

Abstract
Exosomes mediate secretion of crystallin alphaB (cryAB), a well characterized molecular chaperone with anti-apoptotic activity. However, the mechanisms governing its packaging and secretion remained unexplored. In glioma cells, notwithstanding extensive phosphorylation of cryAB at Ser59 followed by Ser45 (Ser19 is largely unphosphorylated), we discovered that the majority of secreted exosomal cryAB is nonphosphorylated. Transient ectopic expression of a yellow fluorescent protein (YFP) tagged triple phosphomimic (3-SD) cryAB construct in cryAB absent glioma cells led to the formation of large cytosolic inclusions. Our findings demonstrate that mimicking phosphorylation significantly reduces cryAB secretion via exosomes. Moreover, decreased colocalization of 3-SD YFP-cryAB with multivesicular endosome (MVE) and exosome marker, CD63 or Rab27, a small GTPase regulating exocytosis of MVEs, suggests that phosphorylation deters packaging of cryAB in vesicles bound for secretion as exosomes. Additionally, we found that preventing O-GlcNAcylation on cryAB also curtailed its colocalization with CD63 and Rab27 resulting in reduced exosomal secretion. Thus, our study points to O-GlcNAcylation and lack of phosphorylation as being the selective processes involved in the packaging and secretion of cryAB via exosomes.
Keywords: Exosomes, cryAB, Phosphorylation, Glycosylation, Glioblastoma multiforme
1. Introduction
In the human central nervous system (CNS), cryAB/HspB5 is constitutively expressed in astrocytes and oligodendrocytes [1]. Under pathological conditions like glioblastoma multiforme (GBM), the levels of cryAB in brain are elevated [2,3], where it inhibits apoptosis by binding directly to and inhibiting caspase-3 activity [2–4]. cryAB levels are also elevated in numerous inflammation driven neurodegenerative disorders such as Parkinson’s disease (PD), Alexander’s disease, multiple sclerosis (MS), amyotrophic lateral sclerosis (ALS), age-related macular degeneration (AMD) and traumatic brain injury accumulating in astrocytes and oligodendrocytes of the CNS [5,6]. The presence of high levels of cryAB and anti-cryAB antibodies in serum and cerebrospinal fluid of MS patients [7], in the extracellular matrix surrounding retinal cells [8,9] and the ability of cryAB to present epitopes to activate T-cells [10,11] suggest extracellular roles to what is an intracellular protein. Studies show that cryAB is secreted and its secretion is mediated via a set of small discrete secretory vesicles, namely exosomes [9,12–15].
CryAB polypeptide of 175 amino acids consists of a hydrophobic N-term domain (NTD), a conserved central alpha-crystallin domain (ACD) and a C-term domain (CTD) containing a hydrophilic and flexible 12 residue tail [16]. Among post-translational modifications (PTMs) decorating cryAB, phosphorylation occurs at three Ser residues at positions 19,45 and 59, all in the hydrophobic NTD [17] and O-linked β-N-acetylglucosamine (O-GlcNAc) glycosylation at Thr170 in the flexible hydrophilic CTD [18]. Phosphorylation at Ser45 and Ser59 [19,20] and glycosylation at Thr170 [18–21] regulate cryAB activity and cellular localization. Ser45 is phosphorylated by p42/p44 ERK1/2 MAP kinase and Ser59 by the p-38 dependent MAPKAPK2 kinase; the kinase for Ser19 phosphorylation is unknown [22]. Phosphorylation is a dynamic process and varies depending on the environment in which the expressing cell finds itself. CryAB phosphorylation modulates dynamics between monomeric and different oligomeric states and thus its activity [16,23–25]. Similar to phosphorylation, O-GlcNAcylation on cryAB has been shown to be a dynamic process [18]. Studies on role of O-GlcNAc at Thr170 on cryAB in cellular localization and more importantly on its oligomerization are scant. However, it has been reported that O-GlcNAcylation occurs on both phosphorylated and nonphosphorylated cryAB [26] and has been implicated in the translocation of cryAB into the nucleus and reduced cryAB phosphorylation [21].
Despite the fact that cryAB is secreted via exosomes, an analysis of its sequence by web-based analytical tools like SecretomeP 2.0 [27] and SignalP 4.1 [28] shows that signal peptides or localization sequences are wanting. The molecular mechanisms targeting cryAB for exosomal secretion remained, until now, unknown. We speculated that phosphorylation and glycosylation of cryAB may be affecting its exosomal secretion.
2. Methods and materials
2.1. Cell culture reagents
U118MG cells were purchased from ATCC (Manassas, VA), cell culture media (EMEM and DMEM) and phosphate buffered saline (PBS) were from Mediatech Inc. (Manassas, VA). Opti-MEM media, Lipofectamine 2000 and penicillin/streptomycin were from Invitrogen (Carlsbad, CA) and fetal bovine serum (FBS) was from Atlas Biologicals Inc. (Fort Collins, CO). IL-1β, TNF-α and poly-L-lysine were from Sigma (St. Louis, MO). Protease and phosphatase inhibitor cocktail, Halt™, was obtained from Thermo Scientific (Rockford, IL). RIPA lysis buffer (Chemcruz™) was obtained from Santa Cruz Biotechnology (Dallas, TX). 100 mm cell culture dishes were from Corning Inc. (Corning, NY). U373 cells were a generous gift from Dr. Suraj Bhat, JEI, UCLA.
2.2. Cell culture and transfection
U373 cells were cultured in complete EMEM media (containing 10% FBS & 100 units/ml of penicillin/streptomycin) at 37 °C in a 5% CO2 incubator. Overnight cultures of cells were grown to 80% confluence in 100 mm culture dishes. Cells were washed 3× with PBS and fresh serum free EMEM media was added to each dish and the cells incubated for 24 h and media collected for isolating exosomes. Cells were harvested by lysing in RIPA lysis buffer containing protease and phosphatase inhibitors.
U118MG cells were cultured in complete DMEM Media (containing 10% FBS and 100 units/ml of penicillin/streptomycin) at 37 °C in a 5% CO2 incubator according to the ATCC instructions. For transfection, overnight cultures of cells grown to 90% confluence in 100 mm dishes coated with poly-L-lysine were transfected with Lipofectamine 2000 according to the manufacturer’s instructions. Briefly, cells were transfected with total 6 μg/100 mm dish of pZsYellow1-C1 (YFP) plasmids encoding the cryAB gene (or its mutants) along with 30 μl of Lipofectamine 2000. After 6 h, transfection media was removed and replaced with fresh complete DMEM medium. After overnight incubation, fresh serum free DMEM medium was added and cells allowed to incubate for 24 h after which media was collected for exosome isolation. The cells were harvested by lysing using the RIPA lysis buffer containing protease and phosphatase inhibitors.
2.3. Exosome isolation
Conditioned media from transfected, treated/untreated cells was collected and pooled and exosomes isolated by sequential centrifugation, all steps done at 4 °C. In brief, collected media was subjected to 600 g for 10 min, 3000 g for 10 min, and 10,000 g for 30 min. Any pellets formed were discarded. The supernatant media was then ultracentrifuged at 150,000 g for 3–4 h. The resulting pellet was re-suspended in PBS for EM analysis and in PBS containing a cocktail of protease and phosphatase inhibitors for WB analysis.
2.4. Electron microscopy
In brief, the exosomal pellets isolated were coated onto 200 mesh fomvar coated grid and dried at room temp. These grids were washed with DI water and stained with 1% uranyl acetate solution for 5 min. The stain was washed off with 70% ethanol followed by 4 washes with molecular grade water. The grids were loaded onto sample holder of the electron microscope (FEI Tecnai F20 200 keV microscope) and images were captured after exposing sample to 80 keV electron beam.
