

Abstract
Bone cancer metastasis is extremely painful and decreases the quality of life of the affected patients. Available pharmacological treatments are not able to sufficiently ameliorate the pain and as cancer patients are living longer new treatments for pain management are needed. Decitabine (5-aza-2’-deoxycytidine), a DNA methyltransferases inhibitor, has analgesic properties in pre-clinical models of post-surgical and soft tissue oral cancer pain by inducing an up-regulation of endogenous opioids. In this study, we report that daily treatment with decitabine (2μg/g, i.p.) attenuated nociceptive behavior in the 4T1-luc2 mouse model of bone cancer pain. We hypothesized that the analgesic mechanism of decitabine involved activation of the endogenous opioid system through demethylation and reexpression of the transcriptionally silenced endothelin B receptor gene, Ednrb. Indeed, Ednrb was hypermethylated and transcriptionally silenced in the mouse model of bone cancer pain. We demonstrated that expression of Ednrb in the cancer cells lead to release of β-endorphin in the cell supernatant which reduced the number of responsive DRG neurons in an opioid-dependent manner. Our study supports a role of demethylating drugs, such as decitabine, as unique pharmacological agents targeting the pain in the cancer microenvironment.
Decitabine attenuates nociception in a mouse model of bone cancer pain through demethylation and re-expression of the endothelin B receptor gene in the cancer-microenvironment.
Introduction
Pain is a common, feared and debilitating complication of cancer [37]. Bone pain occurs in approximately 75% of patients with advanced cancers that have metastasized to bone [26,34]. One hypothesis to explain bone cancer pain is that nociceptors in the bone microenvironment are sensitized by tumor-released factors such as bradykinin, proteases, cytokines and endothelins [15,34,53]. Currently, there is a lack of effective treatment for bone cancer pain. Opioids are widely used as a therapeutic approach, but are associated with dose-limiting side effects [19]. Therefore, investigation of the underlying mechanisms of bone cancer pain is needed to develop new mechanistic-based treatments with fewer side effects.
One possible pharmacologic approach is inhibition of DNA methylation. Demethylating drugs, such as decitabine, are analgesic in oral cancer pain [67]. Decitabine (5-aza-2’-deoxycytidine), is an active cytosine analog, which when incorporated into newly synthesized DNA strands, inhibits DNA methyltransferases, causing DNA hypomethylation. It is a cytotoxic drug and clinically approved for the treatment of myelodysplastic syndromes [12]. Moreover, it has been reported to inhibit metastatic spread and cell proliferation in the 1833 xenograft model of breast cancer metastasis to bone [4]. However, the analgesic effect of decitabine on bone cancer pain has not been tested. We therefore hypothesized that decitabine has an anti-nociceptive effect in the 4T1-luc2 mouse model of bone cancer pain.
One of the genes known to be hypermethylated in cancers is the endothelin B receptor (ETBR) gene, EDNRB [68,71]. The ETBR and the endothelin A receptor (ETAR) mediate the effect of endothelin-1 (ET-1)[55], a 21-amino-acid peptide, which is overexpressed by several cancers metastasizing to the bone, including breast [51,73], lung [75] and prostate [40]. ET-1 has an algogenic effect in different nociceptive models [18,29,56,62], including the NCTC 2472 osteosarcoma model of bone cancer pain [44], through the activation of ETARs on the nociceptors innervating the bone [35]. Moreover, ET-1 can potentiate the effect of other pro-nociceptive mediators such as capsaicin and arachidonic acid via the ETAR [47]. Conversely, ET-1 activation of the ETBR produces analgesia [27,46,68]. ET-1 binding to ETBR leads to release of endogenous opioids, which activate opioid receptors on nociceptors [27,50,73]. ETBR is expressed by non-myelinating Schwann cells, dorsal root ganglia (DRG) satellite cells [44] and keratinocytes [27,28]. In non-malignant tissue, the ETBR-mediated analgesia opposes the nociception generated by activation of ETAR. However, the ETBR-mediated analgesic effect is lost in the cancer microenvironment due to low ETBR expression caused by DNA methylation of the promoter region of EDNRB [10,38,43,68,71]. We hypothesized that Ednrb is hypermethylated in the 4T1-luc2 mouse model of bone cancer and that decitabine would express Ednrb and activate the endogenous opioid system in the cancer-microenvironment, thereby producing an analgesic effect.
2. Methods
2.1. Animals
Adult (7–8 weeks old) female BALB/c mice weighing 16–20 g (Strain code: 028; Charles River Laboratories, Scanbur, Karlslunde, DK) were housed in a climate-controlled room (20–24°C) on a 12:12-hour light-dark cycle (light beginning at 07:00 hours). Female mice were used as the syngeneic cell line, 4T1-luc2, is a mammary carcinoma cell line and the model reflects metastatic breast cancer, which is most prevalent in women [24]. There were 5 mice in each individually ventilated cage (IVC, 523 cm2 floor space) containing Tapvei 2HV bedding (Harjumaa, EE). The mice had unrestricted access to food (Altromin 1314, Brogaarden, Lynge, DK) and water. Animals were acclimatized to the facility for one week before study initiation. The general health of the mice was checked regularly and the body weight was measured every other day. Experiments were approved by the New York University Committee on Animal Research, the New York University Institutional Animal Care and Use Committee, as well as the Danish Animal Experiments Inspectorate, The Danish Veterinary and Food Administration, Ministry of Environment and Food (license no. 2014–15-0201–00031 C4). Researchers were trained under the Animal Welfare Assurance Program at New York University. All procedures were conducted under the guidelines of the International Study of Pain [76] and performed in accordance with National Institutes of Health guidelines for the care and use of laboratory animals in research. All experiments and data analysis were blinded for the researchers.
2.2. Cell culture
Cancer cells:
Culture of the 4T1-luc2 cell line (Caliper, Teralfene, Belgium) was performed as previously described [16] in RPMI medium 1640 (Gibco, Waltham, MA, USA) supplemented with 10% charcoal-treated fetal bovine serum (FBS, Gibco, Waltham, MA, USA) and penicillin/streptomycin (50 U/mL, Gibco, Waltham, MA, USA) at 37°C in 5% CO2. The melanoma cell line, SK-MEL-28 (ATCC® HTB-72™), was cultured in Dulbecco’s Modification of Eagle’s Medium (DMEM, Gibco, Waltham, MA, USA) with the same supplements. Before cancer cell inoculation, 4T1-luc2 cells were rinsed with PBS (no Ca2+, no Mg2+), detached with 0.25% trypsin-EDTA, centrifuged for 3 min at 1000 × g and resuspended in RPMI 1640 medium to a final concentration of 106 cells/ml.
Viral transduction:
Mouse cDNA of Ednrb with a GFP-tagged c-terminal (Cat no. MG223067; OriGene, Rockville, MD, USA) was subcloned into the cytomegalovirus (CMV) shuttle plasmid Acc65I/EcoRV. Subcloning, recombination, amplification and purification were completed through ViraQuest Inc. (North Liberty, IA, USA). Adenovirus transduction of cells with adEdnrb or adGFP (VQAd CMV eGFP; ViraQuest Inc., North Liberty, IA, USA) was performed at 200 multiplicities of infection in RPMI with 2% FBS. After 24 hours, the transduction media was replaced with normal RPMI 1640 culture medium containing 10 % FBS, 5% penicillin/streptomycin.
Conditioned medium:
Collection of conditioned cell medium for calcium imaging was performed when cells in a 10-cm cell culture dish reached 70% to 80% confluence. Cells were washed with PBS (no Ca2+, no Mg2+) and 3 ml colorless culture medium (RPMI 1640 or DMEM) was added. After 48 hours incubation, conditioned medium was collected and frozen at −20˚C until use.
Treatment with decitabine:
When the 4T1-luc2 cells reached a confluence level of 60%, the culture medium was replaced with medium containing either 1μM or 5μM decitabine. After 48 h of decitabine treatment, cells were rinsed with PBS, trypsinized, and centrifuged; the cell pellet was snap frozen in liquid nitrogen. The in vitro doses were based on previous studies [66] and the results from the cell viability assay.
2.3. Induction of bone cancer
The bone cancer surgery was performed as previously described [16]. Briefly, mice were anesthetized with 2–4% isoflurane (Baxter A/S; 100%, Nomeco, Copenhagen, DK) followed by an intraperitoneal (i.p.) injection of a mixture of ketamine (85.5 mg/kg, Ketaminol vet, MSD, Copenhagen, DK) and xylazine (12.5 mg/kg, Rompun Vet., Bayer Animal Health, DK) in 0.9% NaCl. The mice were placed on a heating pad and eye ointment was applied (Ophtha A/S, Actavis group, Gentofte, DK). The femur was shaved with veterinary clippers, disinfected with 70% ethanol and swabbed with a PDI™ Povidone-Iodine Swabstick (Fisher Scientific Company, Pittsburgh, PA, USA). A superficial incision in the skin overlying the patellar tendon was made with scissors. The lateral side of the patellar tendon and retinaculum tendon were loosened, the patellar tendon was carefully pushed aside and the distal femoral epiphysis was exposed. Using a 30G needle, a hole was drilled in the distal epiphysis of the femur and 10μl medium containing 106 cells/ml or vehicle (cell-free RPMI 1640 medium) was injected into the medullary cavity. The hole was sealed with bone wax (Harvard Apparatus, Holliston, MA, USA); the wound was irrigated with a sterile 0.9% NaCl solution; the patellar was placed back in normal position; and, the wound closed with two Michel wound clips (7.5×1.75mm; AgnTho’s AB, Lidingö, SE). A lidocaine gel was applied on the wound (Xylocain gel, 2% lidocaine, AstraZeneca, Copenhagen, DK); 500 μl 0.9% sterile saline was injected subcutaneously (s.c.) to prevent dehydration and the mice were left to recover on a heating pad. For post-surgical analgesia mice were administered buprenorphine s.c. (0.03 mg/kg Temgesic, Indivior UK Limited, Berkshire, UK) before surgery and regularly for 48 hours post-surgery.
