
Fig. (3).
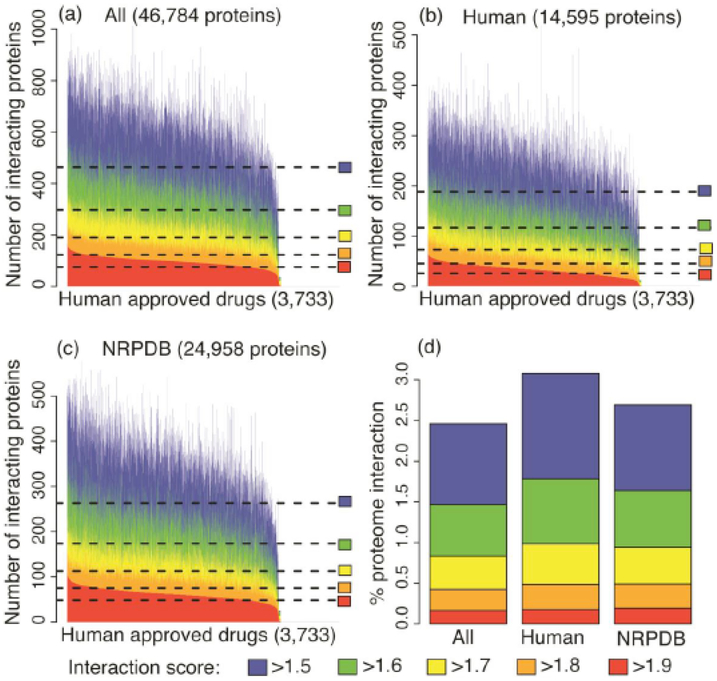
Extent of polypharmacology predicted by the CANDO platform. Panels (a)-(c) indicate the number of proteins predicted to interact with each of the 3733 human use drugs in the CANDO library for different universal proteome sets considered (All: 46,784 proteins; Human: 14,595 proteins; and NRPDB: 24,958 proteins) at different interaction score cutoffs (ranging from >1.5 to to >1.9; with scores ranging from 0-2.0). The data at the most stringest cutoffs (signifying the highest likelihood of an accurate prediction) indicate that the fractions of the proteome involved in strong interactions with our drug library is similar across all the sets considered. A two-sample Kolmogorov-Smirnov test, which compares two distributions with the null-hypothesis that they are not similar, was performed (the lower the p-value, the greater the likelihood of the null hypothesis) indicating that these distributions are very different at each score cutoff (All to Human p-value 2.2e-16 for all cutoffs and All to NRPDB p-value 2.2e-16 for cutoffs >1.9 through >1.6, and 11e-15 for the >1.5 cutoff). While a number of predicted interactions may be incorrect or may not be functionally important, the current data is what is used to obtain the benchmarking and prospective validation results obtained in [11, 15]. Taken together with those results, the above figures illustrate that a large number of protein structures with a broad distribution of the fold space with significant and specific predicted polypharmacology is necessary for making accurate predictions using our proteomic and evolutionary drug discovery and repurposing platform.