2.5. Site directed mutagenesis (SDM)
Yellow (pZsYellow1-Cl) expression vector was obtained from Clontech (Palo Alto, CA), plasmid DNA extraction kits were from Qiagen (Valencia, CA). QuickChange SDM kit was from Agilent Technologies Inc. (Clara, CA). Yellow (pZsYellow1-Cl) expression vector with the WT human cryAB gene was used as template for PCR to generate phosphomimic and phosphodeficient of cryAB. Appropriate SDM primers of human cryAB for the phospho mutants (S19A, S45A, S59A, S19D, S45D, S59D) and non-O-GlcNAcylatable T170A were designed and ordered from IDT (Integrated DNA technologies, Coralville, IO) and used for SDM reactions according to the instructions provided in the kit, with reactions carried out under the following conditions: 95 °C for 50 s, 95 °C for 30 s, 60 °C for 50 s and 68 °C for 5 min for 16 cycles and followed by overall extension at 68 °C for 7 min. Double and triple phosphodeficient and phosphomimetic constructs were prepared sequentially using the single phosphomimetic and phosphodeficient mutants. The unmutated plasmid DNA was digested with Dpn I for 1 h at 37 °C and 3 μl of PCR product was transformed with XL-10 Gold competent cells. The transformants were selected on LB agar medium plates containing 50 μg/ml Kanamycin. The mutant constructs were sequenced and confirmed by DNA sequence analysis at the UAMS Sequencing Core Facility using a 3130XL Genetic Analyzer (Applied Biosystems, Foster City, CA).
2.6. SDS-PAGE and Western blotting
Equal amounts of exosomal pellets and cell lysates, as determined by BCA protein estimation, were boiled with 4× Laemmli’s buffer containing 10% beta-mercaptoethanol and run on a 12% SDS-PAGE denaturing gel. The proteins were transferred to PVDF membranes using conventional transfer apparatus. Blots were blocked with 5% BSA dissolved in Tris Buffered Saline (TBS) containing 0.01% Tween-20 (TBS-t) for 1 h at room temp. Blots were probed with primary Abs [anti-cryAB monoclonal antibody (1:1000) (Santa Cruz Biotech, Dallas, TX), antiphospho-Ser19, antiphospho-Ser45 or anti-phospho-Ser59 pAbs (all at 1:1000), anti-CD63 (SBI Biosciences, Mountain View, CA), anti-GM130 (BD Biosciences, San Jose, CA), anti-RCFP pAb for YFP tag (Clontech Laboratories Inc., San Francisco, CA) or anti-β-actin (1:5000) (Cell Signaling Technology, Beverly, MA)] diluted in blocking buffer for 1 h at room temp. Blots were washed 3× with TBS-t and then incubated with HRP conjugated anti-rabbit or anti-mouse Ab (1:10,000) (Santa Cruz Biotech, Dallas, TX) diluted in blocking buffer at room temperature for 1 h. Blots were washed thoroughly 3× with TBS-t and developed with a chemiluminescence reagent – Luminaforte developing reagent from Millipore (Billerica, MA).The blots were stripped using Restore Western blot Stripping Buffer (Pierce Scientific, Fisher Scientific, Pittsburgh, PA). The blots were washed 3× with TBS-t and reprobed for total cryAB, CD63, GM130 or β-actin.
2.7. Confocal microscopy
Cells cultured in a 4 chambered Lab-Tek Borosilicate Coverglass system slide were transfected with YFP-cryAB or its mutants. 48 h later, the cells were visualized under LSM 510 confocal microscope and live cell images were captured with ×63 oil immersion lens. Quantification of cytosolic inclusions was done by calculating an average of 25 cells per chamber from four different experiments for each mutant. Cells with two or cytosolic inclusions greater than 1 μm were marked as cells containing inclusions.
To study colocalization of YFP-cryAB with CD63 and Rab27, cells were fixed with 4% paraformaldehyde and then permeabilized with 0.1% Triton X-100 for 20 min. Cells were washed with PBS, blocked with 5% normal goat serum or 3% BSA in tris buffered saline containing 1% Triton X-100 and then incubated with primary mAb against CD63 (BD Biosciences, San Jose, CA) or primary pAb against Rab27 (Synaptic Systems, Gottingen, Germany) overnight, at 4 °C at a dilution of 1:500. Cells were washed with PBS and incubated with secondary goat anti-mouse Ab or goat anti-rabbit Ab, respectively, conjugated to Alexa Flour 647 (Life Technologies, Carlsbad, CA) at a dilution of 1:500 for 1 h at room temp. The cells were washed with PBS and images were acquired using a dual channel filter setting for YFP and Alexaflour 647 under Olympus Fluoview FV1000 confocal microscope using ×100 oil immersion lens. Colocalization analysis for Pearson correlation coefficients and Manders colocalization coefficients were obtained using the JACoP plugin in ImageJ software (Version 1.48; Research Service Branch, National Institutes of Health, Bethesda, MD) [29]. In short, the confocal images were opened in ImageJ software. A single cell expressing YFP-cryAB was outlined as the region of interest (ROI). The background was subtracted and the area outside the ROI was cleared. The image was then spilt into the RGB channels. The blue channel image was deleted. The JCoP plugin was then run within the ImageJ analysis software and colocalization values for Pearson’s correlation coefficients and Manders colocalization coefficients were derived.
2.8. Statistical analyses
To determine statistical significance in rations of phosphorylated cryAB or quantification of secreted YFP-cryAB in exosomes, significance was calculated using GraphPad Prism’s (Version 5.03) one way ANOVA followed by Bonferroni’s multiple comparison test. For each group, the average of three independent experiments was determined and results were expressed as mean ± SEM. Value of p < 0.05 was considered as statistically significant. To determine statistical significance in quantification of % cells containing inclusions of cryAB and its mutants, significance was calculated using GraphPad Prism’s one way ANOVA followed by Bonferroni’s multiple comparison test. For each group, the average of four independent experiments was determined and results were expressed as mean ± SEM. Value of p < 0.0001 was considered as statistically significant.
3. Results
3.1. CryAB phosphorylation in U373 human glioma cells vs in secreted exosomes
Exosomes isolated from culture media of U373 glioma cells using previously reported protocol [15], were characterized by electron microscopy analysis and their morphology, size (70–80 nm) and absence of cellular debris were confirmed (Fig. 1 A-D). In a comparative Western blot analysis of equivalent quantities of U373 cell lysate and exosome preparation, the exosomal marker protein CD63 was found enriched in the exosome lane. (Fig. 1 E).
Fig. 1.
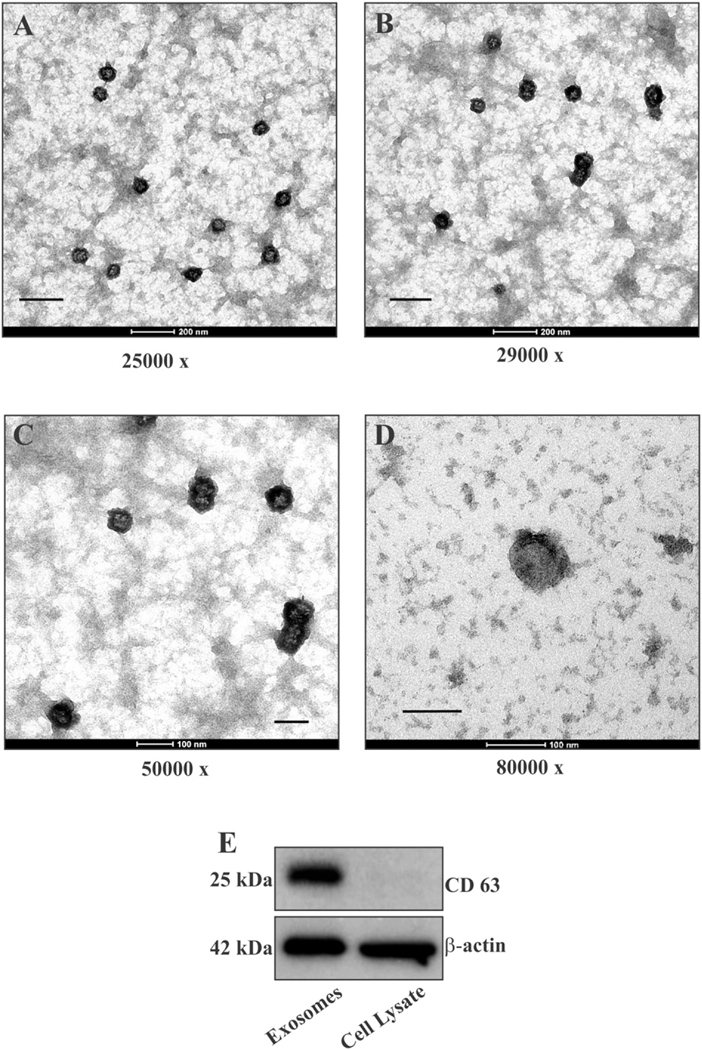
Characterization of exosomal preparation. (A–D) Exosomes isolated from media of U373 human glioma cells were analyzed by TEM at 80 keV. Images of different fields at different magnifications consistently showed a set of uniform and distinct vesicles with diameter ranging between 70 and 80 nm (scale bar: A = 500 nm, B = 250 nm, C = 100 nm, D = 100 nm). (B) Western blot of equivalent quantities of exosomes and U373 cell lysate showing enrichment of CD63 in exosomal preparation.