2.4. Administration of decitabine, pain behavior testing, in vivo imaging and euthanasia
The animals were introduced to the behavioral tests twice before baseline measurements. Behavioral tests and imaging were performed every second day from day 4 post-surgery. Mice were randomized into four groups. Decitabine (Sigma Aldrich, Saint Louis, MO, USA; Product #A3656) was dissolved in isotonic saline and administered i.p. (2μg/g body weight) every day (n=9) or every other day (q.a.d.; n=8), starting from day 4 post cancer-cell inoculation. Cancer-operated (n=9) and sham-operated (n=4) controls received vehicle treatment. Decitabine dosage was based on previous studies on BALB/c mice [30,67]. Decitabine was administered 1 hour before behavioral testing to allow the mice to acclimatize to the procedure room. Animals were euthanized by an overdose of isoflurane and cervical dislocation; thereafter, the femur was dissected, cleaned of connective tissue and snap frozen in liquid nitrogen. Humane endpoints were defined as complete lack of use of the cancer-bearing limb or weight loss above 20%.
Acute administration of naloxone methiodide: 22 mice were inoculated with cancer and 17 mice were administered decitabine daily (2μg/g body weight, i.p.); 5 mice were administered vehicle starting from day 4 post cancer inoculation. Animals were weighed and assessed daily to monitor general health and disease progression. On day 12 post cancer inoculation the mice were randomized according to the weight bearing and limb use scores of the day (baseline values) into two groups; naloxone methiodide (100 mg/kg, i.p., n=8) and saline (i.e. naloxone methiodide control, n=9). Naloxone methiodide or saline was injected by a blinded experimenter and behavioral tests were performed 15 and 45 minutes post injection. Following behavioral testing animals were euthanized by an overdose of isoflurane and cervical dislocation.
2.4.1. Behavioral analysis
Limb use:
All mice from one home cage were allowed to walk freely and acclimatize to a plastic cage without bedding for 10 min. The gait of each animal was thereafter individually observed for 3 min and the gait scored on a scale from 0–4, where 4 is normal use of the cancer-bearing limb; 3 is insignificant limping; 2 is significant limping; 1 is significant limping and partial lack of use of cancer-bearing limb; and 0 is total lack of use of the cancer-bearing limb [16].
Weight-bearing:
The weight-bearing ratio was measured using an incapacitance tester (Columbus Instruments, Columbus, OH, USA). The mouse was placed with the hind limbs on two separate scales on the incapacitance tester and the weight placed on each hind limb was measured for 4 sec. The measurements were performed in triplicates and the weight-bearing ratio calculated as the weight placed on the ipsilateral hind limb divided by the total weight put on both hind limbs.
2.4.2. Bioluminescent imaging
Nine minutes before bioluminescence imaging, the mice were administered with D-Luciferin (Caliper Life Sciences, Teralfene, Belgium) dissolved in PBS (150 mg/kg; i.p.). After 4.5 min mice were placed in an induction chamber and anesthetized with isoflurane (Baxter A/S; 100%, Nomeco, Copenhagen, DK). Bioluminescent images were captured in the Lumina XR instrument (Caliper Life Sciences, Teralfene, Belgium) and maintenance levels of isoflurane were delivered through a nose cone. The following image settings were used: binning: M(4), F/stop: 4 and exposure time from 10 seconds (sec) to 120 sec, according to signal count. Bioluminescence images were captured in triplicate and the animals were repositioned in-between each image. The bioluminescent signals were analyzed using IVIS Imaging Software (Living Image©, version 4.0.0.9801; Caliper Life Sciences, Teralfene, Belgium). The threshold for the region of interest (ROI) was set to 35% of the signal, and the readout was measured in radiance (photons/s).
2.4.3. X-ray imaging and calculation of relative bone density
X-ray densiometry was performed following bioluminescence imaging. Using ImageJ (NIH, USA) the gray scale value of a fixed ROI within the trabecular bone of the distal femur was measured and the average of two corresponding background regions in the tissue adjacent to the distal femur was subtracted. The gray-scale value was normalized to an aluminum wedge and used as an estimate of the relative bone density of the distal femur.
2.5. Quantitative real-time reverse transcription polymerase chain reaction (RT-qPCR)
Total RNA was isolated from fresh frozen tissue or 5×106 cells using the AllPrep DNA/RNA Mini Kits (Qiagen, Germantown, MD, USA) according to the manufacturer’s instructions. Tumor-bearing femurs were pulverized to a fine powder using liquid nitrogen, mortar and pestle and RNA extracted using TRIzol Reagent (Thermo Fisher, Rockford, IL, USA) according to the manufacturer’s manual. cDNA was synthesis was performed using qScript™ cDNA SuperMix (Quanta Quanta BioSciences, Gaithersburg, MD, USA). PCR reaction mixtures were prepared using PerfeCTa Fast Mix (Quanta Quanta BioSciences, Gaithersburg, MD, USA), followed by qRT-PCR on a Stratagene MX3005P (Agilent, Santa Clara, CA, USA). Primers were purchased from Integrated DNA Technologies (Coralville, IA, USA). The following primers were used: Ednrb forward: 5’-GGAATCACAGTGCTGAGTC-3’; reverse, 5’-AAAAGATGCCTTGATGCTATTGC-3’ [1]. Gapdh forward: 5’-CCCTTAAGAGGGATGCTGCC-3’; reverse: 5’- ACTGTGCCGTTGAATTTGCC-3’. Genes were considered ‘not expressed’ in the event that one sample failed to detect expression or the Ct was above 35. The melting curve was used to confirm that the correct gene was amplified. Expression levels were normalized to the housekeeping gene Gapdh. For each treatment three samples were analyzed. Relative quantification analysis of gene expression data was calculated using the 2−ΔΔCt method.
2.6. MethyLight Assay
Genomic DNA was isolated from tumor-bearing femurs or 5×106 cells using the AllPrep DNA/RNA Mini Kits (Qiagen, Germantown, MD, USA). Sodium bisulfite conversion was performed according to manufacturer’s recommendations, using the EZ DNA Methylation Gold Kit (Zymo Research, Irvine, CA, USA). Quantification of methylation levels was performed as previously described [7,42,64]. MethyLight primers and the probe for the promoter region of Ednrb were designed specifically for the bisulfite converted DNA sequence with Beacon Designer (Premier Biosoft, Palo Alto, CA, USA). The primers and the probe were designed to examine methylation levels at 5 CpG island sites (underlined in primer and probe sequence). Ednrb forward: 5’-TAAATACATTAACGTATACCCTTAACCG-3’; Reverse: 5’-TTTTAGTTTTGTCGTAGATTAGTCGG-3’; Probe: 6FAM-5’-AAATACCTTCACTCCGAATCCCCAAACTT-TAMRA. Lhx1 does not contain any CpG sites and was used as the internal control gene as previously described [64]. Lhx1 forward: 5’-AGAGTGTTTGGAAGTTAGGTGAAGGT-3’; reverse: 5’-CACATTCATAAACACAAATTCACACAAC-3’; Probe: 6FAM-5’-CACAATCAACATCCCAAACATATTCAC CCA-3’-Cy5. The total PCR reaction volume was 30 μL/well and the well mixture contained 10 μL DNA, 15.5 μL TaqMan™ Universal Master Mix II, no UNG (Cat no.4324018, Life Technologies, Carlsbad, CA, USA), 4.5 μL of OligoMix (forward and reverse primer concentrations: 2 μM; Probe concentration: 0.67 μM), and 0.5 μL H2O. PCR reactions were carried out as follows: one cycle at 95°C for 10 min followed by 50 cycles at 95°C for 15 s, and 60°C for 1 min. Universal Methylated Mouse DNA Standard (Zymo Research, Irvine, CA, USA) was used as positive control and water as negative control. Samples were run in duplicate. The percentage of methylated reference (PMR), i.e., degree of methylation, was calculated as previously described [70]: 100 × (Ednrb/Lhx1)sample/Ednrb/Lhx1)Universal Methylated DNA standard, for each sample. The gene is classified as fully methylated if the PMR is above 10.
2.7. Cell viability assay
Viability of proliferating cells was investigated in vitro using the CellTiter 96® AQueous Non-Radioactive Cell Proliferation Assay (MTS) (Promega, Nacka, Sweden). The assay was performed as described by the manufacturer. For that purpose, 10,000 cells/well was plated in a 96-well-plate in 100 μL RPMI-1640 medium, without phenol red (Gibco, Waltham, MA, USA). After 24 hours of incubation at 37 °C with 5% CO2, medium was replaced by 100 μL of decitabine (dissolved in RPMI 1640) in increasing concentrations from 0.001 μM to 1000 μM. After 48 hours of incubation 20 μL MTS/PMS dye was added to each well and the plate incubated an additional 2 hours before absorbance was recorded at 490 nm. The MTS assay was performed two times, and all measurements were performed in triplicate. The EC50 values were calculated using GraphPad Prism® software, version 6 (GraphPad Software, La Jolla, CA, USA)
2.8. Enzyme-linked immunosorbent assay (ELISA) for β-endorphin and ET-1
Viral transduction:
Cells were seeded into 6-well tissue culture plates and cultured in 3 mL RPMI 1640 with 10% FBS and 5% pen/strep until 60% confluence. Cells were washed with PBS (no Mg2+, no Ca2+) and transduced with adEdnrb or adGFP at 200 multiplicities of infection (MOI) in RPMI 1640 with 2% FBS (no pen/strep). After 24 hours the transduction media was replaced with 1mL RPMI 1640 without phenol-red, FBS, penicillin or streptomycin.
