
Figure 4. Proteogenomics exploration for protein‐level detection of isoforms, single amino acid variants and alternative translation sites.
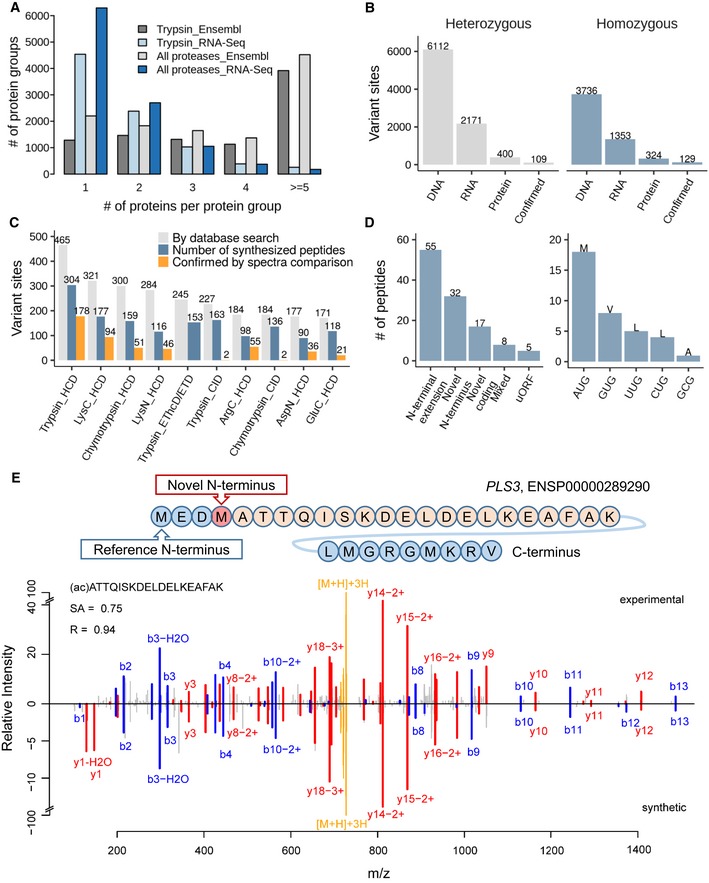
- Searching the tonsil proteomic data (trypsin alone or all enzymes) against a tissue‐specific sequence database constructed from RNA‐Seq data drastically reduces the number of individual protein sequences in protein groups compared to searches against Ensembl, allowing for the more efficient detection of protein isoforms.
- Number of single amino acid variants detected by whole exome sequencing and DNA, by RNA‐Seq at the mRNA and by mass spectrometry at the protein level as well as confirmed candidates by validation using synthetic peptide spectra comparisons. It is apparent that only a very small fraction of all variants detected at the DNA or RNA level can be detected at the proteome level using current technology.
- Analysis of which proteomic workflow contributed to the detection and confirmation of single amino acid variants.
- Results of the detection of non‐canonical coding regions using proteomics data (left panel) and different alternative start codons identified by acetylated N‐terminal peptides (right panel). The majority of cases are N‐terminal extensions of annotated genes. All but one of the detected alternative translation start sites correspond to point mutations of the first base of the classical AUG codon.
- Validation of a novel translation start site for the protein PLS3. The upper panel shows the novel translation site position within the amino acid sequence context, and the lower panel shows a mirror plot of the tandem mass spectra of the endogenous N‐terminally acetylated peptide (peaks pointing upwards) and the corresponding synthetic peptide spectrum (peaks pointing downwards). Y‐type sequence ions are coloured in red, b‐type ions in blue, and the intact peptide as well as neutral losses thereof are marked in yellow.