A comparative analysis of phosphorylated cryAB on equivalent quantities of total exosomal proteins and total cell lysate was carried out by Western blotting (WB) using specific anti-phosphoSer19, anti-phosphoSer45 and anti-phosphoSer59 polyclonal antibodies (Abs).
U373 cells, at the time of harvesting, show presence of varying but significant levels of phosphorylated cryAB, while in exosomes, only small levels of phosphorylation on Ser45 and negligible levels of cryAB phosphorylated on Ser19 and Ser59 were detected (Fig. 2 A). Reprobing the same blots for total cryAB showed significant quantities of cryAB present in exosomes. Quantifying ratios of phosphorylated cryAB to total cryAB revealed that significant amounts of cryAB were phosphorylated in cells with 25% of total cryAB being phosphorylated at Ser19, 35% of total cryAB phosphorylated at Ser45 and 35% of total cryAB found phosphorylated at Ser59. In comparison, immunoblots detected low levels of cryAB phosphorylation on Ser45 (13%) and negligible levels of cryAB phosphorylated on Ser19 (2%) and Ser59 (3%) in secreted exosomes. The ratio of phosphorylated cryAB to nonphosphorylated cryAB was consistently and significantly lower in exosomes (Fig. 2 B). Thus the majority of cryAB secreted via exosomes is nonphosphorylated, suggesting that phosphorylation may actually hinder packaging and secretion of cryAB via exosomes.
Fig. 2.
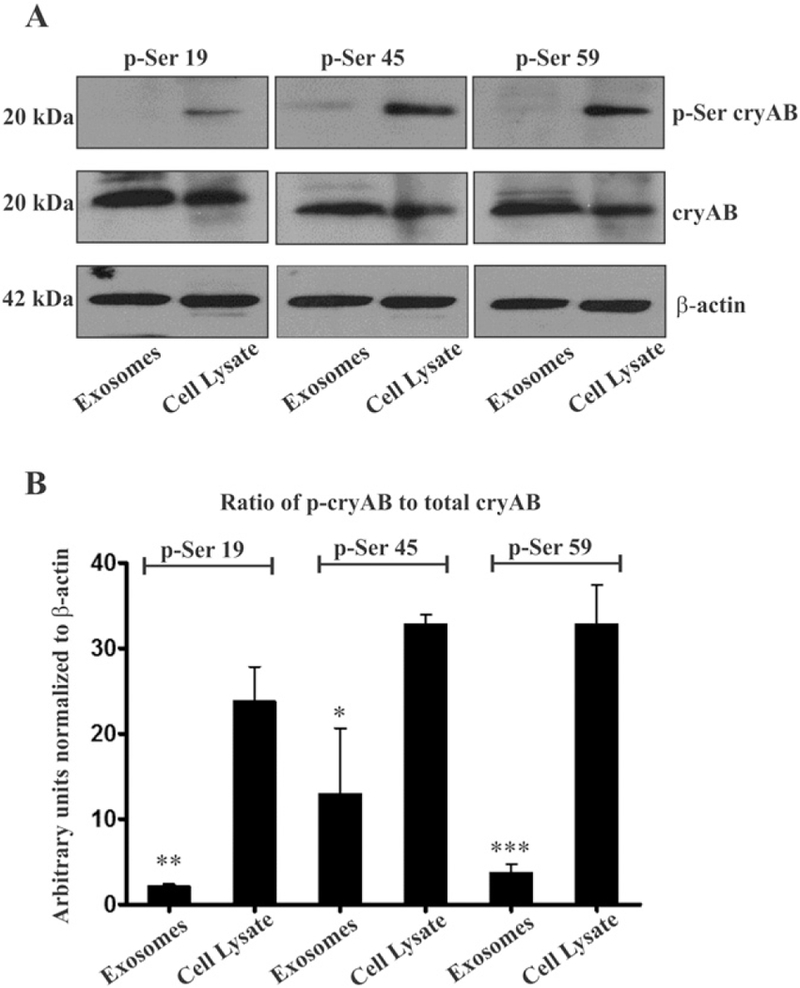
Phosphorylated cryAB in U373 cells vs secreted exosomes. (A) In three independent experiments, WB detected cryAB being phosphorylated at all the three serine residues in U373 cells. In contrast, in exosomes, Abs failed to detect any cryAB phosphorylated at Ser19 or Ser59, while a small quantity of cryAB phosphorylated at Ser45 was detected (top panel). Reprobing blots for full length cryAB with a mAb (middle panel) showed abundant quantities of cryAB being present in both U373 cells and exosomes. β-Actin was used as loading control. (B) Ratio of phosphorylated cryAB to the total cryAB in U373cells and exosomes. Statistical significance determined by one way ANOVA, values indicate mean ± SEM (n = 3), (* = p < 0.05, ** = p < 0.01,*** = p < 0.001).
In order to investigate the role of phosphorylation in regulating cellular distribution and secretion of cryAB via the exosomal secretory pathway, we generated phosphomimic and phosphodeficient constructs of cryAB (Fig. 3 A) by replacing Ser to Asp (SD) and Ser to Ala (SA), respectively, at positions 19, 45 and 59 on a cryAB wild type (WT) construct with a N-term YFP tag (YFP-cryAB). Similarly, based on the report that O-GlcNAcylation plays a role in cellular localization of cryAB, a non-O-GlcNAcylatable, Thr to Ala (T170A) mutant of N-terminal YFP tagged cryAB was generated to investigate whether preventing glycosylation affects cellular distribution and exosomal secretion of cryAB. The YFP-cryAB mutants were confirmed by sequencing.
Fig. 3.
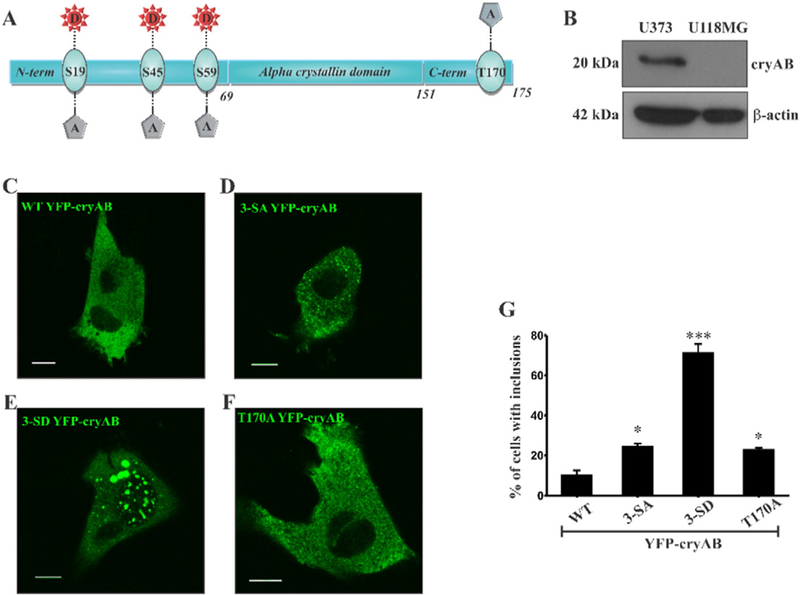
Expression of YFP-cryAB constructs in U118MG cells. U118MG cells do not express cryAB. (A) Full length cryAB depicting Ser phosphorylation sites at positions 19,45, and 59 in the NTD and the O-GlcNAc site at Thr170 in the flexible CTD. (B) Western blot confirms presence of cryAB in U373 cells but not in U118MG cells. (C–F) Confocal images ofU118MG cells ectopically expressing WTYFP-cryAB (C), 3-SAYFP-cryAB (D), 3-SD YFP-cryAB (E) and T170AYFP-cryAB (F) in U118MG cells. Scale bars = 10 μm. (G) Percent of cells with large cytosolic inclusions. 25 cells for each YFP-cryAB construct with inclusions (at least 2 larger than 1 μm) were counted from random fields in 4 experiments. Expression of 3-SD YFP-cryAB mutant significantly increased the number of cells with inclusions. Statistical significance determined by one way ANOVA, values indicate mean ± SEM (n = 4), (* = p < 0.05, ** = p < 0.01, *** = p < 0.0001 ).