Decitabine treatment:
Cells were seeded into 6-well tissue culture plates and cultured in 3 mL RPMI 1640 with 10% FBS and 5% penicillin/streptomycin; upon reaching 25% confluence, decitabine (5 μM) was added. When cells reached 90–100% confluence the decitabine containing media was replaced with 1mL RPMI without phenol-red, 10% FBS, 5% penicillin/streptomycin. Cells without decitabine treatment were used as controls.
Collection of conditioned medium:
After 4 hours or 48 hours of incubation the conditioned medium was collected, treated with 10 μl HALT Protease Inhibitor Cocktail (Pierce, Rockford, IL), vortexed and centrifuged at 20°C for 3 min at 2600 rpm and stored at −80°C until use.
Lysate:
Mammary tissue from female BALB/c mice was isolated as previously described [48]. 3×106 cells or isolated ~10 mg mammary tissue were lysed in 500 μl ice-cold RIPA buffer (Thermo scientific, Rockford, IL, USA) containing 5 μl HALT Protease Inhibitor Cocktail (Pierce, Rockford, IL). The lysate was centrifuged at 13,000 rpm for 15 min and the supernatant removed, aliquoted and stored at −80°C until use.
The BCA protein assay (Pierce, Rockford, IL) was used to normalize protein concentration before performing the ELISA. ELISA was performed to detect beta-endorphin (MD Biosciences, St. Paul, MN) and endothelin-1 (Enzo, Farmingdale, NY, USA) according to the manufacturer’s protocols.
2.9. Primary culture of DRG neurons
Naïve female BALB/c mice were anesthetized with isoflurane and transcardially perfused with cold Hanks balanced salt solution (no Ca2+, no Mg2+) (Invitrogen, Carlsbad, CA, USA). Bilateral DRGs from L3-L5 were dissected, enzymatically treated, mechanically dissociated and cultured as previously described in the protocol by Malin and colleagues [33]. Neurons were plated on precoated poly-L-ornithine (Sigma-Aldrich, Saint Louis, MO, USA) coverslips and 2 hours later 2 ml DMEM media (Gibco, Waltham, MA, USA) containing 10% FBS was added. Calcium imaging was performed within 8 hours after DRG harvest.
2.10. Calcium imaging
DRG neurons were incubated with 5 μM Ca2+ indicator fura-2 AM (Invitrogen, Carlsbad, CA, USA) at room temperature for 30 min in DMEM without phenol red (colorless). Coverslips were placed in a recording chamber and continuously infused with colorless DMEM at room temperature. To measure the neuronal response to the cancer cell line supernatant, conditioned medium was applied for 90 sec followed by a 90 sec wash with colorless DMEM. In the naloxone experiment, DRG neurons were exposed to conditioned medium from 4T1-luc2 cells transduced with adGFP or adEdnrb for 30 sec, DMEM for 90 sec, 10 μM naloxone (Sigma-Aldrich, Saint Louis, MO, USA) dissolved in DMEM for 3 min and conditioned medium for 30 sec. To test for cell viability, 30 mM KCl was applied for 4 sec at the end of each experiment. Conditioned medium from cancer cells, naloxone and KCl were applied with a fast-step perfusion system (switching time <20 ms; Warner Instrument Co, Model SF-77B). A change in intracellular Ca2+ concentration ([Ca2+]i) ≥ 30% of baseline was considered as a response to treatment. The magnitude of the response was calculated as (Peak [Ca2+]i) – (Baseline [Ca2+]i). Fluorescence data was acquired by a Nikon Eclipse Ti microscope at 340 nm and 380 nm excitation wavelengths and analyzed with the TI Element Software (Nikon, Melville, NY, USA). [Ca2+]i was determined from fura-2AM ratio. Calibration of imaging software to convert Fura-2 ratio following in situ calibration experiments was performed as previously described [21,52].
2.14. Statistical analysis
GraphPad Prism (version 7; GraphPad Software, CA, USA) was used for statistical analyses. A confidence interval of 95% was chosen as a measure of statistical significance, p<0.05. The asterisks denote the following level of significance: * P < 0.05; ** P < 0.01; *** P < 0.001; **** P < 0.0001. Data are displayed as means and standard error of the mean (±SEM). Behavioral data were analyzed by a Two-way repeated-measures (RM) analysis of variance (ANOVA) followed by Bonferroni’s multiple comparison test. The difference in the number of mice that experienced a decrease in limb use score in the acute naloxone methiodide group were compared with the saline group using an χ2 test (Excel, Microsoft, WA, USA). Animals with an initial limb use score ≤ 1 were excluded from the limb use and weight-bearing analyses (n=1). In vitro data were analyzed using Student t test or one-way ANOVA followed by Bonferroni’s post-hoc test, as appropriate. The last-measurement carried forward approach was used for data analyses, i.e., an animal reaching a humane endpoint was euthanized and the last measurements carried forward.
3. Results
3.1. Chronic decitabine treatment attenuated nociceptive behavior in the 4T1-luc2 mouse model of bone cancer pain
Bone cancer-bearing animals treated with vehicle exhibited a significant increase in nociceptive behavior, as measured by limb use score (Two-way RM ANOVA, P < 0.0001; Figure 1A) and weight-bearing ratio (P < 0.0001; Figure 1B) 10 days post inoculation compared with the sham-operated control group. Cancer-bearing mice treated daily with decitabine (2 μg/g, i.p.) exhibited significantly less nociceptive behavior compared with the cancer-bearing mice that received vehicle on days 10 to 14 ( P < 0.0001), and no significant difference was observed between the daily decitabine-treated group and the sham-operated vehicle-treated control group on any of the test days (P > 0.05).
Figure 1:

Daily administration of 2μg/g decitabine attenuated the development of bone cancer nociception. A) Daily decitabine administration (2 μg/g, i.p.) significantly decreased nociceptive behavior compared with the cancer vehicle group (P < 0.0001) in the limb use test on days 10 to 14. B) The weight-bearing ratio of the daily decitabine-treated group differed significantly from the cancer vehicle group on days 10 to 14 (P < 0.0001). C) Daily administration of 2μg/g decitabine produced significant weight loss compared with the three other groups (P < 0.0001). Data were analyzed using a Two-way repeated-measurement ANOVA followed by Bonferroni’s multiple comparison test. n=8–9, # = Cancer vehicle vs Sham Vehicle; * = Cancer Vehicle vs. Cancer 2μg/g decitabine daily; ¤ = Cancer Vehicle vs. Cancer 2μg/g decitabine q.a.d; $ = Cancer 2μg/g decitabine q.a.d vs. Cancer 2μg/g decitabine daily.
Treatment with decitabine (2 μg/g, i.p.) every other day (q.a.d.) significantly delayed the onset of nociceptive behavior compared with vehicle-treated controls, reflected by a significantly increased weight-bearing ratio (P < 0.001) and limb use score (P < 0.05), at 10 and 12 days post inoculation (Figure 1A,B). A significant difference was observed between daily and q.a.d. administration in both the limb use (P < 0.01) and weight-bearing (P < 0.05) tests from day 12. No adverse effects were observed in the vehicle and q.a.d. decitabine treatment groups but the mice treated with daily decitabine had significant weight loss towards the end of the experiment (Figure 1C).
3.2. Decitabine reduced cancer-induced bone degradation and bioluminescent signal in vivo
Bone cancer-bearing animals treated with vehicle displayed a decrease in relative bone density from day 8 post inoculation compared with sham-operated controls (Two-way RM ANOVA; P < 0.05; Figure 2A). However, daily decitabine treatment significantly attenuated the loss of relative bone density on day 12 (P < 0.05) compared with vehicle treatment. Furthermore, daily decitabine treatment also resulted in a significant increase in relative bone density on day 12 (P < 0.05) and day 14 (P < 0.01) compared with q.a.d. decitabine treatment (Figure 2A,B).
