
Figure 1.
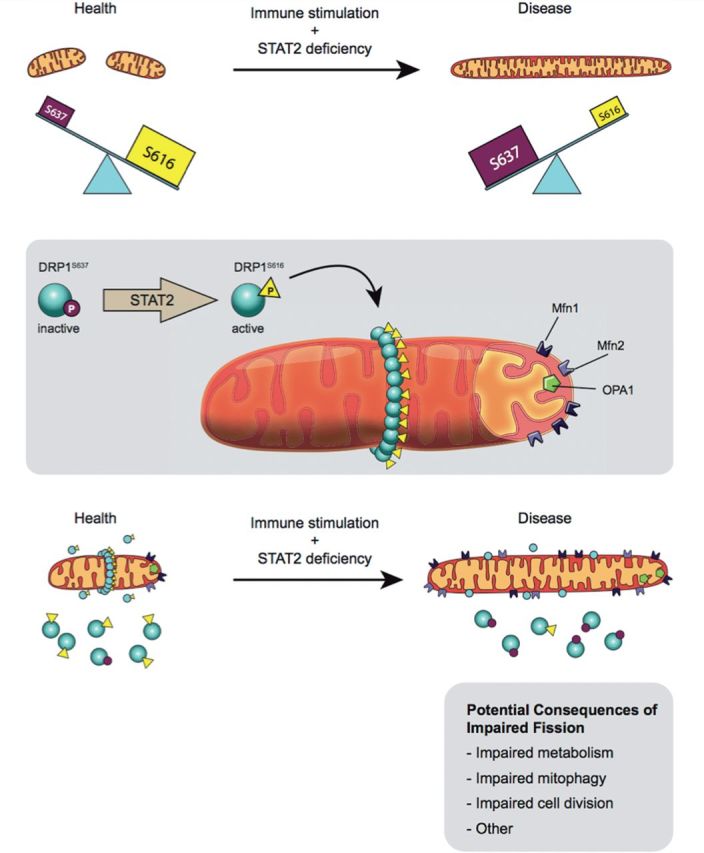
Proposed mechanism for induction of fusion as a consequence of STAT2 mutation. Top: Shahni et al. (2015) postulate that a non-canonical function of STAT2 is phosphorylation of DRP1 at serine 616, which causes a basal level of mitochondrial fission that is deemed to be beneficial. In the three children reported, a STAT2 mutation decreases STAT2 expression and eliminates DRP1 phosphorylation at serine 616, thereby creating elongated mitochondria. This pro-fusion environment caused by STAT2 mutation is compounded by an increase in the inhibitory form of DRP1, which is phosphorylated at serine 637. The details of the link to the immune system require further research, as does the mechanism by which mitochondrial hyperfusion leads to systemic illness or seizures. Middle: Normally, a basal level of fission is maintained by translocation of DRP1 phosphorylated at serine 616 to the outer mitochondrial membrane. Here, DRP1 multimerizes and, with its binding partners, forms the fission apparatus that divides the mitochondrion. The fusogenic GTPases, MFN1, MFN2 and OPA1 oppose fission. Bottom: DRP1 was present in the mitochondria of both patients and controls; however there was a deficiency of activated DRP1 serine 616 and a lack of fission in patient cells. The fusion mediators MFN1, MFN2 and OPA1 were upregulated in STAT2-deficient cells, thus it is likely that fusion was also disordered in these subjects. In addition, the patients showed increased levels of mitochondrial cytochrome c oxidase subunit II (MTCO2) and translocase of outer mitochondrial membrane 20 kDa (TOM20), suggestive of an increase in mitochondrial mass. Figure drawn by Julia Herr, M.Sc. Queen’s University.