3.2. Expression of YFP-cryAB constructs in U118MG (cryAB absent) glioma cells
In order to test our hypothesis and eliminate any influence of endogenous cryAB, we employed a cryAB absent glioma cell line, U118MG glioma cells.
U118MG cells do not express cryAB [30] which was confirmed by WB (Fig. 3 B). Inflammatory cytokines, interleukin 1-beta (IL-1β) or tumor necrosis factor-alpha (TNF-α), did not induce cryAB expression in U118MG cells (Sup Fig. 1A & B). U118MG secrete exosomes as demonstrated in immunoblots by the presence of highly enriched exosomal marker protein CD63 in exosomes isolated from culture media (Sup Fig. 1B) compared to small amounts detected in the cells (Sup Fig. 1A).
To abrogate variability brought about by single or double phosphomimic cryAB which could potentially trigger phosphorylation of remaining Ser residues, we limited our study to triple phosphomimic (3-SD), triple phosphodeficient (3-SA) and non-O-GlcNAcylatable (T170A) cryAB mutants. U118MG cells were transiently transfected with WT, 3-SA, 3-SD and T170A YFP-cryAB. The YFP tag at N-terminus (size of expressed protein increased to ~47 kDa) helped in visualizing its cellular localization and secretion by both confocal microscopy and WB.
Live cell images using confocal microscopy (Fig. 3C–F) of U118MG cells ectopically expressing WT YFP-cryAB showed a uniform cytosolic distribution for the wild type (WT) protein (Fig. 3C). Phosphodeficient 3-SA cryAB, exhibited a diffuse punctate distribution appearing uniformly throughout the cytosol (Fig. 3D). A similar diffuse cytosolic distribution but with smaller puncta, was observed with non-O-GlcNAcylatable T170A cryAB (Fig. 3 F). However, majority of cells expressing the phosphomimic 3-SD cryAB displayed a low level of uniform cellular distribution interspersed with few large, bright cytosolic inclusions (Fig. 3E). The number of cells with two or more cytosolic inclusions, 1 μm or more in size, was thus significantly higher (>75% of U118MG cells) for those expressing the 3-SD construct (Fig. 3G).
We identified the specific phosphorylation site that resulted in formation of cytosolic inclusions. An analysis of confocal images showed that U118MG cells expressing double (2-SD) and single (1-SD) phosphomimic cryAB constructs (Sup Fig. 2, panels C & D), Ser19 phosphorylation on cryAB was solely responsible for formation of cytoplasmic inclusions. Double (2-SA) and single (1-SA) phosphodeficient mutants did not significantly alter cellular distribution. Thus, our results show that mimicking phosphorylation alters the cellular distribution of cryAB with phosphorylation at Ser19 being specifically responsible for generating cytosolic inclusions.
3.3. Exosomal secretion of YFP-cryAB mutants by UH8MG cells
In order to investigate the effect of mimicking phosphorylation or preventing glycosylation on cryAB secretion, exosomes were isolated from culture media of U118MG cells ectopically expressing WT YFP-cryAB or its 3-SD, 3-SA or T170A mutants and characterized by WB for exosomal marker CD63 (Fig. 4A & B). Absence of Golgi apparatus marker protein, GM130, ruled out contamination with other cellular vesicles or debris. Exosomal marker CD63 was found abundant in the exosome lanes.
Fig. 4.
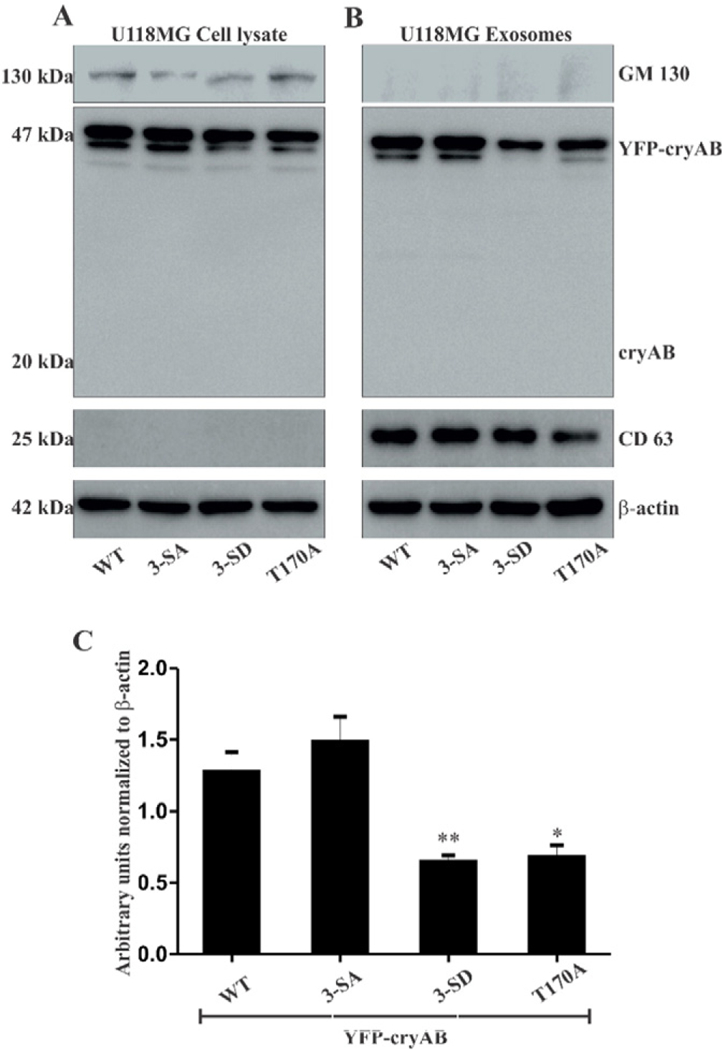
(A) WB of U118MG cells transiently transfected with WT, 3-SA, 3-SD and T170A YFP-cryAB constructs. (B) WB of exosomes isolated from culture media of transfected U118MG cells. YFP-cryAB constructs expressed in U118MG cells were secreted via exosomes, WT and 3-SA appeared to be secreted in fairly large quantities while levels of 3-SD secreted were reduced. Lack of a cryAB band at 20 kDa mark suggests that the YFP tag in YFP-cryAB was not cleaved off. GM130, a Golgi marker protein, was detected in the (A) U118MG cells, but (B) absent in the exosome lanes. Exosome marker protein, (B) CD63 was found enriched in the exosome lanes. β-Actin was used as a loading control. (C) Quantification of the Western blots showed a significant decrease in levels of phosphomimic (3-SD) and non-O-GlcNAcylatable (T170A) YFP-cryAB in isolated exosomes. β-Actin was used as loading control. Statistical significance determined by one way ANOVA, values indicate mean ± SEM (n = 3), (* = p < 0.05, ** = p < 0.01.
Total cryAB secreted in exosomes by transiently transfected U118MG cells was analyzed by Western blots (Fig. 4B). Quantification of these blots (Fig. 4C), from three independent experiments, showed that levels of 3-SD and T170A secreted in exosomes are significantly lower than those for WT or 3-SA YFP-cryAB mutants. Western blots for U118MG cells transiently expressing the pZsYellow1-C1 (YFP) plasmid alone showed a robust expression for YFP in the cells. Immunoblots also detected presence of abundant levels of YFP in the exosomes isolated from these cells (Sup Fig. 3). Although the expressed YFP is secreted via exosomes by U118MG cells and may probably account for the presence of 3-SD and T170A YFP-cryAB in exosomes, the suppressed exosomal secretion of these two cryAB mutants can be conclusively attributed to the specific cryAB mutations.