Figure 2:
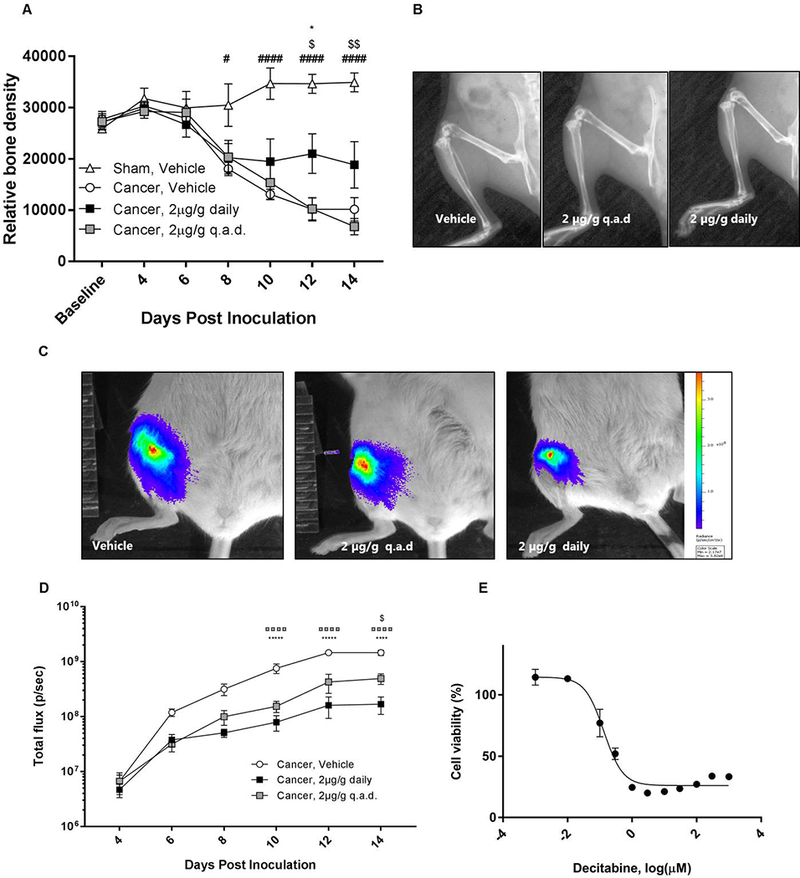
Decitabine reduced bone degradation, inhibited cell proliferation of 4T1-luc2 cells in vitro and reduced the bioluminescent signal from the 4T1-luc2-bearing mice. A) Relative bone density was calculated from x-ray images captured every other day. Daily decitabine treatment decreased cancer-induced bone degradation. B) Examples of x-ray images from the three cancer-bearing groups 12 days post inoculation. C) Examples of bioluminescent imaging data from the three cancer-bearing groups 12 days post inoculation. D) The bioluminescent signal in vivo increased during disease progression in all three cancer groups. E) 4T1-luc2 cells were treated with increasing concentrations of decitabine for 48 hours and the cell viability measured by a MTS assay. Data were normalized to non-treated controls (100% activity) and blank controls (wells without cells, 0% activity). Results were analyzed by Two-way repeated-measurement ANOVA followed by Bonferroni’s multiple comparison test. n=8–9, ¤ = Cancer Vehicle vs. Cancer 2μg/g decitabine q.a.d; # = Cancer vehicle vs Sham Vehicle; * = Cancer Vehicle vs. Cancer 2μg/g decitabine daily; $ = Cancer 2μg/g decitabine q.a.d vs. Cancer 2μg/g decitabine daily.
Decitabine treatment did not cause eradication of the tumor in any of the treatment groups; tumor burden was measured with bioluminescent imaging. However, both the daily and q.a.d. administration of decitabine resulted in significantly less bioluminescent signal detected in the femur on day 10, 12 and 14 (Two-way RM ANOVA; P < 0.0001; Figure 2C,D). Additionally, on day 14 the bioluminescent signal from daily decitabine treatment was significantly less compared with q.a.d. treatment (P < 0.05).
To study if decitabine had an inhibiting effect on tumor proliferation, 4T1-luc2 cells were treated with increasing concentrations of decitabine in vitro for 48 hours and the metabolic activity of the cells were measured with a MTS assay. There was a significant dose-dependent decrease in cell viability in response to decitabine (Figure 2E). The IC50 value for the 4T1-luc2 cell line in the MTS assay was 0.133 ± 1.19 μM.
3.3. Decitabine treatment increased Ednrb mRNA expression in the 4T1-luc2 cell line and decreased Ednrb methylation in cancer-bearing femurs
We sought to investigate if hypermethylation of Ednrb leads to transcriptional silencing of the gene in the 4T1-luc2 model of bone cancer pain and if demethylation with decitabine would re-express Ednrb mRNA. Methylation of Ednrb in the 4T1-luc2 cell line was determined using a MethyLight assay and compared with isolated mammary gland tissue as a control. In the 4T1-luc2 cell line, Ednrb was significantly more methylated than in mammary gland tissue (Unpaired, Student’s t test, P < 0.05; Figure 3A), and the 4T1-luc2 cell line expressed significantly less Ednrb mRNA compared with the control mammary gland tissue (Unpaired, Student’s t-test, P < 0.01; Figure 3B).
Figure 3:
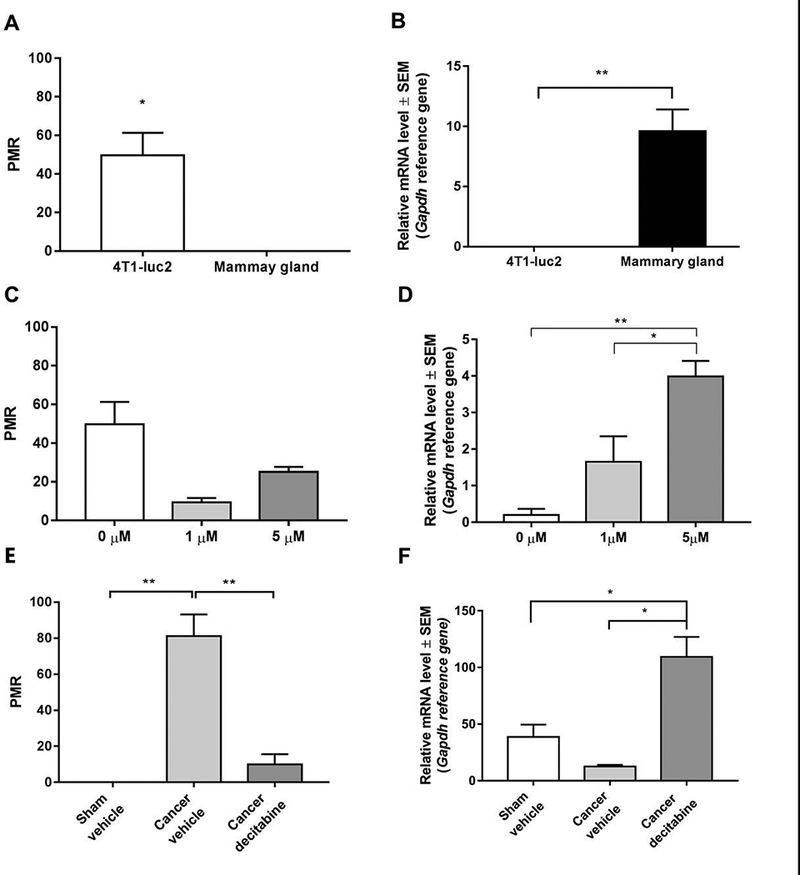
Ednrb is methylated and transcriptionally silenced in 4T1-luc2 cells which can be reversed by decitabine treatment A) The promoter region of Ednrb is hypermethylated in the 4T1-luc2 cell line compared with control mammary gland tissue. B) mRNA expression levels of Ednrb in the 4T1-luc2 cell line and normal mammary gland. C) Decitabine treatment does not demethylate Ednrb in the 4T1-luc2 cells. D) mRNA expression levels of Ednrb in the 4T1-luc2 mammary carcinoma cells after 48 hours of 1 μM and 5 μM decitabine treatment compared with non-treated controls. F) Decitabine treatment demethylates Ednrb in vivo. E) Decitabine treatment leads to an increase in Ednrb mRNA expression in vivo. The gene is classified as fully methylated if the percentage of methylated reference is above 10. n=2–5. * P < 0.5; ** P < 0.01; Unpaired Student’s t-test or one-way ANOVA, Bonferroni post hoc test.
To determine if decitabine treatment could demethylate Ednrb and increase the Ednrb mRNA levels in the 4T1-luc2 cell line, we treated the cells with 1 μM or 5 μM decitabine for 48 hours. Treatment with decitabine had no significant effect on Ednrb methylation levels between decitabine and vehicle-treated 4T1-luc2 cells (One-way ANOVA, P > 0.050; Figure 3C). However, treatment significantly increased Ednrb mRNA expression levels in the 4T1-luc2 cell line compared with non-treated controls (One-way ANOVA, P < 0.01; Figure 3D).
Based on the results from the cell line, we measured mRNA expression levels and degree of Ednrb methylation isolated from cancer-bearing femurs. Cancer-bearing femurs from mice treated daily with decitabine showed a significant decrease in degree of Ednrb methylation (P < 0.01; Figure 3E), compared with sham-operated controls and vehicle-treated cancer-bearing mice accompanied by a significant increase in Ednrb mRNA expression (One-way ANOVA; P < 0.05; Figure 3F).
3.4. Cancer cell-secreted mediators activate DRG neurons
To determine if mediators released from the 4T1-luc2 cells could directly activate dissociated lumbar DRG neurons, calcium imaging was used to quantify the evoked Ca2+ transient in response to conditioned medium from 4T1-luc2 cells compared to the non-painful melanoma cell line SK-MEL-28. Application of conditioned medium from 4T1-luc2 cells resulted in a significant increase in the percent of responsive DRG neurons (Unpaired Student’s t test, P < 0.01; Figure 4A,B) compared with application of conditioned medium from SK-MEL-28 (Figure 4B,C). However, there was no significant difference in the magnitude of the evoked Ca2+ transient in response to conditioned medium from SK-MEL-28 and 4T1-luc2 (Figure 4D).
Figure 4:
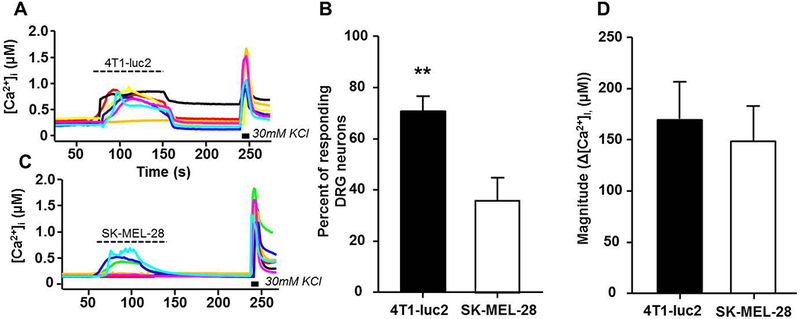
Conditioned medium (CM) from 4T1-luc2 cells activates significantly more DRG neurons compared with conditioned medium from the non-painful melanoma cell line SK-MEL-28. A) Examples of Ca2+ traces in DRG neurons before and after the application of 4T1-luc2 CM. B) Percentage of DRG neurons treated with CM from SK-MEL-28 cells and 4T1-luc2. Treatment with 4T1-luc2 CM resulted in significantly higher percent responders compared with SK-MEL-28 CM. C) Examples of Ca2+ traces in DRG neurons treated with CM from SK-MEL-28 cells. D) The magnitude of the response (Peak [Ca2+]i – Baseline [Ca2+]I, (μM)) remained unchanged between cell line CMs. A Ca2+change ≥ 30% of baseline was considered a “response” to treatment. n=3. ** P < 0.01, Unpaired Student’s t-test.
