-wide and functional annotation of human E3 ubiquitin ligases identifies MULAN, a mitochondrial E3 that regulates the organelle’s dynamics and signaling. PLoS One. 2008. January 23;3(1):e1487. doi: 10.1371/journal.pone.0001487. PubMed PMID: ; PubMed Central PMCID: PMCPMC2198940. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zhang J, Xu P, Wang Y, et al. Astaxanthin prevents pulmonary fibrosis by promoting myofibroblast apoptosis dependent on Drp1-mediated mitochondrial fission. J Cell Mol Med. 2015. September;19(9):2215–31. doi: 10.1111/jcmm.12609. PubMed PMID: ; PubMed Central PMCID: PMCPMC4568926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Radke S, Chander H, Schafer P, et al. Mitochondrial protein quality control by the proteasome involves ubiquitination and the protease Omi. J Biol Chem. 2008. May 9;283(19):12681–5. doi: 10.1074/jbc.C800036200. PubMed PMID: ; PubMed Central PMCID: PMCPMC2442309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Cohen MM, Leboucher GP, Livnat-Levanon N, et al. Ubiquitin-proteasome-dependent degradation of a mitofusin, a critical regulator of mitochondrial fusion. Molecular biology of the cell. 2008. June;19(6):2457–64. doi: 10.1091/mbc.E08-02-0227. PubMed PMID: ; PubMed Central PMCID: PMCPMC2397313. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Tar K, Dange T, Yang C, et al. Proteasomes associated with the Blm10 activator protein antagonize mitochondrial fission through degradation of the fission protein Dnm1. J Biol Chem. 2014. April 25;289(17):12145–56. doi: 10.1074/jbc.M114.554105. PubMed PMID: ; PubMed Central PMCID: PMCPMC4002118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Geng J, Huang X, Li Y, et al. Down-regulation of USP13 mediates phenotype transformation of fibroblasts in idiopathic pulmonary fibrosis. Respiratory Research. 2015. October 9;16:124. doi: 10.1186/s12931-015-0286-3. PubMed PMID: ; PubMed Central PMCID: PMCPMC4600336. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Nho RS, Hergert P, Kahm J, et al. Pathological alteration of FoxO3a activity promotes idiopathic pulmonary fibrosis fibroblast proliferation on type i collagen matrix. Am J Pathol. 2011. November;179(5):2420–30. doi: 10.1016/j.ajpath.2011.07.020. PubMed PMID: ; PubMed Central PMCID: PMCPMC3204034. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kral JB, Kuttke M, Schrottmaier WC, et al. Erratum: Sustained PI3K Activation exacerbates BLM-induced Lung Fibrosis via activation of pro-inflammatory and pro-fibrotic pathways. Sci Rep. 2016. May 20;7:26048. doi: 10.1038/srep26048. PubMed PMID: ; PubMed Central PMCID: PMCPMC4874234. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Tatler AL, Jenkins G. TGF-beta activation and lung fibrosis. Proc Am Thorac Soc. 2012. July;9(3):130–6. doi: 10.1513/pats.201201-003AW. PubMed PMID: . [DOI] [PubMed] [Google Scholar]
- 63.Fernandez IE, Eickelberg O. The impact of TGF-beta on lung fibrosis: from targeting to biomarkers. Proc Am Thorac Soc. 2012. July;9(3):111–6. doi: 10.1513/pats.201203-023AW. PubMed PMID: . [DOI] [PubMed] [Google Scholar]
- 64.Imamura T, Oshima Y, Hikita A. Regulation of TGF-β family signalling by ubiquitination and deubiquitination. The Journal of Biochemistry. 2013;154(6):481–489. doi: 10.1093/jb/mvt097. [DOI] [PubMed] [Google Scholar]
- 65.Kavsak P, Rasmussen RK, Causing CG, et al. Smad7 binds to Smurf2 to form an E3 ubiquitin ligase that targets the TGF beta receptor for degradation. Mol Cell. 2000. December;6(6):1365–75. PubMed PMID: ; eng. [DOI] [PubMed] [Google Scholar]
- 66.Tang LY, Yamashita M, Coussens NP, et al. Ablation of Smurf2 reveals an inhibition in TGF-beta signalling through multiple mono-ubiquitination of Smad3. Embo j. 2011. November 1;30(23):4777–89. doi: 10.1038/emboj.2011.393. PubMed PMID: ; PubMed Central PMCID: PMCPMC3243605. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Gvaramia D, Blaauboer ME, Hanemaaijer R, et al. Role of caveolin-1 in fibrotic diseases. Matrix Biol. 2013. August 08;32(6):307–15. doi: 10.1016/j.matbio.2013.03.005. PubMed PMID: . [DOI] [PubMed] [Google Scholar]
- 68.Mutlu GM, Budinger GR, Wu M, et al. Proteasomal inhibition after injury prevents fibrosis by modulating TGF-beta(1) signalling. Thorax. 2012. February;67(2):139–46. doi: 10.1136/thoraxjnl-2011-200717. PubMed PMID: ; PubMed Central PMCID: PMCPMC3595535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Sundaresan NR, Bindu S, Pillai VB, et al. SIRT3 Blocks Aging-Associated Tissue Fibrosis in Mice by Deacetylating and Activating Glycogen Synthase Kinase 3β. Molecular and Cellular Biology. 2016. March 1;36(5):678–92. doi: 10.1128/mcb.00586-15. PubMed PMID: ; PubMed Central PMCID: PMCPMC4760222. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Sosulski ML, Gongora R, Feghali-Bostwick C, et al. Sirtuin 3 Deregulation Promotes Pulmonary Fibrosis. J Gerontol A Biol Sci Med Sci. 2017. May 1;72(5):595–602. doi: 10.1093/gerona/glw151. PubMed PMID: ; eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Akamata K, Wei J, Bhattacharyya M, et al. SIRT3 is attenuated in systemic sclerosis skin and lungs, and its pharmacologic activation mitigates organ fibrosis. Oncotarget. 2016. October 25;7(43):69321–69336. doi: 10.18632/oncotarget.12504. PubMed PMID: ; PubMed Central PMCID: PMCPMC5342480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Bindu S, Pillai VB, Kanwal A, et al. SIRT3 blocks myofibroblast differentiation and pulmonary fibrosis by preventing mitochondrial DNA damage. Am J Physiol Lung Cell Mol Physiol. 2017. January 1;312(1):L68–L78. doi: 10.1152/ajplung.00188.2016. PubMed PMID: ; PubMed Central PMCID: PMCPMC5283928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Jablonski RP, Kim SJ, Cheresh P, et al. SIRT3 deficiency promotes lung fibrosis by augmenting alveolar epithelial cell mitochondrial DNA damage and apoptosis. FASEB J. 2017. June;31(6):2520–2532. doi: 10.1096/fj.201601077R. PubMed PMID: ; PubMed Central PMCID: PMCPMC5434657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Uhal B, Abdul-Hafez A. Bleomycin Downregulates ACE-2 In Alveolar Epithelial Cells Through A Posttranscriptional Mechanism Inhibitable By Angiotensin 1-7. A58. ANIMAL MODELS OF PULMONARY FIBROSIS. American Thoracic Society International Conference Abstracts: American Thoracic Society; 2010. p. A1979–A1979. [Google Scholar]
- 75.Sanders PM, Russell ST, Tisdale MJ. Angiotensin II directly induces muscle protein catabolism through the ubiquitin-proteasome proteolytic pathway and may play a role in cancer cachexia. British journal of cancer. 2005. August 22;93(4):425–34. doi: 10.1038/sj.bjc.6602725. PubMed PMID: ; PubMed Central PMCID: PMCPMC3217221. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Wynn TA, Ramalingam TR. Mechanisms of fibrosis: therapeutic translation for fibrotic disease. Nat Med. 2012. July 6;18(7):1028–40. doi: 10.1038/nm.2807. PubMed PMID: ; PubMed Central PMCID: PMCPMC3405917. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Manka SW, Carafoli F, Visse R, et al. Structural insights into triple-helical collagen cleavage by matrix metalloproteinase 1. Proc Natl Acad Sci U S A. 2012. July 31;109(31):12461–6. doi: 10.1073/pnas.1204991109. PubMed PMID: ; PubMed Central PMCID: PMCPMC3411981. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Lin PS, Chang HH, Yeh CY, et al. Transforming growth factor beta 1 increases collagen content, and stimulates procollagen I and tissue inhibitor of metalloproteinase-1 production of dental pulp cells: Role of MEK/ERK and activin receptor-like kinase-5/Smad signaling. Journal of the Formosan Medical Association = Taiwan yi zhi. 2017. May;116(5):351–358. doi: 10.1016/j.jfma.2016.07.014. PubMed PMID: ; eng. [DOI] [PubMed] [Google Scholar]
- 79.Goffin L, Seguin-Estevez Q, Alvarez M, et al. Transcriptional regulation of matrix metalloproteinase-1 and collagen 1A2 explains the anti-fibrotic effect exerted by proteasome inhibition in human dermal fibroblasts. Arthritis research & therapy. 2010;12(2):R73. doi: 10.1186/ar2991. PubMed PMID: ; PubMed Central PMCID: PMCPMC2888229. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Ma Y, Chen Y, Yang Y, et al. Proteasome inhibition attenuates heart failure during the late stages of pressure overload through alterations in collagen expression. Biochemical pharmacology. 2013. January 15;85(2):223–33. doi: 10.1016/j.bcp.2012.10.025. PubMed PMID: . [DOI] [PubMed] [Google Scholar]
- 81.Morrow JK, Lin HK, Sun SC, et al. Targeting ubiquitination for cancer therapies. Future medicinal chemistry. 2015;7(17):2333–50. doi: 10.4155/fmc.15.148. PubMed PMID: ; PubMed Central PMCID: PMCPMC4976843. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Shearer RF, Iconomou M, Watts CK, et al. Functional Roles of the E3 Ubiquitin Ligase UBR5 in Cancer. Molecular cancer research : MCR. 2015. December;13(12):1523–32. doi: 10.1158/1541-7786.Mcr-15-0383. PubMed PMID: ; eng. [DOI] [PubMed] [Google Scholar]
- 83.Shen M, Schmitt S, Buac D, et al. Targeting the ubiquitin-proteasome system for cancer therapy. Expert opinion on therapeutic targets. 2013. September;17(9):1091–108. doi: 10.1517/14728222.2013.815728. PubMed PMID: ; PubMed Central PMCID: PMCPMC3773690. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Masumoto K, Kitagawa M. E3 Ubiquitin Ligases as Molecular Targets in Human Oral Cancers. Current cancer drug targets. 2016;16(2):130–5. PubMed PMID: ; eng. [DOI] [PubMed] [Google Scholar]
- 85.Lear T, Chen BB. Therapeutic targets in fibrotic pathways. Cytokine. 2016. December;88:193–195. doi: 10.1016/j.cyto.2016.09.008. PubMed PMID: ; PubMed Central PMCID: PMCPMC5119757. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]