Our results show that cryAB is a secreted protein despite lacking localization or signal sequences. Furthermore, our data demonstrate that while ectopic expression of WT cryAB did result in its secretion via exosomes, mimicking phosphorylation or preventing glycosylation significantly reduced levels of cryAB secreted via exosomes, confirming that phosphorylation negatively regulates exosomal secretion of cryAB, with glycosylation at Thr170 also being essential for cryAB secretion. Our findings suggest a selective process of packaging glycosylated and nonphosphorylated cryAB for exosomal secretion.
3.4. Co-localization of YFP-cryAB mutants with CD63 and Rab27
Having established that mimicking phosphorylation and preventing glycosylation reduced exosomal secretion of cryAB, we wanted to understand how phosphorylation and glycosylation govern cryAB packaging upstream of exosomal secretion. Exosomes arise by the fusion of MVEs with the cell membrane and release of intraluminal vesicles into the extracellular milieu. We studied association of CD63 or Rab27 with various YFP-cryAB constructs ectopically expressed in U118MG cells. CD63 is a tetraspanin protein associated with the endosomal membranes and employed as MVE marker [31]. Two isoforms of Rab27, a small GTPase, are involved in the exocytosis of MVEs with subsequent release of intraluminal vesicles (ILVs) extracellularly as exosomes [32]. We reasoned that colocalization of any YFP-cryAB constructs with CD63 and Rab27 would infer the expressed construct as being packaged into MVEs and targeted for secretion as exosomal cargo.
U118MG cells expressing either WT, 3-SA, 3-SD and T170A constructs of YFP-cryAB (green in confocal images) were fixed, permeabilized and labelled with an anti-CD63 mouse mAb or with an anti-Rab27 rabbit pAb (recognizes both Rab27 isoforms) followed with corresponding secondary Abs conjugated with AlexaFlour 647 (red in confocal images).
Confocal images were acquired using the Olympus Fluoview1000 confocal microscope. Qualitative analysis of colocalization, Pearson’s correlation coefficient (PCC) and Manders overlap coefficients (M1 = fraction of CD63 or Rab27 overlapping with YFP-cryAB constructs, M2 = fraction of YFP-cryAB constructs overlapping CD63 or Rab27), was carried out using the JACoP plugin in ImageJ [29].
Captured images showed large quantities of WT (Fig. 5, panels A & E) and 3-SA (Fig. 5, panels B & F) colocalizing with CD63 and Rab27. In contrast, significant decrease in colocalization was seen for 3-SD including the cytosolic inclusions (Fig. 5, panels C & G) and T170A with CD63 and Rab27 (Fig. 5, panels D & H). Consistent with confocal images, the JACoP analysis indicated significant colocalization between CD63 and WT (PCC = 0.8, M1 = 0.76 & M2 = 0.4) and between CD63 and 3-SA (PCC = 0.74, M1 = 0.70 & M2 = 0.33). Mimicking phosphorylation or preventing glycosylation reduced localization of CD63 with 3-SD (PCC = 0.68, M1 = 0.32 & M2 = 0.17) and with T170A (PCC = 0.63, M1 = 0.68 & M2 = 0.l7) respectively. Thus mimicking phosphorylation or preventing glycosylation negatively regulated packaging of cryAB into vesicles which constitute MVEs. JACoP analysis showed a similar pattern with WT (PCC = 0.70, M1 = 0.50 & M2 = 0.33) and 3-SA (PCC = 0.72, M1 = 0.45 & M2 = 0.44) colocalizing with Rab27. Mimicking phosphorylation or preventing glycosylation reduced localization of Rab27 with 3-SD (PCC = 0.46, M1 = 0.33 & M2 = 0.24) and with T170A (PCC = 0.46, M1 = 0.46 & M2 = 0.21). PCC values and reduced fractions of Rab27 colocalizing with 3-SD and Tl70A suggest that only a small fraction of these constructs get targeted for secretion via exosomes.
Fig. 5.
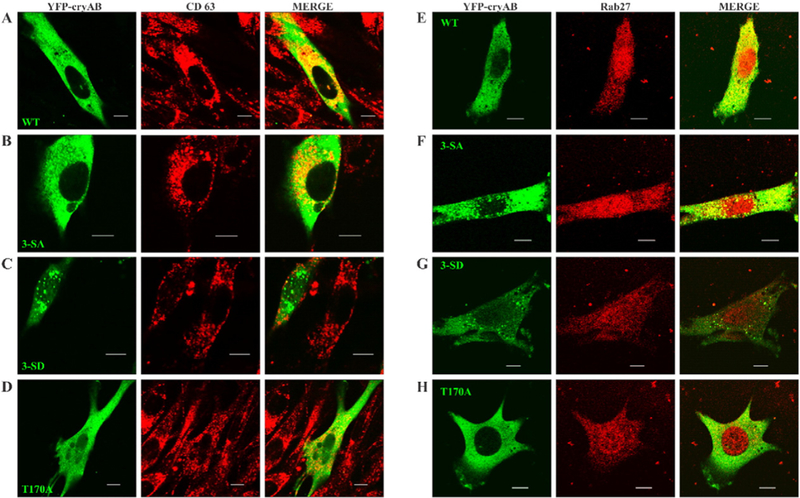
YFP-cryAB colocalization with CD63 &Rab27. U118MG Cells transfected with WT (panel A &E), phosphodeficient 3-SA (panel B&F), phosphomimetic 3-SD (panel C&G) andnon-O-GlcNAcylatable T170A (panel D&H) constructs of YFP-cryAB were labelled with CD63 (red, panels A-D), or Rab27 (red, panels E-H). Confocal images showed large quantities of the WT and 3-SA colocalizing with CD63 (panels A & B) and Rab27 (panels D & E), while expression of 3-SD and T170A YFP-cryAB constructs markedly reduced colocalization with CD63 (panel C) and Rab27 (panel F). Colocalization analysis of these images using JACoP plugin of ImageJ supported the observations. Scale bars = 10 μm.
By virtue of its association with CD63 and Rab27, significant quantities of the WT and 3-SA YFP-cryAB are packaged into vesicles bound for secretion as exosomes. In contrast, mimicking phosphorylation or preventing glycosylation on cryAB hinders its association with CD63 enriched endosomal compartments or Rab27 associated vesicles targeted for secretion. This data, coupled with earlier secretion results, demonstrates that the nonphosphorylated cryAB is selectively packaged into vesicles for secretion as exosomal cargo. Additionally, our results also indicate the importance of O-GlcNAc atThr170 for packaging and exosomal secretion of cryAB.
4. Discussion
Exosomes constitute the non-classical secretory pathway for many cytosolic proteins. Although exosomes, irrespective of their cellular origin, carry a conserved set of proteins [33], many cells secrete certain proteins which are specific to those cell types. Our previous study has shown that inflammatory cytokines elevate levels of cryAB in astrocytes and secreted exosomes. Additionally, exosomal secretion of some proteins involved in glioma invasiveness and progression, is also increased [15]. The molecular mechanisms targeting these proteins to vesicles fated for secretion as exosomes is unexplored. We sought to address this enigma using cryAB.
The results of our study support the following important findings. Notwithstanding the considerable amount of cryAB carried in exosomes [15], a majority of the constitutively secreted exosomal cryAB is nonphosphorylated (Fig. 2). Ser59 is the major site for cryAB phosphorylation in glioma cells, followed by phosphorylation at Ser45. In exosomes, Ser59 in cryAB is devoid of phosphorylation while very small quantities of phosphorylated Ser45 still exist. Ser19 phosphorylation is found virtually absent on cryAB in glioma cells and exosomes.