3.5. Conditioned medium from 4T1-luc2 cells contained ET-1 and adenoviral transduction with adEdnrb increased β-endorphin
To quantify the amount of ET-1 and β-endorphin in the conditioned medium from 4T1-luc2 cancer cells we used enzyme-linked immunosorbent assays (ELISA). The 4T1-luc2 cells synthesized and released increasing levels of ET-1 over time (One-Way ANOVA; P < 0.001; Figure 5A). After 48 hours conditioned medium from 4T1-luc2 cells contained significantly more ET-1 than conditioned medium from the non-painful melanoma cell line SK-MEL-28 (P < 0.01; Figure 5A). To test whether 4T1-luc2 cells release the endogenous opioid β-endorphin, conditioned medium from the 4T1-luc2 cell line was tested. β-endorphin was detectable in conditioned medium from 4T1-luc2 cells (Figure 5B). To investigate the role of Ednrb in the cancer cell line, we transduced 4T1-luc2 mammary carcinoma cells with an adenovirus expressing Ednrb (adEdnrb). Transduction of the 4T1-luc2 cell line with Ednrb significantly increased β-endorphin release to the media compared with non-transduced and control GFP-transduced cells (One-way ANOVA, P < 0.05; Figure 5B).
Figure 5:
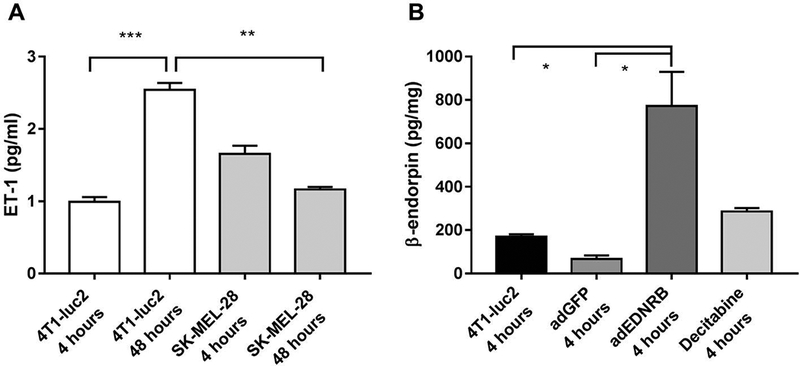
ELISA quantification of ET-1 and β-endorphin in conditioned medium from 4T1-luc2 cells, SK-MEL-28 cells as well as 4T1-luc2 cells transduced with Ednrb (adEdnrb) or GFP (adGFP). A) 4T1-luc2 cells released increasing levels of ET-1 over 48 hours. After 48 h, conditioned medium from 4T1-luc2 cells contained significantly more ET-1 compared with conditioned medium from the non-painful melanoma cell line SK-MEL-28. B) Conditioned medium from the 4T1-luc2 cell line transduced with Ednrb released significantly more β-endorphin, compared with non-transduced and GFP-transduced controls. P < 0.05. Results analyzed by One-Way ANOVA followed by Bonferroni’s post hoc comparisons. n=2–3.
3.6. Conditioned medium from 4T1-luc2 cells transduced with adEdnrb suppressed neuronal activation, and can be reversed by naloxone
To test if the adEdnrb-mediated increase in β-endorphin affected the 4T1-luc2 supernatant-evoked activation of DRG neurons, we performed calcium imaging and applied conditioned medium from adEdnrb and adGFP to the neurons. Transduction with adEdnrb caused a significant decrease in percentage of responsive DRG neurons (One-way ANOVA; P < 0.01; Figure 6A,B). Transduction with adGFP did not affect the percentage of responsive DRG neurons (Figure 6B,C), indicating that the transduction itself did not inflict significant changes in Ca2+ signaling. To test whether the adEdnrb-induced decrease in activated DRG neurons was due to β-endorphin signaling, the nonselective opioid receptor antagonist, naloxone (10 μM, 3 min) was applied (Figure 7A). Naloxone reversed the adEdnrb-induced suppression of evoked Ca2+ responses, seen as an increase in percent responsive DRG neurons (One-way ANOVA; P < 0.01; Figure 7B).
Figure 6:
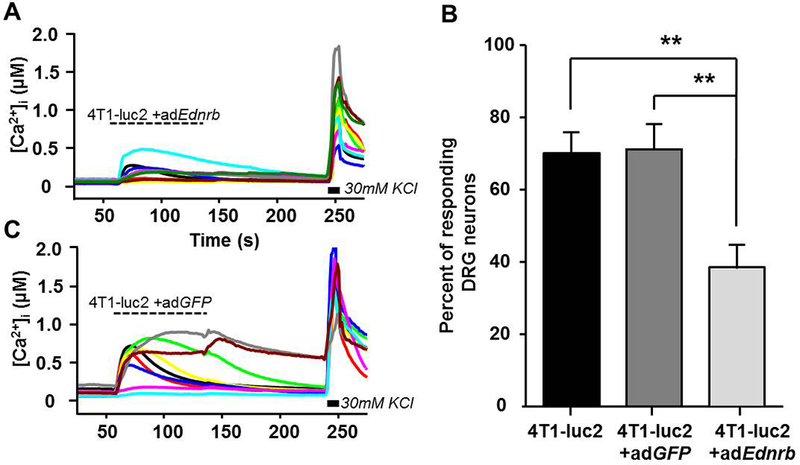
Fewer DRG neurons respond to supernatant from 4T1-luc2 cells with re-expressed Ednrb. A) Representative Ca2+ transients from DRG neurons before and after the application of supernatant from 4T1-luc2 cells transduced with adEdnrb B) Re-expression of Ednrb results in a significant decrease in percent of DRG neurons that respond to supernatant compared with adGFP and non-transduced conditioned medium. C) Examples of Ca2+ traces from DRG neurons before and after the application of supernatant from 4T1-luc2 cells transduced with adGFP. A Ca2+change ≥ 30% of baseline was considered a response to treatment. n=3. ** P < 0.01. Results analyzed by One-Way ANOVA, followed by Bonferroni post hoc comparisons.
Figure 7:
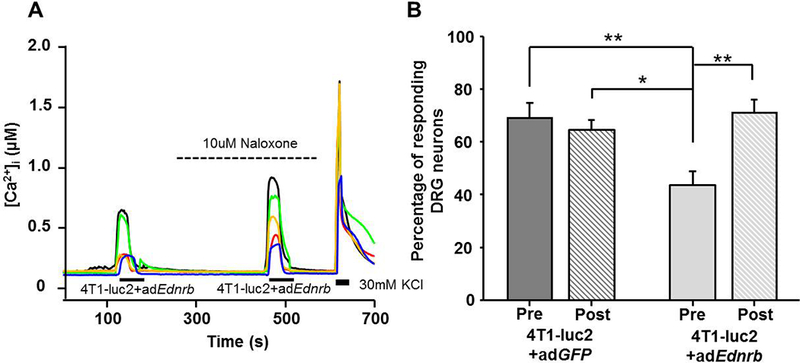
Naloxone reversed the adEdnrb-induced suppression of DRG response to cancer supernatant. A) Examples of Ca2+ traces in DRG applied cancer supernatant from 4T1-luc2 cells transduced with adEdnrb neurons before (pre) and after (post) the application of 10 μM naloxone B) Inhibition of opioid signaling with 10uM naloxone increased the percentage of Ca2+ responsive DRG neurons. n=3. ** P < 0.01. Results analyzed by Two-way ANOVA followed by Bonferroni’s post hoc comparisons.
3.7. Naloxone methiodide increased nociceptive behavior in dectabine-treated cancer-bearing mice
To test whether peripheral opioids, e.g. β-endorphin, that were released in response to decitabine, contributed to the anti-nociceptive effect observed in vivo, the selective peripherally acting opioid receptor antagonist, naloxone methiodide (100 mg/kg, i.p.), was administered on day 12 post cancer inoculation to bone cancer-bearing mice treated daily with decitabine (2 μg/g, i.p.)
Fifteen minutes post administration of naloxone methiodide 5 out of 7 mice treated daily with decitabine experienced a decrease in limb use score compared with 2 out of 9 in the saline administered decitabine-treated control group (χ2 test; P < 0.05; Figure 8A,B). There was no difference between the naloxone methiodide-administered group and the saline group in the weight-bearing test 15 minutes post injection (P > 0.05; Figure 8C). Treatment with naloxone methiodide did not decrease limb use score or weight-bearing ratio 45 minutes after drug administration compared with saline-treated control (Figure 8).
Figure 8:

Naloxone methiodide increased movement evoked nociception in decitabine-treated mice 12 days post inoculation. A) Administration of naloxone methiodide (100 mg/kg, i.p.) led to a decrease in limb use score in 5 out of 7 cancer-bearing mice treated daily with decitabine. B) In the saline-treated control group 2 out of 9 mice experienced a decrease in limb use score. C) Naloxone methiodide did not induce a significant difference in weight-bearing ratio between the naloxone methiodide-administered group and the saline group. Results were analyzed using an χ2 test.