Analysis of confocal images showed that mimicking phosphorylation altered cellular distribution and localization of the protein and promoted formation of large cytosolic inclusions (Fig. 3 G). Our results corroborate previously published study [34] showing that phosphorylation of cryAB at both Ser45 and Ser59 is required for translocation to the nucleus (Sup Fig. 2, panel C). Interestingly, this study also led to the finding that the cytosolic inclusions observed contained at least one Ser19 to Asp substitution (Sup Fig. 2, panel C & D) and that Ser19 phosphorylation on cryAB was primarily responsible for the generation of cytoplasmic inclusions. However, it is unknown whether mimicking phosphorylation at Ser19 is solely responsible for formation of cytoplasmic inclusions or it triggers subsequent phosphorylation of the remaining Ser residues (which remained unaltered in the S19D mutant) in NTD of cryAB.
Ectopically expressed WT YFP-cryAB was secreted in exosomes (Fig. 4 A & B). Our findings clearly show that, despite wanting signal peptides, cryAB is a secreted protein. Moreover, mimicking phosphorylation (Ser19, Ser45 & Ser59) on cryAB result in decreased levels of this protein being secreted via exosomes (Fig. 4 C), implying that the nonphosphorylated cryAB is selectively secreted via exosomes. Additionally, we find that exosomal secretion of non-O-GlcNAcylatable cryAB is also significantly reduced (Fig. 4 C) revealing the importance of O-GlcNAc modification for exosomal secretion of cryAB.
Finally, the propensity of phosphomimic cryAB mutant to form large cytosolic inclusions and its decreased colocalization with either CD63 or Rab27, strongly suggested that mimicking phosphorylation hindered the packaging of cryAB into MVEs and subsequent secretion via exosomes. Interestingly, preventing glycosylation on cryAB also had a similar effect with interfering in its association with CD63 and Rab27 leading to reduced levels of the mutant being secreted via exosomes. Based on our data we conclude that lack of phosphorylation of Ser residues, primarily at position 59 and to some extent at position 45 on cryAB promote its packaging and secretion via exosomes. Additionally, O-GlcNAcylation at Thr170 is also an essential modification for packaging and secretion of cryAB via exosomes.
Previous structural studies have shown that cryAB exists as a polydisperse species of 24–32mer oligomers in its native state. Oligomerization of cryAB follows a ‘hierarchical’ assembly of its monomers. The fundamental building block of cryAB oligomers is a curved dimer formed by the interactions of (β-strands of the core alpha-crystallin domain (ACD) [16]. Domain swapping of one dimer’s flexible CTD with another, through CTD’s interactions with the groove created by the (β-strand region of the dimer forms a stable hexameric subunit [16,35–37]. Subsequent formation of higher ordered oligomers of cryAB depend upon the interactions and conformation of the hydrophobic NTD [16,38–41]. Phosphorylation plays an important role in the dynamics of oligomeric state and polydispersity of cryAB, thus affecting its function and cellular distribution [16,23–25,39]. Studies show that phosphorylation at Ser59 and Ser45 lead to its translocation to the nucleus and association with S35 nuclear speckles [34].
It could be speculated that, upon phosphorylation, addition of negatively charged phosphate groups on Ser residues in the NTD of cryAB renders the hydrophobic NTD more hydrophilic and cause the interacting NTDs to mutually repel each other. This change would disrupt the hydrophobic NTD interactions holding together multiple hexameric subunits forming large oligomers, resulting in unravelling the oligomeric structures into smaller oligomers or monomers. These small cryAB oligomers or monomers could then recruit other smaller oligomers and form random disordered assemblies through the interactions of ACD β-strands. With the phosphorylated NTDs being unable to interact to form higher ordered oligomers, such assemblies would result in formation of cytosolic inclusions. These cytosolic inclusions may invariably hinder association of phosphorylated cryAB with endosomal vesicles marked for secretion as exosomes. While phosphorylation has been shown to disrupt or break down large oligomers of cryAB into smaller oligomers or monomers, dephosphorylation may trigger the hierarchical assembly of cryAB molecules into structured, higher order oligomers by promoting and stabilizing hydrophobic interactions of cryAB’s NTDs. The appearance of uniformly distributed puncta observed with 3-SA YFP-cryAB expression implies presence of such assemblies of highly ordered, very large oligomeric structures of cryAB. It can be theorized here that these structured large oligomers of cryAB may be specifically targeted for packaging into endosomal vesicles destined for secretion as exosomes.
Additionally, O-GlcNAcylation reduced levels of phosphorylated cryAB [21]. Association of native cryAB with detergent resistant microdomains in cells [13] and its stress-induced translocation to insoluble fraction of cell lysates with O-GlcNAc modification [21] suggests that O-GlcNAcylation is essential for its association with raft-like microdomains in the endosomal vesicles. It is not known whether O-GlcNAc modification modulates the dynamics of cryAB oligomerization. However, since preventing O-GlcNAcylation suppresses cryAB’s exosomal secretion and the assumption that cryAB secretion via exosomes depends on the non-phosphorylated protein being assembled into higher order oligomers, one could speculate about the role of O-GlcNAcylation in modulating the dynamics of cryAB oligomerization. The O-GlcNAc modification occurs on Thr170 in the flexible CTD of cryAB. This domain, as noted earlier, is responsible for stabilizing the hexameric structure of cryAB in its hierarchical assembly into larger ordered oligomers. Since O-GlcNAcylation is necessary for exosomal secretion of cryAB oligomers, it can be speculated that the O-GlcNAc modification probably stabilizes the formation of cryAB hexameric subunits by occurring on Thr170 in CTD domains, swapped between adjacent dimers, interacting with grooves in the ACD formed by β-strands. With the stabilization of the hexameric cryAB, the NTDs of multiple hexamers would then interact to form variable higher order oligomers.
The experimental evidence presented confirms that phosphorylation negatively regulates exosomal secretion of cryAB and O-GlcNAcylation at Thr170 is an essential modification required for cryAB secretion. It can be speculated that dephosphorylation and O-GlcNAcylation stabilize the oligomeric structure of cryAB ensuring its secretion via exosomes. These findings suggest a selective process of packaging O-GlcNAcylated and nonphosphorylated cryAB for exosomal secretion.
Based on published data on phosphorylation affecting oligomerization of cryAB and our experimental results, we propose a consolidated model to explain the packaging and secretion of cryAB via exosomes (Fig. 6). The dynamic state of cytoplasmic cryAB phosphorylation depends upon the cellular response to extracellular signals or the microenvironment. Phosphorylation of cryAB on Ser residues disrupts the NTD interactions that stabilize large cryAB oligomers. Disruption of these interactions results in unravelling of the oligomeric form into smaller oligomers or monomers. Phosphorylation at Ser19 leads to formation of cytosolic inclusions (I). However, with dephosphorylation and removal of negative phosphate charges from the hydrophobic NTD, cryAB coalesces into larger structured oligomers, where Thr170 in the flexible CTD may or may not be O-GlcNAcylated (II). The glycosylated and de-phosphorylated cryAB then gets selectively packaged into CD63 containing lipid rich MVEs which (III), with the assistance of Rab27 GTPases (IV), are then targeted for fusion with the cell membrane and subsequent release of ILVs as exosomes. However, a small amount of non-O-GlcNAcylatable cryAB was also found associated with CD63. One of the functions of MVEs is to sort protein cargo to lysosomes for degradation. It may be postulated that since preventing glycosylation hinders exosomal secretion of cryAB, the association of T170A mutant with CD63 may possibly be the process of marking endocytic vesicles containing non-O-GlcNAcylatable cryAB for degradation and sorting into MVEs fated for fusion with lysosomes (V).
Fig. 6.
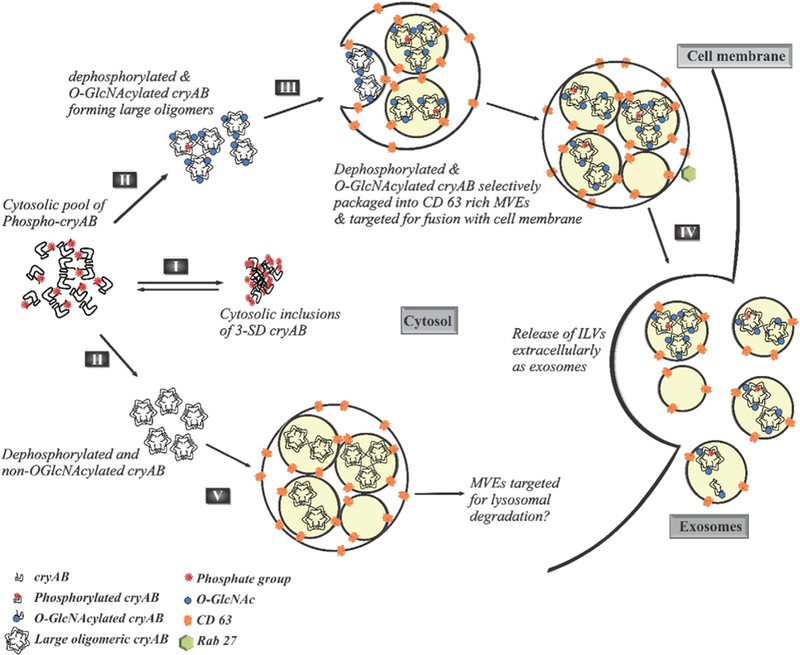
PTMs of cryAB dictate its exosomal secretion. Phosphorylation of cryAB at all its three Ser sites (l9, 45 & 59) result in formation of cytosolic inclusions (I). However, dephosphorylation of cryAB, which may or may not be coupled with O-linked glycosylation at Thr170, results in formation of larger structured oligomers (II). The glycosylated and nonphosphorylated oligomeric cryAB is selectively packaged into CD63 rich MVEs (III) which, with the assistance of Rab27, are then targeted for exocytosis (IV) and subsequent release of cryAB containing ILVs as exosomes. Some MVEs containing non-O-GlcNAcylatable cryAB are sorted for fusion with lysosomes (V).
The significance of cryAB phosphorylation and its role as an anti-apoptotic protein comes from a study which showed that phosphorylated cryAB inhibits tumor formation and growth in mice orthotopically implanted with nontumorigenic glioma cell line expressing phosphomimic cryAB mutant [2]. In GBM, cryAB levels are elevated [3,42–44]. Our recent study established that inflammatory cytokines increase levels of cryAB in astrocytes and secreted exosomes [15]. The anti-apoptotic function of cryAB [2,4] has been shown to be a contributing factor in the intense resistance to apoptosis observed during treatment of GBM. It may be argued that packaging of O-GlcNAcylated and nonphosphorylated cryAB for secretion via exosomes may provide target or recipient cells with a readily available, large pool of cryAB which may be modified according to the requirements of the target cell. The exosomal secretion of glycosylated and highly oligomeric nonphosphorylated cryAB by glioma cells in GBM may help confer resistance to apoptosis in surrounding cells following radiation and chemotherapy.
Supplementary Material
Acknowledgments
This study was supported by NIH/NEI Grant R01EY011352–17. We would like to thank Dr. Suraj Bhat from JSEI, UCLA for generously providing the U373 glioma cell line. We would like to acknowledge Jeffrey A. Kamykowski of UAMS Digital Microscopy Core for the electron microscopy images. We would also like to acknowledge the generous contribution of Dr. Fusan Kilic in providing aliquots of Abs against GM130 and Rab27 for our studies.
Footnotes
Transparency document
The Transparency document associated with this article can be found, in online version.
Appendix A. Supplementary data
Supplementary data to this article can be found online at http://dx.doi.org/10.1016/j.bbamcr.2015.11.027.
References
- [1].Iwaki T, Kumeiwaki A, Goldman JE, Cellular-distribution of alpha-B-crystallin in Non-lenticular tissues, J. Histochem. Cytochem. 38 (1990) 31–39. [DOI] [PubMed] [Google Scholar]
- [2].Stegh AH, Kesari S,Mahoney JE, Jenq HT, Forloney KL, Protopopov A, Louis DN, Chin L, DePinho RA, Bcl2L12-mediated inhibition of effector caspase-3 and caspase-7 via distinct mechanisms in glioblastoma, Proc. Natl. Acad. Sci. U. S. A. 105 (2008) 10703–10708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Goplen D, Bougnaud S, Rajcevic U, Boe SO, Skaftnesmo KO,Voges J, Enger PO,Wang J, Tysnes BB, Laerum OD, Niclou S, Bjerkvig R, AlphaB-crystallin is elevated in highly infiltrative apoptosis-resistant glioblastoma cells, Am. J. Pathol. 177 (2010) 1618–1628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Kamradt MC, Chen F, Sam S, Cryns VL, The small heat shock protein alpha B-crystallin negatively regulates apoptosis during myogenic differentiation by inhibiting caspase-3 activation, J. Biol. Chem. 277 (2002) 38731–38736. [DOI] [PubMed] [Google Scholar]
- [5].Iwaki T, Wisniewski T, Iwaki A, Corbin E, Tomokane N, Tateishi J, Goldman JE, Accumulation of alpha-B-crystallin in central-nervous-system glia and neurons in pathological conditions, Am. J. Pathol. 140 (1992) 345–356. [PMC free article] [PubMed] [Google Scholar]
- [6].Celet B, Akman-Demir G, Serdaroglu P, Yentur SP, Tasci B, van Noort JM, Eraksoy M, Saruhan-Direskeneli G, Anti-alpha B-crystallin immunoreactivity in inflammatory nervous system diseases, J. Neurol. 247 (2000) 935–939. [DOI] [PubMed] [Google Scholar]
- [7].Stoevring B, Vang O, Christiansen M, (Alpha)B-crystallin in cerebrospinal fluid of patients with multiple sclerosis, Clin. Chim. Acta; Int. J. Clin. Chem. 356 (2005) 95–101. [DOI] [PubMed] [Google Scholar]
- [8].Steiner-Champliaud MF, Sahel J, Hicks D, Retinoschisin forms a multi-molecular complex with extracellular matrix and cytoplasmic proteins: interactions with beta2 laminin and alphaB-crystallin, Mol. Vis. 12 (2006) 892–901. [PubMed] [Google Scholar]
- [9].Sreekumar PG, Kannan R, Kitamura M, Spee C, Barron E, Ryan SJ, Hinton DR, AlphaB crystallin is apically secreted within exosomes by polarized human retinal pigment epithelium and provides neuroprotection to adjacent cells, PloS One 5 (2010) e12578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Bajramovic JJ, Plomp AC, Goes A, Koevoets C, Newcombe J, Cuzner ML, van Noort JM, Presentation of alpha B-crystallin to T cells in active multiple sclerosis lesions: an early event following inflammatory demyelination, J. Immunol. 164 (2000) 4359–4366. [DOI] [PubMed] [Google Scholar]
- [11].Chou YK, Burrows GG, LaTocha D, Wang C, Subramanian S, Bourdette DN, Vandenbark AA, CD4 T-cell epitopes of human alpha B-crystallin, J. Neurosci. Res. 75 (2004) 516–523. [DOI] [PubMed] [Google Scholar]
- [12].Gonzales PA MG, Pisitkun T, Ruttenberg B, Knepper MA, Urinary Exosome Protein Database, in, NHLBI Laboratory of Kidney and Electrolyte Metabolism, 2009. [Google Scholar]
- [13].Gangalum RK, Atanasov IC, Zhou ZH, Bhat SP, AlphaB-crystallin is found in detergent-resistant membrane microdomains and is secreted via exosomes from human retinal pigment epithelial cells, J. Biol. Chem. 286 (2011) 3261–3269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Fitzner D, Schnaars M, van Rossum D, Krishnamoorthy G, Dibaj P, Bakhti M, Regen T, Hanisch UK, Simons M, Selective transfer of exosomes from oligodendrocytes to microglia by macropinocytosis, J. Cell Sci. 124 (2011) 447–458. [DOI] [PubMed] [Google Scholar]
- [15].Kore RA, Abraham EC, Inflammatory cytokines, interleukin-1 beta and tumor necrosis factor-alpha, upregulated in glioblastoma multiforme, raise the levels of CRYAB in exosomes secreted by U373 glioma cells, Biochem. Biophys. Res. Commun. (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Jehle S, Rajagopal P, Bardiaux B, Markovic S, Kuhne R, Stout JR, Higman VA, Klevit RE, van Rossum BJ, Oschkinat H, Solid-state NMR and SAXS studies provide a structural basis for the activation of alphaB-crystallin oligomers, Nat. Struct. Mol. Biol. 17 (2010) 1037–1042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Smith JB, Sun Y, Smith DL, Green B, Identification of the posttranslational modifications of bovine lens alpha B-crystallins by mass spectrometry, Protein Sci. : Public. Protein Soc. 1 (1992) 601–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Roquemore EP, Chevrier MR, Cotter RJ, Hart GW, Dynamic O-GlcNAcylation of the small heat shock protein alpha B-crystallin, Biochemistry 35 (1996) 3578–3586. [DOI] [PubMed] [Google Scholar]