4. Discussion
We demonstrated that decitabine attenuates nociceptive behavior in a mouse model of bone cancer. Pharmacological management of bone cancer pain primarily involves opioids; however, over 25% of patients with painful bone metastases experience inadequate pain relief [9,26,72]. In addition, opioids are associated with tolerance, abuse and adverse effects following activation of opioid receptors in the central nervous system (CNS) and gastrointestinal system [60]. Systemic administration of demethylating drugs has an anti-nociceptive effect in pre-clinical models of post-surgical [61] and soft tissue oral cancer pain [67] through activation of the endogenous opioid system. In our bone cancer model we propose that decitabine produces anti-nociception through a local secretion of opioids, inducing a direct anti-nociceptive effect. We hypothesized that the analgesic mechanism of decitabine involved demethylation of Ednrb [67,68]. We found that Ednrb is hypermethylated and transcriptionally silenced in the mouse model of bone cancer pain and that decitabine treatment demethylates Ednrb in vivo. We demonstrated that re-expression of Ednrb in the cancer cells lead to release of β-endorphin, which subsequently reduced the number of responsive DRG neurons in an opioid-dependent manner. Thus, the analgesia observed in this study of bone cancer pain could be due to decreased proliferation of the cancer, direct analgesic pharmacological effect or activation of endogenous analgesia.
Decitabine inhibits cancer proliferation; decreased bone cancer proliferation may explain its analgesic effect. Decitabine can induce growth retardation or apoptosis in vitro and in vivo in osteosarcoma cells [2] prostate cancer [11,49], multiple myeloma [32] and melanoma [3,31] through epigenetic reactivation of apoptotic genes and/or tumor suppressor genes or through direct DNA damage. We report an antineoplastic effect of decitabine using an in vitro proliferation assay. The cancer-induced bioluminescent signal was diminished compared to vehicle-treated controls; however, the signal increased throughout the 10 days of treatment, indicating that decitabine does not eradicate the tumor. Thus, the analgesic effect of decitabine is unlikely to be solely explained by its anti-proliferative effect. Radiographic imaging was performed to evaluate the effect of decitabine on bone mass. Pre-clinical data show that decitabine treatment decreases the invasiveness of bone metastasis and reduces osteolysis [4,36]. Bendinelli and colleagues demonstrated that decitabine treatment reduces expression of hepatocyte growth factor, which can substitute for macrophage-colony stimulating factor (M-CSF) in osteoclast formation, and thereby inhibit osteoclast differentiation and preserve bone volume [4]. We report a bone preserving effect in cancer-bearing animals treated daily with 2 μg/g decitabine, but not the every other day treatment paradigm. Administration of decitabine every other day had an anti-nociceptive effect in vivo even though the amount of bone degradation was similar to the vehicle-treated controls. This difference in nociceptive behavior suggests an analgesic effect of decitabine, possibly by activation of the endogenous opioid system.
Endogenous analgesia can be mediated through central pathways, for example, descending pain modulation [13], noxious stimulus-induced analgesia [17,20] and stress-induced analgesia [65]. Beyond expression in the CNS, μ-, δ- and κ- opioid receptors are also expressed on peripheral terminals of sensory neurons [59]. Exclusive stimulation of opioid receptors located in the periphery can convey anti-nociception [6,41]. Based on previous reports of endogenous analgesia in cancer [50,67], we investigated mechanism of endogenous-dependent pain modulation in the bone cancer-microenvironment. Endogenous opioid peptides, such as β-endorphin, dynorphin and enkephalins, are integral components of most tumors [74] and inhibit pancreatic and oral cancer pain [54,68] by suppressing transmission of the nociceptive input. β-endorphin induced thermal hypoalgesia in a murine model of osteosarcoma, which was suppressed by the peripherally acting non-selective opioid receptor antagonist, naloxone methiodide [6]. The role of endogenous analgesia through ETBR in keratinocytes was first described by Khodorova and colleagues [27,28]. ET-1 is released after injury and binds to ETBR on keratinocytes triggering release of β-endorphin. β-endorphin activates μ- and κ-opioid receptors on nociceptors to induce membrane hyperpolarization, which inhibits further ET-1 induced depolarization through ETAR activation. Painful carcinomas, e.g. prostate, bone and oral cancer produce high amounts of ET-1 compared with non-painful melanoma [39,44,45] and ET-1 is reported to induce breakthrough cancer pain [62]. Viet et al. demonstrated that oral squamous cell carcinomas, derived from keratinocytes, release high levels of ET-1 that activates ETBR and mediates release of β-endorphin [68] causing reduced nociceptive behavior in vivo. However, this mechanism has not been described in bone cancer pain following adenocarcinoma metastasis. In primary breast cancers, Ednrb mRNA expression levels vary [5,69,73]; however, very low levels are reported after the breast cancer has metastasized to bone [5]. We report a low level of Ednrb transcript in the 4T1-luc2 breast cancer cell line and demonstrate that through demethylation by decitabine, Ednrb transcript expression increased both in vitro and in the bone cancer metastasis model. Furthermore, our results using adenoviral Ednrb transduction suggest that overexpression of Ednrb in cancer cells leads to the release of β-endorphin in the cancer microenvironment. 4T1-luc2 cells transduced with adEdnrb evoked Ca2+ transients in significantly fewer DRG neurons in vitro and administration of the non-selective opioid receptor antagonist, naloxone, reversed that effect suggesting an opioid-mediated mechanism is inhibiting DRG activation. Altogether, our data suggest that the analgesic effects of decitabine treatment observed in vivo are due to re-expression of Ednrb through demethylation leading to release of β-endorphin in the cancer microenvironment acting on primary afferent neurons to decrease nociceptive behavior.
We investigated whether a local increase in opioid secretion contributed to the anti-nociceptive effect of decitabine. Acute administration of naloxone methiodide led to a subtle increase in movement-evoked nociception but not weight-bearing induced nociception. Our result is supported by previous work that demonstrated that the limb use test is more sensitive than the weight-bearing test for the measurement of nociception in animal models of bone cancer pain [14,22,23]. Another explanation may be that the two tests measure different components of cancer-induced bone pain mediated by different mechanisms with different opioid sensitivity [25]. The limb use test might be more sensitive to opioids compared with weight-bearing induced nociception. Our in vivo and in vitro results support a possible role of local opioid secretion; however, the incomplete reversal of anti-nociception following naloxone methiodide indicates that preserved bone mass and reduced tumor viability, as demonstrated by a reduction in bioluminescent signal following daily decitabine administration, likely contribute to the anti-nociceptive effect.
Opioid agonists have been used to relieve post-operative pain after knee joint surgery by intraarticular injections [58]. The effect is mediated through local opioid receptors and can be countered by intraarticular naloxone [57–59]. Peripherally acting μ-opioid receptor antagonists such as methylnaltrexone bromide and naloxegol are used to alleviate opioid-induced constipation. The opioid receptor antagonists target μ-opioid receptors in the gastrointestinal tract without affecting analgesia [8]. In a 2-week, double-blind, randomized, placebo-controlled clinical study methylnaltrexone bromide did not induce changes in overall pain scores compared with the placebo [63]. Based on these studies we do not anticipate that the opioid receptor antagonists that target the gastrointestinal tract would impact on decitabine-mediated analgesia in the setting of bone cancer pain. Bone is the most common site of metastasis and bone pain is the most common cause of pain in cancer patients [15]. Cancer-induced bone pain is usually severe and hard to control. Demethylating drugs, such as decitabine, could lead the way for unique treatments targeting the pain in the cancer microenvironment. Decitabine is clinically available and being used or considered for different cancers. We have previously demonstrated that decitabine produces analgesia in a pre-clinical soft tissue oral cancer pain model. Here, we show that decitabine might have analgesic efficacy in a metastatic bone cancer through activation of an endogenous analgesic mechanism. As patients with metastatic bone cancer are living longer, decitabine may show promise for management of pain related to the malignancy.
Acknowledgement
The authors thank Ratna Veeramachaneni, Yogin Patel and Anders Bue Klein for their technical assistance. This work was supported by National Institutes of Health Grant R01DE025393 and travel grants from the Danish Cancer Society R153-A9693, the Oticon Foundation 16–1353, the Academic Guest House of the Bikuben foundation, and the Graduate School at the Faculty of Health and Medical Sciences, University of Copenhagen. The authors have no conflict of interests.