- [19].Palminiello S, Jarzabek K, Kaur K, Walus M, Rabe A, Albertini G, Golabek AA, Kida E, Upregulation of phosphorylated alphaB-crystallin in the brain of children and young adults with down syndrome, Brain Res. 1268 (2009) 162–173. [DOI] [PubMed] [Google Scholar]
- [20].Launay N, Tarze A, Vicart P, Lilienbaum A, Serine 59 phosphorylation of {alpha}B-crystallin down-regulates its anti-apoptotic function by binding and sequestering Bcl-2 in breast cancer cells, J. Biol. Chem. 285 (2010) 37324–37332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Krishnamoorthy V, Donofrio AJ, Martin JL, O-GlcNAcylation of alphaB-crystallin regulates its stress-induced translocation and cytoprotection, Mol. Cell. Biochem. (2013). [DOI] [PubMed] [Google Scholar]
- [22].Kato K, Ito H, Kamei K, Inaguma Y, Iwamoto I, Saga S, Phosphorylation of alpha beta-crystallin in mitotic cells and identification enzymatic activities responsible for phosphorylation, J. Biol. Chem. 273 (1998) 28346–28354. [DOI] [PubMed] [Google Scholar]
- [23].Ito H, Kamei K, Iwamoto I, Inaguma Y, Nohara D, Kato K, Phosphorylation-induced change of the oligomerization state of alpha B-crystallin, J. Biol. Chem. 276 (2001) 5346–5352. [DOI] [PubMed] [Google Scholar]
- [24].Aquilina JA, Benesch JL, Ding LL, Yaron O, Horwitz J, Robinson CV, Phosphorylation of alphaB-crystallin alters chaperone function through loss of dimeric substructure, J. Biol. Chem. 279 (2004) 28675–28680. [DOI] [PubMed] [Google Scholar]
- [25].Ecroyd H, Meehan S, Horwitz J, A Aquilina J, Benesch JL, Robinson CV, Macphee CE, Carver JA, Mimicking phosphorylation of alphaB-crystallin affects its chaperone activity, Biochem. J. 401 (2007) 129–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Roquemore EP, Dell A, Morris HR, Panico M, Reason AJ, Savoy LA, Wistow GJ, Zigler JS Jr., Earles BJ, Hart GW, Vertebrate lens alpha-crystallins are modified by O-linked N-acetylglucosamine, J. Biol. Chem. 267 (1992) 555–563. [PubMed] [Google Scholar]
- [27].Bendtsen JD, Jensen LJ, Blom N, Von Heijne G, Brunak S, Feature-based prediction of non-classical and leaderless protein secretion, Protein Eng., Des. Sel. : PEDS 17 (2004) 349–356. [DOI] [PubMed] [Google Scholar]
- [28].Petersen TN, Brunak S, von Heijne G, Nielsen H, SignalP 4.0: discriminating signal peptides from transmembrane regions, Nat. Methods 8 (2011) 785–786. [DOI] [PubMed] [Google Scholar]
- [29].Bolte S, Cordelieres FP, A guided tour into subcellular colocalization analysis in light microscopy, J. Microsc. 224 (2006) 213–232. [DOI] [PubMed] [Google Scholar]
- [30].Lee JS, Kim HY, Jeong NY, Lee SY, Yoon YG, Choi YH, Yan C, Chu IS, Koh H, Park HT, Yoo YH, Expression of alphaB-crystallin overrides the anti-apoptotic activity of XIAP, Neuro-Oncology 14 (2012) 1332–1345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Vanlandingham PA, Ceresa BP, Rab7 regulates late endocytic trafficking downstream of multivesicular body biogenesis and cargo sequestration, J. Biol. Chem. 284(2009) 12110–12124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Ostrowski M, Carmo NB, Krumeich S, Fanget I, Raposo G, Savina A, Moita CF, Schauer K, Hume AN, Freitas RP, Goud B, Benaroch P, Hacohen N, Fukuda M, Desnos C, Seabra MC, Darchen F, Amigorena S, Moita LF, Thery C, Rab27a and Rab27b control different steps of the exosome secretion pathway, Nat. Cell Biol. 12 (2010) 11–13 [DOI] [PubMed] [Google Scholar]
- [33].Mathivanan S, Simpson RJ, ExoCarta: a compendium of exosomal proteins and RNA, Proteomics 9 (2009) 4997–5000. [DOI] [PubMed] [Google Scholar]
- [34].den Engelsman J, Gerrits D, de Jong WW, Robbins J, Kato K, Boelens WC, Nuclear import of {alpha}B-crystallin is phosphorylation-dependent and hampered by hyperphosphorylation of the myopathy-related mutant R120G, J. Biol. Chem. 280 (2005) 37139–37148. [DOI] [PubMed] [Google Scholar]
- [35].Pasta SY, Raman B, Ramakrishna T, Rao Ch M, The IXI/V motif in the C-terminal extension of alpha-crystallins: alternative interactions and oligomeric assemblies, Mol. Vis. 10 (2004) 655–662. [PubMed] [Google Scholar]
- [36].Hilton GR, Hochberg GK, Laganowsky A, McGinnigle SI, Baldwin AJ, Benesch JL, C-terminal interactions mediate the quaternary dynamics ofalphaB-crystallin, Phil-osophical transactions of the Royal Society of London, Ser. B, Biol. Sci. 368 (2013) [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Delbecq SP, Jehle S, Klevit R, Binding determinants of the small heat shock protein, alphaB-crystallin: recognition of the ‘IxI’ motif, EMBO J. 31 (2012) 4587–4594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Jehle S, van Rossum B, Stout JR, Noguchi SM, Falber K, Rehbein K, Oschkinat H, Klevit RE, Rajagopal P, AlphaB-crystallin: a hybrid solid-state/solution-state NMR investigation reveals structural aspects of the heterogeneous oligomer, J. Mol. Biol. 385 (2009) 1481–1497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Peschek J, Braun N, Rohrberg J, Back KC, Kriehuber T, Kastenmuller A, Weinkauf S, Buchner J, Regulated structural transitions unleash the chaperone activity of alphaB-crystallin, Proc. Natl. Acad. Sci. U. S. A. 110 (2013) E3780–E3789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Jehle S, Vollmar BS, Bardiaux B, Dove KK, Rajagopal P, Gonen T, Oschkinat H, Klevit RE, N-terminal domain of alphaB-crystallin provides a conformational switch for multimerization and structural heterogeneity, Proc. Natl. Acad. Sci. U. S. A. 108 (2011) 6409–6414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Delbecq SP, Klevit RE, One size does not fit all: the oligomeric states of alphaB crystallin, FEBS Lett. 587 (2013) 1073–1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Aoyama A, Steiger RH, Frohli E, Schafer R, von Deimling A, Wiestler OD, Klemenz R, Expression of alpha B-crystallin in human brain tumors, Int. J. Cancer. J. Int. du Cancer 55 (1993) 760–764. [DOI] [PubMed] [Google Scholar]
- [43].Iwaki T, Iwaki A, Miyazono M, Goldman JE, Preferential expression of alpha B- crystallin in astrocytic elements of neuroectodermal tumors, Cancer 68 (1991) 2230–2240. [DOI] [PubMed] [Google Scholar]
- [44].Hitotsumatsu T, Iwaki T, Fukui M, Tateishi J, Distinctive immunohistochemical profiles of small heat shock proteins (heat shock protein 27 and alpha B- crystallin) in human brain tumors, Cancer 77 (1996) 352–361. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.