References
- [1].Akashi K, Saegusa J, Sendo S, Nishimura K, Okano T, Yagi K, Yanagisawa M, Emoto N, Morinobu A. Knockout of endothelin type B receptor signaling attenuates bleomycin-induced skin sclerosis in mice. Arthritis research & therapy 2016;18(1):113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Al-Romaih K, Somers GR, Bayani J, Hughes S, Prasad M, Cutz JC, Xue H, Zielenska M, Wang Y, Squire JA. Modulation by decitabine of gene expression and growth of osteosarcoma U2OS cells in vitro and in xenografts: identification of apoptotic genes as targets for demethylation. Cancer cell international 2007;7:14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Alcazar O, Achberger S, Aldrich W, Hu Z, Negrotto S, Saunthararajah Y, Triozzi P. Epigenetic regulation by decitabine of melanoma differentiation in vitro and in vivo. International journal of cancer Journal international du cancer 2012;131(1):18–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Bendinelli P, Maroni P, Matteucci E, Desiderio MA. Epigenetic regulation of HGF/Met receptor axis is critical for the outgrowth of bone metastasis from breast carcinoma. Cell death & disease 2017;8(2):e2578. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- [5].Bendinelli P, Maroni P, Matteucci E, Luzzati A, Perrucchini G, Desiderio MA. Microenvironmental stimuli affect Endothelin-1 signaling responsible for invasiveness and osteomimicry of bone metastasis from breast cancer. Biochimica et biophysica acta 2014;1843(4):815–826. [DOI] [PubMed] [Google Scholar]
- [6].Baamonde A, Lastra A, Juarez L, Garcia-Suarez O, Meana A, Hidalgo A, Menendez L. Endogenous beta-endorphin induces thermal analgesia at the initial stages of a murine osteosarcoma. Peptides 2006;27(11):2778–2785. [DOI] [PubMed] [Google Scholar]
- [7].Campan M, Weisenberger DJ, Trinh B, Laird PW. MethyLight. Methods in molecular biology (Clifton, NJ) 2009;507:325–337. [DOI] [PubMed] [Google Scholar]
- [8].Candy B, Jones L, Vickerstaff V, Larkin PJ, Stone P. Mu-opioid antagonists for opioid-induced bowel dysfunction in people with cancer and people receiving palliative care. The Cochrane database of systematic reviews 2018;6:CD006332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Caraceni A, Hanks G, Kaasa S, Bennett MI, Brunelli C, Cherny N, Dale O, De Conno F, Fallon M, Hanna M, Haugen DF, Juhl G, King S, Klepstad P, Laugsand EA, Maltoni M, Mercadante S, Nabal M, Pigni A, Radbruch L, Reid C, Sjogren P, Stone PC, Tassinari D, Zeppetella G. Use of opioid analgesics in the treatment of cancer pain: evidence-based recommendations from the EAPC. The Lancet Oncology 2012;13(2):e58–68. [DOI] [PubMed] [Google Scholar]
- [10].Chen C, Wang L, Liao Q, Huang Y, Ye H, Chen F, Xu L, Ye M, Duan S. Hypermethylation of EDNRB promoter contributes to the risk of colorectal cancer. Diagnostic pathology 2013;8:199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Chiam K, Centenera MM, Butler LM, Tilley WD, Bianco-Miotto T. GSTP1 DNA methylation and expression status is indicative of 5-aza-2’-deoxycytidine efficacy in human prostate cancer cells. PloS one 2011;6(9):e25634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Chuang JC, Yoo CB, Kwan JM, Li TW, Liang G, Yang AS, Jones PA. Comparison of biological effects of non-nucleoside DNA methylation inhibitors versus 5-aza-2’-deoxycytidine. Molecular cancer therapeutics 2005;4(10):1515–1520. [DOI] [PubMed] [Google Scholar]
- [13].D’Mello R, Dickenson AH. Spinal cord mechanisms of pain. British journal of anaesthesia 2008;101(1):8–16. [DOI] [PubMed] [Google Scholar]
- [14].Diaz-delCastillo M, Christiansen SH, Appel CK, Falk S, Woldbye DPD, Heegaard AM. Neuropeptide Y is Up-regulated and Induces Antinociception in Cancer-induced Bone Pain. Neuroscience 2018;384:111–119. [DOI] [PubMed] [Google Scholar]
- [15].Falk S, Dickenson AH. Pain and nociception: mechanisms of cancer-induced bone pain. Journal of clinical oncology : official journal of the American Society of Clinical Oncology 2014;32(16):1647–1654. [DOI] [PubMed] [Google Scholar]
- [16].Falk S, Uldall M, Appel C, Ding M, Heegaard AM. Influence of sex differences on the progression of cancer-induced bone pain. Anticancer research 2013;33(5):1963–1969. [PubMed] [Google Scholar]
- [17].Ferrari LF, Gear RW, Levine JD. Attenuation of activity in an endogenous analgesia circuit by ongoing pain in the rat. The Journal of neuroscience : the official journal of the Society for Neuroscience 2010;30(41):13699–13706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Ferreira SH, Romitelli M, de Nucci G. Endothelin-1 participation in overt and inflammatory pain. Journal of cardiovascular pharmacology 1989;13 Suppl 5:S220–222. [DOI] [PubMed] [Google Scholar]
- [19].Frost CO, Hansen RR, Heegaard AM. Bone pain: current and future treatments. Current opinion in pharmacology 2016;28:31–37. [DOI] [PubMed] [Google Scholar]
- [20].Gear RW, Aley KO, Levine JD. Pain-induced analgesia mediated by mesolimbic reward circuits. The Journal of neuroscience : the official journal of the Society for Neuroscience 1999;19(16):7175–7181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Grynkiewicz G, Poenie M, Tsien RY. A new generation of Ca2+ indicators with greatly improved fluorescence properties. The Journal of biological chemistry 1985;260(6):3440–3450. [PubMed] [Google Scholar]
- [22].Guedon JM, Longo G, Majuta LA, Thomspon ML, Fealk MN, Mantyh PW. Dissociation between the relief of skeletal pain behaviors and skin hypersensitivity in a model of bone cancer pain. Pain 2016;157(6):1239–1247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Hansen RR, Nielsen CK, Nasser A, Thomsen SI, Eghorn LF, Pham Y, Schulenburg C, Syberg S, Ding M, Stojilkovic SS, Jorgensen NR, Heegaard AM. P2X7 receptor-deficient mice are susceptible to bone cancer pain. Pain 2011;152(8):1766–1776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Hernandez RK, Wade SW, Reich A, Pirolli M, Liede A, Lyman GH. Incidence of bone metastases in patients with solid tumors: analysis of oncology electronic medical records in the United States. BMC cancer 2018;18:44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Kabadi R, Kouya F, Cohen HW, Banik RK. Spontaneous pain-like behaviors are more sensitive to morphine and buprenorphine than mechanically evoked behaviors in a rat model of acute postoperative pain. Anesthesia and analgesia 2015;120(2):472–478. [DOI] [PubMed] [Google Scholar]
- [26].Kane CM, Hoskin P, Bennett MI. Cancer induced bone pain. BMJ (Clinical research ed) 2015;350:h315. [DOI] [PubMed] [Google Scholar]
- [27].Khodorova A, Fareed MU, Gokin A, Strichartz GR, Davar G. Local injection of a selective endothelin-B receptor agonist inhibits endothelin-1-induced pain-like behavior and excitation of nociceptors in a naloxone-sensitive manner. The Journal of neuroscience : the official journal of the Society for Neuroscience 2002;22(17):7788–7796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Khodorova A, Navarro B, Jouaville LS, Murphy JE, Rice FL, Mazurkiewicz JE, Long-Woodward D, Stoffel M, Strichartz GR, Yukhananov R, Davar G. Endothelin-B receptor activation triggers an endogenous analgesic cascade at sites of peripheral injury. Nature medicine 2003;9(8):1055–1061. [DOI] [PubMed] [Google Scholar]
- [29].Klass M, Hord A, Wilcox M, Denson D, Csete M. A role for endothelin in neuropathic pain after chronic constriction injury of the sciatic nerve. Anesthesia and analgesia 2005;101(6):1757–1762. [DOI] [PubMed] [Google Scholar]
- [30].Lemaire M, Chabot GG, Raynal NJ, Momparler LF, Hurtubise A, Bernstein ML, Momparler RL. Importance of dose-schedule of 5-aza-2’-deoxycytidine for epigenetic therapy of cancer. BMC cancer 2008;8:128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Liu QY, Chen DW, Xie LP, Zhang RQ, Wang HZ. Decitabine, independent of apoptosis, exerts its cytotoxic effects on cell growth in melanoma cells. Environmental toxicology and pharmacology 2011;32(3):423–429. [DOI] [PubMed] [Google Scholar]
- [32].Maes K, De Smedt E, Lemaire M, De Raeve H, Menu E, Van Valckenborgh E, McClue S, Vanderkerken K, De Bruyne E. The role of DNA damage and repair in decitabine-mediated apoptosis in multiple myeloma. Oncotarget 2014;5(10):3115–3129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Malin SA, Davis BM, Molliver DC. Production of dissociated sensory neuron cultures and considerations for their use in studying neuronal function and plasticity. Nature protocols 2007;2(1):152–160. [DOI] [PubMed] [Google Scholar]
- [34].Mantyh P Bone cancer pain: causes, consequences, and therapeutic opportunities. Pain 2013;154 Suppl 1:S54–62. [DOI] [PubMed] [Google Scholar]
- [35].Mantyh PW. Bone cancer pain: from mechanism to therapy. Current opinion in supportive and palliative care 2014;8(2):83–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Matteucci E, Maroni P, Disanza A, Bendinelli P, Desiderio MA. Coordinate regulation of microenvironmental stimuli and role of methylation in bone metastasis from breast carcinoma. Biochimica et biophysica acta 2016;1863(1):64–76. [DOI] [PubMed] [Google Scholar]
- [37].Mercadante S Malignant bone pain: pathophysiology and treatment. Pain 1997;69(1–2):1–18. [DOI] [PubMed] [Google Scholar]
- [38].Nelson JB, Chan-Tack K, Hedican SP, Magnuson SR, Opgenorth TJ, Bova GS, Simons JW. Endothelin-1 production and decreased endothelin B receptor expression in advanced prostate cancer. Cancer research 1996;56(4):663–668. [PubMed] [Google Scholar]
- [39].Nelson JB, Hedican SP, George DJ, Reddi AH, Piantadosi S, Eisenberger MA, Simons JW. Identification of endothelin-1 in the pathophysiology of metastatic adenocarcinoma of the prostate. Nature medicine 1995;1(9):944–949. [DOI] [PubMed] [Google Scholar]
- [40].Nelson JB, Nguyen SH, Wu-Wong JR, Opgenorth TJ, Dixon DB, Chung LW, Inoue N. New bone formation in an osteoblastic tumor model is increased by endothelin-1 overexpression and decreased by endothelin A receptor blockade. Urology 1999;53(5):1063–1069. [DOI] [PubMed] [Google Scholar]
- [41].Obara I, Przewlocki R, Przewlocka B. Local peripheral effects of mu-opioid receptor agonists in neuropathic pain in rats. Neuroscience letters 2004;360(1–2):85–89. [DOI] [PubMed] [Google Scholar]
- [42].Ogino S, Kawasaki T, Brahmandam M, Cantor M, Kirkner GJ, Spiegelman D, Makrigiorgos GM, Weisenberger DJ, Laird PW, Loda M, Fuchs CS. Precision and performance characteristics of bisulfite conversion and real-time PCR (MethyLight) for quantitative DNA methylation analysis. The Journal of molecular diagnostics : JMD 2006;8(2):209–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Pao MM, Tsutsumi M, Liang G, Uzvolgyi E, Gonzales FA, Jones PA. The endothelin receptor B (EDNRB) promoter displays heterogeneous, site specific methylation patterns in normal and tumor cells. Human molecular genetics 2001;10(9):903–910. [DOI] [PubMed] [Google Scholar]
- [44].Peters CM, Lindsay TH, Pomonis JD, Luger NM, Ghilardi JR, Sevcik MA, Mantyh PW. Endothelin and the tumorigenic component of bone cancer pain. Neuroscience 2004;126(4):1043–1052. [DOI] [PubMed] [Google Scholar]
- [45].Pickering V, Jay Gupta R, Quang P, Jordan RC, Schmidt BL. Effect of peripheral endothelin-1 concentration on carcinoma-induced pain in mice. European journal of pain (London, England) 2008;12(3):293–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Piovezan AP, D’Orleans-Juste P, Souza GE, Rae GA. Endothelin-1-induced ET(A) receptor-mediated nociception, hyperalgesia and oedema in the mouse hind-paw: modulation by simultaneous ET(B) receptor activation. British journal of pharmacology 2000;129(5):961–968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Piovezan AP, D’Orleans-Juste P, Tonussi CR, Rae GA. Endothelins potentiate formalin-induced nociception and paw edema in mice. Canadian journal of physiology and pharmacology 1997;75(6):596–600. [PubMed] [Google Scholar]
- [48].Plante I, Stewart MK, Laird DW. Evaluation of mammary gland development and function in mouse models. Journal of visualized experiments : JoVE 2011(53). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Pulukuri SM, Rao JS. Activation of p53/p21Waf1/Cip1 pathway by 5-aza-2’-deoxycytidine inhibits cell proliferation, induces pro-apoptotic genes and mitogen-activated protein kinases in human prostate cancer cells. International journal of oncology 2005;26(4):863–871. [PubMed] [Google Scholar]
- [50].Quang PN, Schmidt BL. Peripheral endothelin B receptor agonist-induced antinociception involves endogenous opioids in mice. Pain 2010;149(2):254–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Ratna A, Das SK. Endothelin: Ominous Player in Breast Cancer. Journal of cancer clinical trials 2016;1(1). [PMC free article] [PubMed] [Google Scholar]
- [52].Scheff NN, Lu SG, Gold MS. Contribution of endoplasmic reticulum Ca2+ regulatory mechanisms to the inflammation-induced increase in the evoked Ca2+ transient in rat cutaneous dorsal root ganglion neurons. Cell calcium 2013;54(1):46–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Scheff NN, Ye Y, Bhattacharya A, MacRae J, Hickman DN, Sharma AK, Dolan JC, Schmidt BL. Tumor necrosis factor alpha secreted from oral squamous cell carcinoma contributes to cancer pain and associated inflammation. Pain 2017;158(12):2396–2409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Sevcik MA, Jonas BM, Lindsay TH, Halvorson KG, Ghilardi JR, Kuskowski MA, Mukherjee P, Maggio JE, Mantyh PW. Endogenous opioids inhibit early-stage pancreatic pain in a mouse model of pancreatic cancer. Gastroenterology 2006;131(3):900–910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Shihoya W, Nishizawa T, Okuta A, Tani K, Dohmae N, Fujiyoshi Y, Nureki O, Doi T. Activation mechanism of endothelin ETB receptor by endothelin-1. Nature 2016;537(7620):363–368. [DOI] [PubMed] [Google Scholar]
- [56].Smith TP, Haymond T, Smith SN, Sweitzer SM. Evidence for the endothelin system as an emerging therapeutic target for the treatment of chronic pain. Journal of pain research 2014;7:531–545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Stein C The control of pain in peripheral tissue by opioids. The New England journal of medicine 1995;332(25):1685–1690. [DOI] [PubMed] [Google Scholar]
- [58].Stein C, Comisel K, Haimerl E, Yassouridis A, Lehrberger K, Herz A, Peter K. Analgesic effect of intraarticular morphine after arthroscopic knee surgery. The New England journal of medicine 1991;325(16):1123–1126. [DOI] [PubMed] [Google Scholar]
- [59].Stein C, Lang LJ. Peripheral mechanisms of opioid analgesia. Current opinion in pharmacology 2009;9(1):3–8. [DOI] [PubMed] [Google Scholar]
- [60].Stone P, Minton O. European Palliative Care Research collaborative pain guidelines. Central side-effects management: what is the evidence to support best practice in the management of sedation, cognitive impairment and myoclonus? Palliative medicine 2011;25(5):431–441. [DOI] [PubMed] [Google Scholar]
- [61].Sun Y, Sahbaie P, Liang D, Li W, Shi X, Kingery P, Clark JD. DNA Methylation Modulates Nociceptive Sensitization after Incision. PloS one 2015;10(11):e0142046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Tang Y, Peng H, Liao Q, Gan L, Zhang R, Huang L, Ding Z, Yang H, Yan X, Gu Y, Zang X, Huang D, Cao S. Study of breakthrough cancer pain in an animal model induced by endothelin-1. Neuroscience letters 2016;617:108–115. [DOI] [PubMed] [Google Scholar]
- [63].Thomas J, Karver S, Cooney GA, Chamberlain BH, Watt CK, Slatkin NE, Stambler N, Kremer AB, Israel RJ. Methylnaltrexone for opioid-induced constipation in advanced illness. The New England journal of medicine 2008;358(22):2332–2343. [DOI] [PubMed] [Google Scholar]
- [64].Trinh BN, Long TI, Nickel AE, Shibata D, Laird PW. DNA methyltransferase deficiency modifies cancer susceptibility in mice lacking DNA mismatch repair. Molecular and cellular biology 2002;22(9):2906–2917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Vachon-Presseau E, Martel MO, Roy M, Caron E, Albouy G, Marin MF, Plante I, Sullivan MJ, Lupien SJ, Rainville P. Acute stress contributes to individual differences in pain and pain-related brain activity in healthy and chronic pain patients. The Journal of neuroscience : the official journal of the Society for Neuroscience 2013;33(16):6826–6833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Viet CT, Dang D, Achdjian S, Ye Y, Katz SG, Schmidt BL. Decitabine rescues cisplatin resistance in head and neck squamous cell carcinoma. PloS one 2014;9(11):e112880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Viet CT, Dang D, Ye Y, Ono K, Campbell RR, Schmidt BL. Demethylating drugs as novel analgesics for cancer pain. Clinical cancer research : an official journal of the American Association for Cancer Research 2014;20(18):4882–4893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Viet CT, Ye Y, Dang D, Lam DK, Achdjian S, Zhang J, Schmidt BL. Re-expression of the methylated EDNRB gene in oral squamous cell carcinoma attenuates cancer-induced pain. Pain 2011;152(10):2323–2332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Watanabe J, Kaneko Y, Kurosumi M, Kobayashi Y, Sakamoto M, Yoshida MA, Akiyama M, Matsushima Y. High-incidence spontaneous tumors in JF1/Ms mice: relevance of hypomorphic germline mutation and subsequent promoter methylation of Ednrb. Journal of cancer research and clinical oncology 2014;140(1):99–107. [DOI] [PubMed] [Google Scholar]
- [70].Weisenberger DJ, Siegmund KD, Campan M, Young J, Long TI, Faasse MA, Kang GH, Widschwendter M, Weener D, Buchanan D, Koh H, Simms L, Barker M, Leggett B, Levine J, Kim M, French AJ, Thibodeau SN, Jass J, Haile R, Laird PW. CpG island methylator phenotype underlies sporadic microsatellite instability and is tightly associated with BRAF mutation in colorectal cancer. Nature genetics 2006;38(7):787–793. [DOI] [PubMed] [Google Scholar]
- [71].Welch A, Jacobs M, Wingo C, Cain B. Early progress in epigenetic regulation of endothelin pathway genes. British journal of pharmacology 2013;168(2):327–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].WHO. WHO’s cancer pain ladder for adults: World Health Organization. [Google Scholar]
- [73].Wulfing P, Diallo R, Kersting C, Wulfing C, Poremba C, Rody A, Greb RR, Bocker W, Kiesel L. Expression of endothelin-1, endothelin-A, and endothelin-B receptor in human breast cancer and correlation with long-term follow-up. Clinical cancer research : an official journal of the American Association for Cancer Research 2003;9(11):4125–4131. [PubMed] [Google Scholar]
- [74].Zagon IS, McLaughlin PJ, Goodman SR, Rhodes RE. Opioid receptors and endogenous opioids in diverse human and animal cancers. Journal of the National Cancer Institute 1987;79(5):1059–1065. [PubMed] [Google Scholar]
- [75].Zhang WM, Zhou J, Ye QJ. Endothelin-1 enhances proliferation of lung cancer cells by increasing intracellular free Ca2+. Life sciences 2008;82(13–14):764–771. [DOI] [PubMed] [Google Scholar]
- [76].Zimmermann M Ethical guidelines for investigations of experimental pain in conscious animals. Pain 1983;16(2):109–110. [DOI] [PubMed] [Google Scholar]